
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effective removal of cesium by pristine graphene oxide: performance, characterizations and mechanisms†
Patcharanat Kaewmeea,
Jedsada Manyamb,
Pakorn Opaprakasit a,
Giang Thi Truc Lea,
Narong Chanlekc and
Paiboon Sreearunothai*a
a,
Giang Thi Truc Lea,
Narong Chanlekc and
Paiboon Sreearunothai*a
aSchool of Bio-Chemical Engineering and Technology, Sirindhorn International Institute of Technology (SIIT), Thammasat University, Pathum Thani 12121, Thailand. E-mail: paiboon_sree@siit.tu.ac.th
bNANOTEC, National Science and Technology Development Agency (NSTDA), 111 Thailand Science Park, Khlong Luang, PathumThani 12120, Thailand
cSynchrotron Light Research Institute, 111 University Avenue, Muang, Nakhon Ratchasima, 30000, Thailand
First published on 8th August 2017
Abstract
Radioactive Cs is a major by-product of nuclear power plants, with high radioactivity and long half-life. It is highly soluble in water and is difficult to remove. In this study, pristine graphene oxide (GO) synthesized via a Hummer's method has been demonstrated as a very efficient Cs sorbent with the maximum adsorption capacity of GO found to be 180, 465, 528 mg Cs/g sorbent at pH of 3, 7, and 12, respectively. The results from Fourier-transform infrared (FTIR) spectroscopy of GO before and after Cs sorption at various pH values reveal the mechanism of Cs sorption by GO. Several functional groups which are carboxyls, phenols, and hole defects containing multi-ether groups, are shown to play an important role in Cs capture. GO's affinity for other major cations found in seawater, namely, Na, K, and Mg was also evaluated, and the effect of these cations in competing with Cs for adsorption on GO was also studied. This reveals GO's exceptional ability in capturing Cs even in the presence of high concentrations of competitive cations and its high potential for use in Cs decontamination, as well as other heavy metal removal applications.
1. Introduction
Cesium is one of the main radioactive nuclides that was released following the Fukushima nuclear accident and is considered very hazardous due to its long half-life of 30 years and high radioactivity.1,2 It is also one of the major fission products generated during operation of most current nuclear power plants and has been involved in many nuclear accidents in the past.3 Much effort has been invested in cleaning up radioactive Cs from contaminated environments.4 Cesium is a group-I alkali metal and is highly soluble in water, making it difficult to remove from contaminated water.Several Cs sorbents with high specificity have already been extensively investigated, such as zeolites,5 calixarene,6,7 aluminum molybdophosphate,8 crystalline silico-titanate,9,10 and Prussian-blue type hexacyanoferrate.11 However, these sorbents such as those of calixarene and crown ethers are quite difficult and expensive to synthesize. Zeolites and clay minerals tend to have low to moderate sorption capacity. Prussian blue which shows good Cs sorption capacity, tends to decompose in an alkaline environment at a pH above 10.12 Crystalline silico-titanate sorbents also took long synthesis time mainly due to its highly crystalline structure.9 In view of this, simple and efficient sorbents that can function across a broad pH range are still needed.
Graphene oxide (GO), which can be easily synthesized from the oxidation of graphite, offers a high potential for use in adsorption applications. GO contains a large amount of oxygenated functional groups, such as epoxide, carbonyl, carboxyl, hydroxyl, and phenol groups and should therefore be ideal for sorption of metal cations.13,14 Its aromatic basal plane region is also suitable for trapping of hydrophobic organic molecules.15 Graphene oxide with a layered structure has been shown to have good ability to bind with many heavy metals, such as Pb(II), Cd(II), Cr(III),15–18 and several polyvalent radionuclides, such as Sr(II), Am(III), Pu(IV), and U(VI).19–21 GO demonstrates a very high ability to capture polyvalent cations with sorption capacity in the range of few mmol Cs/g.
The use of GO in capturing group-I alkali cations such as Cs(I) has also been demonstrated although typically in combination with other sorbent materials or with chemically modified GO.22–25 Sun et al. demonstrate the use of polyaniline-grafted GO to have Cs sorption capacity of about 1.39 mmol Cs/g of the composite sorbent. The high capacity was attributed to the grafting of polyaniline which reduced GO's aggregation. However, the grafting is well known to have also reduced the reactive sites of GO that can potentially be used for adsorption. The active sites on GO that can adsorb the cations may also serve as the active sites for grafting, or for binding with the composite material, which would decrease the number of useful sites for cation adsorption. GO has also been synthesized with Prussian blue (PB) and magnetic nanoparticles (MNP), where the iron of the PB or MNP were bound to the functional groups on the GO sheets.22–25 The Cs adsorption of such composite GO was usually shown to be quite small, on the order of a few tens of mg Cs/g sorbent which could be because of the blocking of Cs sorption sites upon composite's formation. In fact, pristine GO itself may have quite high ability to adsorb Cs, if its active functional groups are not occupied by grafting or by bonding with the composite materials of other compounds. The use of pristine GO without the complications of adding other compounds also makes the synthesis and large scale production of sorbent materials to be very simple and economical.
In this work, pristine graphene oxide prepared via a simple modified Hummer's method is demonstrated to possess a very high Cs sorption capacity of more than a few hundred mg Cs/g GO over a broad pH range. The GO was characterized and tested for physical and Cs adsorption properties using a range of techniques, which are XRD, SEM, FTIR, zeta potential analysis and ICP. FTIR spectra of GO sorbent taken before and after Cs sorption at various pHs help to reveal the mechanism of Cs sorption by GO. GO's affinity for other major cations found in seawater, namely, Na, K, and Mg were also evaluated, and the effect of these cations in competition with Cs was also studied both in simulated binary solution and also in seawater matrix.
2. Materials and methods
2.1 Materials
All reagents used in the synthesis were of analytical grade and used as received. Graphite powder was obtained from Acros Organics (synthetic origin, >99% purity). Graphene oxide was prepared according to that of the modified Hummers' method26 In brief, 3 grams of graphite powder and 1.5 grams of NaNO3 were mixed together followed by the addition of 69 mL of concentrated H2SO4 (98% w/w). The mixture was then placed in an ice bath and 9 grams of KMnO4 was added into the mixture under vigorous stirring for 30 min. De-ionized water (130 mL) was added slowly causing the temperature of the mixture to increase. The mixture was left to cool down to room temperature followed by addition of the hydrogen peroxide in aqueous solution (6% w/v). The oxidized graphite was then separated from the solution by centrifugation at 4000 rpm for 10 min. The obtained gel-like graphene oxide was washed by centrifugation with the concentrated hydrochloric acid (HCl, 35.4% w/w) once followed by copious amount of de-ionized water. The obtained GO gel was then dried in a vacuum oven at 40 °C for 48 h.2.2 Characterizations
The dried GO films were characterized for their morphology using the field-emission scanning electron microscope (FE-SEM, JEOL JSM-7800F) operating at a voltage of 5.0 kV. Fourier-transform infrared (FTIR) spectra of the GO films were taken in an attenuated total reflection (ATR) mode by co-adding of 32 scans over the spectral range of 500–4000 cm−1 at a resolution of 4 cm−1 (Nicolet iS5 equipped with a diamond crystal ATR). For each sample, at least 3 spectra from different locations of the GO films were measured to ensure a good representative of the whole film. The zeta potentials of the GO suspension were measured using the zeta sizer (Malvern Nano-ZS90). The measurement parameters were as follows: medium refractive index of 1.330, medium viscosity of 1.0 mPa s, and a relative dielectric constant of 80.4. The X-ray photoelectron spectroscopy (XPS) measurements of the GO samples before and after contact with Cs were performed using PHI 5000 Versa Probe II XPS system (ULVAC-PHI, Japan) with Al K-alpha radiation at the SUT-NANOTEC-SLRI joint research facility, Synchrotron Light Research Institute (SLRI), Thailand. The Raman spectra of the GO films were obtained using a Perkin Elmer System 2000 Fourier-transform spectrometer equipped with an FT-Raman accessory. The spectra was recorded in the range of 400 to 4500 cm−1 with 2 cm−1 resolution using a 532 nm laser excitation with the excitation power kept low to avoid any laser induced damage to the samples.For adsorption experiments, the metal ion concentrations were analysed using an Inductively-Coupled Plasma (ICP) spectroscopy (PerkinElmer Optima 8000, emission mode).
2.3 Sorption experiment
The GO were tested for Cs sorption performance in batch mode by dispersing 20 mg of the GO flakes in 10 mL of Cs solution prepared from CsCl at various concentrations. The Cs stock solution was prepared by dissolving CsCl (Merck, 99.99%) in deionized water at various concentrations from 10 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm. The samples were placed in a polypropylene vial for 24 h at room temperature (27 °C) using a rotary shaker under an agitation speed of 200 rpm. The GO adsorbent was separated from the solution using centrifugation at 4000 rpm for 5 min, and the supernatant was collected for metal concentration determination using Inductively Couple Plasma (ICP) spectroscopy (PerkinElmer Optima 8000, optical emission). The measurement was done in triplicate using samples prepared in three separate polypropylene vials and the average value was reported. For reference, stock solutions without GO sorbent were prepared and treated under the same conditions and run in parallel in every batch. The amount of Cs adsorbed to the wall of these vials was found to be negligible at all concentrations studied.
000 ppm. The samples were placed in a polypropylene vial for 24 h at room temperature (27 °C) using a rotary shaker under an agitation speed of 200 rpm. The GO adsorbent was separated from the solution using centrifugation at 4000 rpm for 5 min, and the supernatant was collected for metal concentration determination using Inductively Couple Plasma (ICP) spectroscopy (PerkinElmer Optima 8000, optical emission). The measurement was done in triplicate using samples prepared in three separate polypropylene vials and the average value was reported. For reference, stock solutions without GO sorbent were prepared and treated under the same conditions and run in parallel in every batch. The amount of Cs adsorbed to the wall of these vials was found to be negligible at all concentrations studied.
The adsorption isotherm was constructed from a plot between Qe versus Ce, where Qe is the equilibrium amount of Cs adsorbed onto the sorbent (mg Cs/g GO) and Ce is the equilibrium amount of Cs in the solution (mg L−1). Qe was calculated from Qe = (C0 − Ce)V/m where m is the mass of the dried sorbent, V is volume of the solution, C0 and Ce are the initial and final Cs concentrations in the solution after sorption. For contact time study, the supernatant was taken for Cs concentration determination after an elapse time of 5 min, 0.5, 1, 2, 4, 8, and 24 h. Effect of pH on Cs sorption efficiency was studied from pH 1 to 12 with an incubation time of 24 h. The pH of the solutions was adjusted using either 0.1 or 0.5 M HCl or NaOH solutions. The stock CsCl solutions without addition of GO were also adjusted using the same amount of HCl or NaOH and were used as references for initial Cs concentration.
The adsorption affinity of GO for Cs and other metal cations was evaluated and presented in terms of the distribution coefficient, Kd (mL g−1), which is the ratio between the concentration of the cation adsorbed on the sorbent to the concentration of the cation remained in the solution at equilibrium. The Kd value can be calculated following the expression: Kd = (C0 − Ce)V/(mCe), where parameters are the same as above.
For competitive cation experiment, chloride salts: NaCl, KCl, and MgCl2 were employed. The concentration of Cs was fixed at 1 mM, while the concentrations of competitive cations varied from 1 mM up to 10 mM for K, 50 mM for Mg, and 1000 mM for Na, in accordance with their mole abundance in natural sea water. The Kd values of Cs in the presence of these competitive cations (binary solution) were then measured and reported. The Kd values of each cation in the binary cation solution at their initial equimolar ratio can be used for comparing the GO's cation selectivity. The ratio of their Kd values in a binary cation solution, known as the separation factor,  , between the two cations A and B in solution yields directly the selectivity of GO for one cation as compared to the other cation.
, between the two cations A and B in solution yields directly the selectivity of GO for one cation as compared to the other cation.
The Cs sorption performance of GO was also evaluated in the natural sea water matrix. The seawater was collected from the sea in Petchburi province of Thailand. The pH of the seawater was 8.0 and the major cation composition determined using ICP was: Na (430 mM), Mg (54 mM), K (16 mM), and Ca (10 mM). The seawater was filtrated by 0.2 μm syringe filter, spiked with Cs and contacted with GO for 24 h.
3. Results and discussion
3.1 Morphology of the synthesized graphene oxide
The synthesized GO has a gel-like appearance, which upon drying forms a film. The morphology of the film, investigated using SEM, is shown in Fig. 1(a). The film consists of several graphene oxide sheets packed together. This can be seen in (b) at higher magnification on some of the rough edges on the surface where the GO sheets have protruded out of the surface, revealing their underlying sheet-like structure. These micron-size sheets then packed together forming a film, as seen in Fig. 1(a). The X-ray diffraction pattern (XRD) of the synthesized GO is shown in the ESI Fig. S1.† A broad peak around a 2θ angle of 10.1 degree, corresponding to the average spacing between the graphene oxide layers of about 0.87 nm, was observed. The typical graphite XRD peak at 26.5° could not be observed, indicating that the oxidation and exfoliation of the initial graphite has been quite complete, and most of the morphology seen in the SEM image here is due to the graphene oxide rather than the initial graphite powder. | ||
| Fig. 1 Morphology of the GO film formed upon drying. | ||
3.2 FTIR spectra
To further characterize the synthesized GO, FTIR spectra of GO at various pH values were examined. Fig. 2 shows FTIR spectra of the GO in solutions of pH 3, 7, and 12. At pH 3, the GO clearly shows distinctive bands at 1718, 1620, 1412, 1225, and 1067 cm−1. The band at 1718 cm−1 is assigned to undissociated carboxylic acids and aryl ketone functional groups.27 This band can still be observed at pH 7, but disappeared completely at pH 12, indicating the deprotonation of the carboxyl groups at an alkaline pH. This is in concert with an appearance of a shoulder band at 1580 cm−1 as a result of the increase in the pH value. This band is associated with the asymmetric stretching mode of sodium carboxylate groups, generated from the conversion of carboxylic acids by reacting with NaOH. The shift of a weaker band at 1412 cm−1, associated with the symmetric stretching mode of the carboxyls, to a lower frequency around 1365 cm−1 upon increasing pH also indicated the deprotonation of the carboxyl and the formation of the corresponding carboxylate salts. It is also noted that the 1580 cm−1 band may also be due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration, indicating that the conjugation or aromaticity of the GO is increased as the pH is raised.28 In addition, a broad band located at 1620 cm−1 originates from several overlapping vibrational modes, but the dominant one here is that due to the –OH bending mode of water molecules trapped in the GO film.
C vibration, indicating that the conjugation or aromaticity of the GO is increased as the pH is raised.28 In addition, a broad band located at 1620 cm−1 originates from several overlapping vibrational modes, but the dominant one here is that due to the –OH bending mode of water molecules trapped in the GO film.
 | ||
| Fig. 2 FTIR spectra of neat GO film incubated at three different pH values. | ||
The sharp band at 1225 cm−1 is assigned to the C–O stretching vibration of epoxide groups. This vibration mode is highly sensitive to the environment and completely disappears upon increasing pH. This is likely because the epoxide groups on the GO surface may have been dissociated into hydroxyl groups in the presence of a high concentration of electrophilic OH− at alkaline pH.29 The broad band at 1067 cm−1 is assigned either to the C–O stretching vibration of primary alcohol or ether R–O–R vibration. However, most of the hydroxyl groups in GO are attached to the basal plane or to aryl rings (phenol).29 As a result, their C–O vibrational mode should appear at around 1260 cm−1 rather than at 1067 cm−1, which is also observed in the spectrum at pH 3 as a small bump next to the epoxide band. The band at 1067 cm−1 most likely originates from the ether R–O–R vibration in the GO structure, possibly from various oxygen at the edge and hole defects generated on the GO sheets during the oxidation process.28–30 The C–O band at around are 1067 cm−1 is also slightly shifted to a lower frequency as the pH is increased. This may be due to the addition of alkaline Na+ into the system which can interact with the ethereal oxygen causing a slight shift to lower frequency.31 J. Guo et al. have also shown the abundance of holes created on the GO sheets upon oxidation. The hole edges were observed to be decorated with oxygen atoms, forming a crown ether-like structure.32 The C–O vibration of these crown-ethers appear in this region of the FTIR spectrum.31,33
3.3 Zeta potential of GO
Zeta potential (ZP) is an electrical potential arising from the charges on the surface of particles suspended in solution.15,20 The ZP of GO as a function of pH and Cs concentrations are shown in Fig. 3. The ZP of GO is always negative even at a pH as low as 1 with the value of about −15 mV. The ZP of GO becomes more negative as the pH of the solution is raised to 4. This is possibly due to the dissociation of carboxylic acid and its derivatives that possess an acid dissociation constant (Ka) in this region.27 The ZP then gradually becomes more negative at about −80 mV, as the pH is further increased to 12. Upon addition of Cs salt, the ZP becomes less negative. The higher the Cs addition, the more the decrease in the ZP, indicating the adsorption of Cs cations onto the GO negative surface, modifying its zeta potential. | ||
| Fig. 3 Zeta potential of GO as a function of pH and Cs concentrations. | ||
3.4 Evaluation of Cs sorption performance
 | ||
| Fig. 4 Amount of Cs adsorbed onto GO as a function of contact time. | ||
 | ||
| Fig. 5 Effect of pH on the Cs sorption by GO. | ||
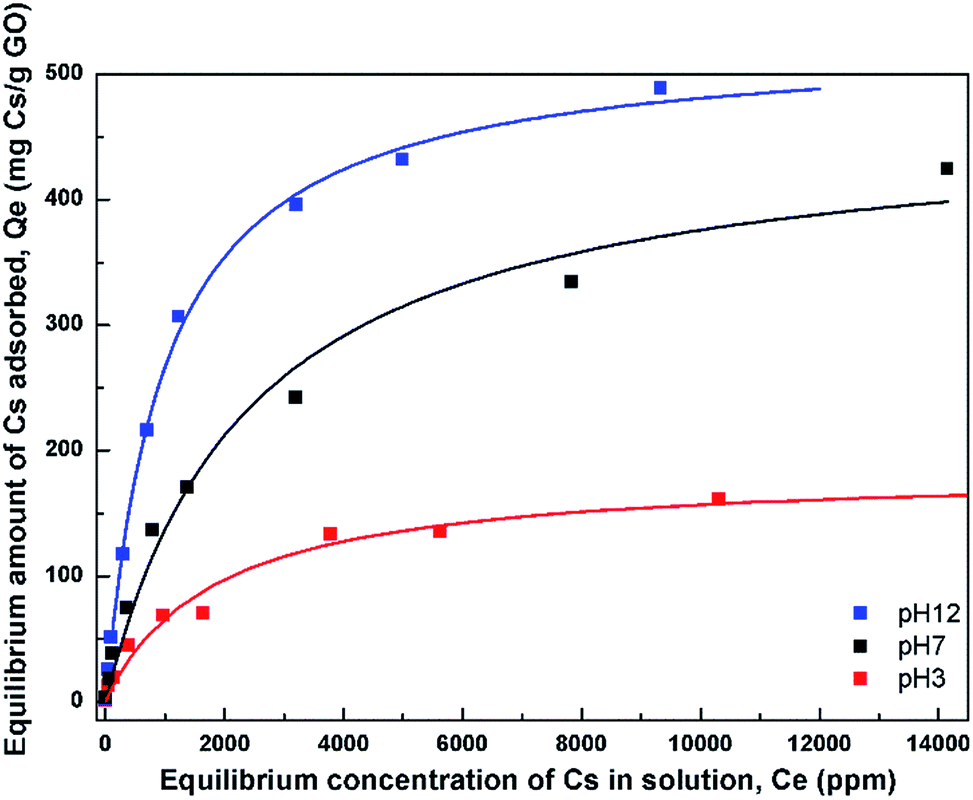 | ||
| Fig. 6 Adsorption isotherms of Cs on GO at 3 different pH values. The fits using the Langmuir model are also shown. | ||
In order to describe the adsorption isotherms further, the Langmuir model which assumes a uniform monolayer adsorption was fitted to the isotherm data. The Langmuir model is expressed as Qe = (QmkCe)/(1 + kCe) where Qm is the maximum adsorption capacity, and k is the affinity constant. Table 1 lists the parameters obtained from the fit of the Langmuir model to the isotherms. The maximum adsorption capacities from the model were obtained to be 180, 465, and 528 mg Cs/g sorbent for the three pH values, respectively.
| Adsorbents | Langmuir isotherm | ||
|---|---|---|---|
| Qmax (mg g−1) | k (L mg−1) | R2 | |
| GO_pH3 | 180 | 6.0 × 10−4 | 0.973 |
| GO_pH7 | 465 | 4.2 × 10−4 | 0.982 |
| GO_pH12 | 528 | 1.0 × 10−3 | 0.997 |
The affinity constant obtained from the fit to Langmuir model is observed to have the highest value at pH 12 with a value of about 1 × 10−3 L mg−1. This value is also quite comparable, or slightly smaller, to that of typical hexacyanoferrate which has affinity constants in the range of about 10−2–10−3 L mg−1, but may not be as good as those of crown ethers, where the affinity has been found to be as high as 2.2 L mg−1.34 However, the synthesis cost of GO may be much cheaper than those of crown ethers.
 | ||
| Fig. 7 FTIR spectra of GO before and after contact with Cs solutions (a) pH 3, (b) pH 7, and (c) pH 12. The corresponding difference spectrum, showing the changes after Cs is added to the solutions is also shown. | ||
Fig. 7(b) and (c) show the spectra of GO incubated at pH 7 and pH 12, respectively. At the neutral pH, a major spectral change after incubation with Cs occurs at around 1340 cm−1, which corresponds to the binding of Cs to carboxylate functional groups. In addition, another positive band at 1270 cm−1 is observed. This is a characteristic band of the C–O stretching mode of tertiary or aromatic alcohols, such as that of phenol.35 Since GO is known to possess a large number of phenol and hydroxyl groups connected to the basal plane,28 this phenol group may start to deprotonate and take part in the adsorption of Cs.
At pH 12, where GO shows the highest capacity for Cs sorption, the difference spectrum shows a bimodal negative-positive pattern at 1600/1580 cm−1, which is characteristic of the asymmetric stretching mode of carboxylate salts. This indicates that the Na cation bound to GO at pH 12 may have been replaced by the heavier Cs cation causing the downshift in vibration frequency. Additionally, other functional groups may also contribute to the band intensity in this region. It has been shown that GO also contains a large number of phenol groups as well as the –OH attached to the basal plane.28 B. Konkena et al. have shown through pKa measurements that GO possess at least 3 pKa regions around pH of 4.3, 6.6 and 9.0. The former two correspond to various carboxylate functional groups on GO, while the latter one corresponds to the hydroxyl or phenol groups. As a result, at pH 12, it is possible that these phenols or the hydroxyl connected to the basal plane may have dissociated into the phenolate anion which has metal cation binding ability.36,37 The binding of a cation to the phenolate anion typically results in the downshift of the aromatic ring vibration to around 1580 cm−1, as has been observed.38 The notable peak shift observed here in the difference spectrum may be the result of contributions from both the carboxylate as well as the phenolate anions. Their symmetric stretching modes are also visibly seen as a broad band in the region of 1365–1200 cm−1.
It should also be noted that across the pH range, the increase in intensity of the 1030 cm−1 band, assigned to the ether C–O–C stretching vibration, is consistently observed. This indicates that the ether C–O–C groups also interact with the Cs cation. This C–O–C may be originated from hole defects on the graphene oxide sheets where the hole edges are decorated with oxygen atoms.30 J. Guo et al. have reported observations of several sizes of crown-ether hole defects on the GO sheets using high resolution scanning transmission electron microscope.32 These crown-ether type hole defects in GO were also shown to be able to bind strongly to metal cations.32 Our observation of the positive difference band centered around 1030 cm−1 may also signify this Cs sorption mechanism by the ether hole defects on the graphene oxide sheets. Based on these FTIR results, a model illustrating various functional groups and defects that may be responsible for the Cs adsorption by GO is depicted in Fig. 8.
 | ||
| Fig. 8 Schematic diagram of GO structure showing possible locations where Cs might be captured by the GO sheets. (i) The carboxylate anion, (ii) the phenolate anion, (iii) the oxygen-decorated hole defect, and (iv) the deprotonated hydroxyl groups on the basal plane. | ||
To investigate the mechanism of Cs binding to GO further, the X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy measurements of the GO samples before and after Cs sorption were also performed and are shown in Fig. 9.
 | ||
| Fig. 9 XPS spectra of GO before and after Cs sorption. (a) Survey spectra showing the presence of Cs in GO after contact, (b) C1s spectrum of GO and its deconvolution, (c) comparison of C1s spectra of GO before and GO after Cs sorption, (d) O1s spectrum of GO and its deconvolution, (e) comparison of O1s spectra of GO before and after Cs sorption. | ||
Fig. 9 shows the XPS survey spectrum of the GO sorbent after Cs sorption, the present of cesium Cs3d peaks can be clearly observed at the binding energies of 723 and 737 eV for the Cs3d3/2 and Cs3d5/2 electrons respectively. Fig. 9(b) shows the C1s spectra of the GO sorbent before and after Cs sorption. The spectrum can be deconvoluted into three major peaks at 288.4 eV, 286.2 and 288.8 eV, corresponding to the C–C, C–O and O–CO species, respectively. It can be seen that the C1s XPS spectra are very similar in both the GO and GO after Cs sorption samples. The center binding energy and the atomic fraction (AF) of each species are also shown in the Table 2. The O1s spectra for GO and GO after Cs contact were also very similar. No significant chemical shifts in the O1s spectra could be observed after Cs sorption. This is also in the same trend as that in the work by S. Tas et al.39 where they have measured XPS spectra of alkali cations adsorbed onto the crown ether grafted polyacrylonitrile fiber, but no significant shift in the C1s or O1s spectra of the spent sorbent could be observed.
| Element | GO | GO after Cs sorption | ||
|---|---|---|---|---|
| BE (eV) | AF% | BE (eV) | AF% | |
| C1s | 284.14 | 31.6 | 284.16 | 28.9 |
| C1s | 286.21 | 33.2 | 286.18 | 32.1 |
| C1s | 287.90 | 4.5 | 287.78 | 4.7 |
| Total C1s | 69.3 | 69 | ||
| O1s | 531.55 | 10.7 | 531.45 | 13 |
| O1s | 532.03 | 20 | 532.01 | 20.9 |
| Total O1s | 30.7 | 33.9 | ||
| Total Cs3d | 0.4 | |||
XPS has been shown to be sensitive enough to detect interaction of many di- and tri-valent transition metal cations adsorbed onto the biomass sorbent e.g. in the work by M. Zhang et al.40 where they have detected slight shift in the XPS C1s and O1s spectra. In this work, the XPS spectra of the Cs-adsorbed GO are rather similar to those of the neat GO and no significant shift in either the C1s or O1s spectra could be clearly observed. This is probably due to the rather large size and facile charge of Cs cation in contrast to those observed in the interactions between GO, or cellulosic sorbent, with the di- and poly-valent cations.
The Raman spectra of the GO before and after Cs sorption are also shown in Fig. 10. There are two major bands in the Raman spectra. The band at around 1597 cm−1 is known as the G band corresponding to the in-plane C–C vibrational mode. The band around 1355 cm−1, or the D band, are associated with the degree of disorder, and the oxidation degree of the GO sheet.41 The intensity ratio of the D- to the G-band, ID/IG, can be used to indicate the degree of disorder in the GO sheets. This ratio changes from 0.88 in the GO sample to 0.93 after contact with Cs. The Cs cation may interact with the oxygen defects in the GO structure causing the slight increase in the intensity of the D-band observed. In addition, the band at 1355 cm−1 is also slightly red-shifted to 1349 cm−1 after Cs sorption signifying that the oxygen defects in GO sheet may have some interactions with the adsorbed cation. The weak change in the Raman spectra as compared to that in the FTIR spectra indicates that GO interaction with Cs cations probably has the dipolar character which will show up strongly in the FTIR spectra but not sensitively detected by the Raman technique42,43.
 | ||
| Fig. 10 Raman spectra of GO before and after Cs sorption. | ||
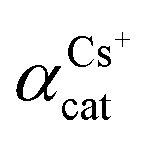 , using the binary mixed cation solution
, using the binary mixed cation solution
| Cations | Kd (mL g−1) |
|
|---|---|---|
| Cs+ | 115 | — |
| Na+ | 19 | 4.9 |
| K+ | 80 | 1.3 |
| Mg2+ | 436 | 0.3 |

Another way to compare the selectivity of the sorbent for any pair of cations is to compare the Kd values of the two cations in a mixed binary cation solution. The ratio between their Kd values is known as the separation factor, αAB = Kd,A/Kd,B, between the two cations A and B in the solution45,46 which yields directly the selectivity of GO for one cation as compared to the other cation given the same initial concentrations of A and B in the solution. The larger the separation factor indicates the more preference of the sorbent towards cation A compared to that of B. Table 3 shows the separation factor of GO between the Cs+ and the other cation in the mixed binary cation solution system.
From both the Kd and the separation factor values, it can be observed that GO has the highest affinity for the divalent cation Mg2+. This is as expected since the strong charge of Mg2+ would be able to interact strongly with the negatively-charged functional groups on the GO structure. It is well known that the interaction energy between the group-II alkaline earth cations is much stronger than that of the group-I alkali metal cation.
For the Na+ and K+ cations which have the charge of +1 similar to that of the Cs+, it can be seen that GO displays affinity for cations in the order of Cs+ > K+ > Na+. This is in the same trend of interaction as that observed in the Hofmeister series47 that describes the interaction between ions and large macromolecules such as proteins. The mechanism underlying the Hofmeister series is still unclear but is thought to be linked to the hydration energies of the two opposite charge species.48 Strongly hydrated cations such as Na+ or K+ have a high energy barrier to break their solvation shells before they can interact with the functional groups on GO, making them less active when compared to Cs+. Another way to view this is that Na+ is strongly hydrated and, as a result, possesses larger hydration radius,49 and smaller surface charge density compared to that of Cs+. Na+ therefore interacts less strongly to the oxygenated functional groups on the GO sheets compared to the Cs+. This result also agrees with the work by Tang and Allen50 who observed the selectivity order in fatty acid salt using vibrational sum frequency probe that K+ can interact directly with the carboxylate group while the Na+ is solvent separated from the carboxylate group causing the binding of Na+ to be less strong than that of the K+. It should also be noted that the selectivity order we observed here is also quite similar to the selectivity order of many polymeric cation-exchange resins,51,52 whose selectivity order has also been explained based on the same trend of hydration radius and charge density of the hydrated cations as above.46 In addition, the ether hole defects in GO sheet may also play important role in the observed selectivity of GO towards Cs cations. J. Guo et al. have also shown that large hole defects on GO sheets can bind Cs+ stronger than that of Na+ or Li+.32
The effect of these cations at higher concentrations in competing with Cs for GO's adsorption is also studied and is shown in Fig. 11. In this experiment, the Cs concentration is fixed at 1 mM, while the concentrations of other cations were varied up to or higher than their natural concentrations in seawater (Na ∼ 470 mM, K ∼ 10 mM, and Mg ∼ 50 mM).53 For Cs with a competitive cation concentration ratio of 1:0 (i.e. no competitive cation), the Kd value of GO for Cs is approximately 156 mL g−1. As the concentration of the competitive cation is increased, the Kd value of Cs is decreased. Mg2+ shows quite detrimental effect on the Cs uptake by GO, causing the Cs distribution coefficient to be reduced to about 40 mL g−1 at a Mg2+ concentration of 50 mM. The effect of Na+ and K+ on the Kd of Cs+ seems to be much less. Even in the presence of Na+ as high as 1000 mM, the Kd of GO for Cs is still quite high at about 80 mL g−1, which is about half of its original value.
 | ||
| Fig. 11 Kd of Cs adsorption onto GOs in the presence of competitive cations. The final ratio corresponds to the concentrations of the competitive cations found in seawater. Initial Cs concentration of 1 mM, while varying the concentrations of other competitive cations. | ||
The performance of GO in the natural seawater has also been evaluated. The major cation composition of the natural seawater used was determined using ICP as follows: Na (430 mM), Mg (54 mM), K (16 mM), and Ca (10 mM). The distribution coefficient of Cs on GO in this natural seawater is still quite high with the Kd value of about 48 mL g−1. This is quite similar to the performance of GO in the simulated binary solution of Cs with Mg indicating that in the natural seawater matrix, the Mg2+ may not be so competitive in binding to the GO's surface probably because of the binding of Mg to organic matters present in the seawater which prevent it from binding to the GOs surface. This indicates that GO can perform exceptionally well even in the real seawater in the presence of high concentration of competitive cations.
In some practical situations, it may also be desirable to separate other di- and polyvalent radioactive cations such as Sr2+ and UO22+ that may be released with the radioactive Cs+. One of the strategies that can be used to separate these polyvalent cations out could be to use the pH adjustment to basic pH prior to the Cs sorption. Sr2+, being an alkaline earth cation, will precipitate out at the pH above 10, while the UO22+ will also undergo further hydrolysis at high pH becoming UO2(OH)3−, or UO2(OH)42−.54 The negatively charged uranyl ions will no longer bind with GO. This should have enabled GO's selectivity towards Cs, which has been shown to be highest at the basic pH.
It is also worth mentioning that the sensitivity of GO to the group-I and group-II competitive cations is in the opposite direction to that of the Prussian-blue (PB) type sorbents. PB are usually not significantly affected by the presence of divalent cations such as Mg2+ and Ca2+ due to its size exclusion effect.55 However, PB is very sensitive to the presence of Na+ and especially K+, which can reduce its Cs adsorption significantly. From the experiment here, the presence of both Na+ and K+ does not strongly affect the Cs sorption, while the presence of Mg2+ moderately degrades the GO performance but could also be separated out using pH adjustment. GO may thus be a good complimentary to the commonly use PB type sorbents in situation where the background group-I cations are high such as that in seawater, or in the highly alkaline pH where PB degrades. GO offers also a high potential for use in emergency responses since it is easy and inexpensive to synthesize compared to the crystalline silico-titanate family of sorbents56 that usually require long crystallization times.
4. Conclusions
Pristine graphene oxide (GO) has been demonstrated in this work to be very effective in the Cs sorption. The maximum adsorption capacity of GO is increased as the pH increases, reaching a maximum adsorption capacity of about 528 mg Cs/g GO at pH 12 due to the full availability of the active sites on GO sheets. GO possesses fast sorption kinetics, reaching 90% of its equilibrium sorption capacity in less than 60 min. FTIR spectra reveal various oxygenated functional groups including carboxyls, phenols, and the hole defects containing multi-ether groups similar to that of crown ethers to play important role in Cs capturing. The GO affinity for other common cations found in natural seawater were also found to be in the order of Mg2+ > Cs+ > K+ > Na+. Although divalent cations such as Mg2+ may inhibit GO ability to adsorb Cs quite strongly, most divalent cations can also be easily removed prior to Cs sorption using a simple alkaline pH adjustment and precipitation. The Cs removal performance of GO in the seawater matrix has also been conducted and showed GO's exceptional ability in Cs removal even in the presence of high concentration of other cations.Acknowledgements
This work is supported by the DPST research grant Project no. 007/2555. PO gratefully acknowledges support from the Thai Government Annual Research Budget through Thammasat University (2557), and the Centre of Excellence in Materials and Plasma Technology, Thammasat University. PK is grateful to the scholarship from the TAIST-Tokyo Tech program.Notes and references
- T. J. Yasunari, A. Stohl, R. S. Hayano, J. F. Burkhart, S. Eckhardt and T. Yasunari, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19530–19534 CrossRef CAS PubMed.
- K. O. Buesseler, S. R. Jayne, N. S. Fisher, I. I. Rypina, H. Baumann, Z. Baumann, C. F. Breier, E. M. Douglass, J. George, A. M. Macdonald, H. Miyamoto, J. Nishikawa, S. M. Pike and S. Yoshida, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5984–5988 CrossRef CAS PubMed.
- S. V. Fesenko, R. M. Alexakhin, M. I. Balonov, I. M. Bogdevitch, B. J. Howard, V. A. Kashparov, N. I. Sanzharova, A. V. Panov, G. Voigt and Y. M. Zhuchenka, Sci. Total Environ., 2007, 383, 1–24 CrossRef CAS PubMed.
- N. A. Beresford and B. J. Howard, Integr. Environ. Assess. Manage., 2011, 7, 382–384 CrossRef CAS PubMed.
- E. H. Borai, R. Harjula, L. Malinen and A. Paajanen, J. Hazard. Mater., 2009, 172, 416–422 CrossRef CAS PubMed.
- D. Cuc, S. Bouguet-Bonnet, N. Morel-Desrosiers, J.-P. Morel, P. Mutzenhardt and D. Canet, J. Phys. Chem. B, 2009, 113, 10800–10807 CrossRef CAS PubMed.
- C. Xiao, A. Zhang and Z. Chai, J. Hazard. Mater., 2014, 267, 109–118 CrossRef CAS PubMed.
- G. Sushma, J. Chromatogr. A, 1996, 733, 57–71 CrossRef.
- A. J. Celestian, J. D. Kubicki, J. Hanson, A. Clearfield and J. B. Parise, J. Am. Chem. Soc., 2008, 130, 11689–11694 CrossRef CAS PubMed.
- D. Yang, H. Liu, Z. Zheng, S. Sarina and H. Zhu, Nanoscale, 2013, 5, 2232–2242 RSC.
- P. J. Faustino, Y. Yang, J. J. Progar, C. R. Brownell, N. Sadrieh, J. C. May, E. Leutzinger, D. A. Place, E. P. Duffy, F. Houn, S. A. Loewke, V. J. Mecozzi, C. D. Ellison, M. A. Khan, A. S. Hussain and R. C. Lyon, J. Pharm. Biomed. Anal., 2008, 47, 114–125 CrossRef CAS PubMed.
- C. Thammawong, P. Opaprakasit, P. Tangboriboonrat and P. Sreearunothai, J. Nanopart. Res., 2013, 15, 1689–1698 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. Shamaila, A. K. L. Sajjad and A. Iqbal, Chem. Eng. J., 2016, 294, 458–477 CrossRef CAS.
- J. Wang and B. Chen, Chem. Eng. J., 2015, 281, 379–388 CrossRef CAS.
- S. Yang, L. Li, Z. Pei, C. Li, J. Lv, J. Xie, B. Wen and S. Zhang, Colloids Surf., A, 2014, 457, 100–106 CrossRef CAS.
- D. Gu and J. B. Fein, Colloids Surf., A, 2015, 481, 319–327 CrossRef CAS.
- J. Zheng, L. Xia and S. Song, RSC Adv., 2017, 7, 23543–23549 RSC.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- H. Xu, G. Li, J. Li, C. Chen and X. Ren, J. Mol. Liq., 2016, 213, 58–68 CrossRef CAS.
- Y. Sun, S. Yang, Y. Chen, C. Ding, W. Cheng and X. Wang, Environ. Sci. Technol., 2015, 49, 4255–4262 CrossRef CAS PubMed.
- D. Li, B. Zhang and F. Xuan, J. Mol. Liq., 2015, 209, 508–514 CrossRef CAS.
- S. C. Jang, Y. Haldorai, G. W. Lee, S. K. Hwang, Y. K. Han, C. Roh and Y. S. Huh, Sci. Rep., 2015, 5, 17510 CrossRef CAS PubMed.
- H. Yang, L. Sun, J. Zhai, H. Li, Y. Zhao and H. Yu, J. Mater. Chem. A, 2014, 2, 326–332 CAS.
- S. Yang, N. Okada and M. Nagatsu, J. Hazard. Mater., 2016, 301, 8–16 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. Mao, H. Pu and J. Chen, RSC Adv., 2012, 2, 2643 RSC.
- D. Pacilé, J. C. Meyer, A. Fraile Rodríguez, M. Papagno, C. Gómez-Navarro, R. S. Sundaram, M. Burghard, K. Kern, C. Carbone and U. Kaiser, Carbon, 2011, 49, 966–972 CrossRef.
- Y. Inokuchi, T. Ebata, T. Ikeda, T. Haino, T. Kimura, H. Guo and Y. Furutani, New J. Chem., 2015, 39, 8673–8680 RSC.
- J. Guo, J. Lee, C. I. Contescu, N. C. Gallego, S. T. Pantelides, S. J. Pennycook, B. A. Moyer and M. F. Chisholm, Nat. Commun., 2014, 5, 5389 CrossRef CAS PubMed.
- T. E. Cooper, D. R. Carl, J. Oomens, J. D. Steill and P. B. Armentrout, J. Phys. Chem. A, 2011, 115, 5408–5422 CrossRef CAS PubMed.
- M. R. Awual, T. Yaita, T. Taguchi, H. Shiwaku, S. Suzuki and Y. Okamoto, J. Hazard. Mater., 2014, 278, 227–235 CrossRef CAS PubMed.
- M. Nonella and H. U. Suter, J. Phys. Chem. A, 1999, 103, 7867–7871 CrossRef CAS.
- M. Nonella and H. U. Suter, J. Phys. Chem. A, 1999, 103, 7867–7871 CrossRef CAS.
- X. Wang, L.-H. Liu, J.-H. Shi, J. Peng, H.-Y. Tu and A.-D. Zhang, Eur. J. Org. Chem., 2013, 2013, 6870–6877 CrossRef CAS.
- R. M. Ahmed, E. I. Yousif, H. A. Hasan and M. J. Al-Jeboori, Sci. World J., 2013, 2013, 289805 Search PubMed.
- S. Tas, O. Kaynan, E. Ozden-Yenigun and K. Nijmeijer, RSC Adv., 2016, 6, 3608–3616 RSC.
- M. Zhang, L. Song, H. Jiang, S. Li, Y. Shao, J. Yang and J. Li, J. Mater. Chem. A, 2017, 5, 3434–3446 CAS.
- B. Ryan, C. Luiz Gustavo and N. Lukas, J. Phys.: Condens. Matter, 2015, 27, 083002–083027 CrossRef PubMed.
- K. Eberhardt, C. Stiebing, C. Matthäus, M. Schmitt and J. Popp, Expert Rev. Mol. Diagn., 2015, 15, 773–787 CAS.
- V. Otero, D. Sanches, C. Montagner, M. Vilarigues, L. Carlyle, J. A. Lopes and M. J. Melo, J. Raman Spectrosc., 2014, 45, 1197–1206 CrossRef CAS.
- P. A. Haas, Sep. Sci. Technol., 1993, 28, 2479–2506 CrossRef CAS.
- E. Pehlivan and T. Altun, J. Hazard. Mater., 2007, 140, 299–307 CrossRef CAS PubMed.
- C. E. Harland, in Ion Exchange: Theory and Practice, The Royal Society of Chemistry, 2nd edn, 1994, pp. 90–133, 10.1039/9781847551184-00090.
- P. Jungwirth, J. Phys. Chem. Lett., 2013, 4, 4258–4259 CrossRef CAS PubMed.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS PubMed.
- B. E. Conway and E. Ayranci, J. Solution Chem., 1999, 28, 163–192 CrossRef CAS.
- C. Y. Tang and H. C. Allen, J. Phys. Chem. A, 2009, 113, 7383–7393 CrossRef CAS PubMed.
- F. W. E. Strelow, R. Rethemeyer and C. J. C. Bothma, Anal. Chem., 1965, 37, 106–111 CrossRef CAS.
- R. Ratner, J. Itzchaki and D. H. Kohn, J. Appl. Chem., 1968, 18, 48–49 CrossRef CAS.
- J. Wright and A. Colling, in Seawater: its Composition, Properties and Behaviour, 2nd edn, Pergamon, 1995, pp. 85–127, DOI:10.1016/B978-0-08-042518-4.50011-6.
- D. Hagberg, G. Karlström, B. O. Roos and L. Gagliardi, J. Am. Chem. Soc., 2005, 127, 14250–14256 CrossRef CAS PubMed.
- M. Ishizaki, S. Akiba, A. Ohtani, Y. Hoshi, K. Ono, M. Matsuba, T. Togashi, K. Kananizuka, M. Sakamoto, A. Takahashi, T. Kawamoto, H. Tanaka, M. Watanabe, M. Arisaka, T. Nankawa and M. Kurihara, Dalton Trans., 2013, 42, 16049–16055 RSC.
- T. A. Todd and V. N. Romanovskiy, Radiochemistry, 2005, 47, 398–402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04868h |
| This journal is © The Royal Society of Chemistry 2017 |