DOI:
10.1039/C7RA04911K
(Paper)
RSC Adv., 2017,
7, 30911-30918
DNase-targeted natural product screening based on a sensitive and selective DNase I detecting system†
Received
2nd May 2017
, Accepted 4th June 2017
First published on 15th June 2017
Abstract
As a widely used deoxyribonuclease, DNase I is involved in many physiological processes including tumor cell proliferation, metastasis and apoptosis. Furthermore, the level of this enzyme in serum can act as a functional biomarker for the therapeutic monitoring of systemic lupus erythematosus and other diseases. We report here a low cost and sensitive DNase I detecting system based on the single-stranded fluorogenic substrate and nanographene oxide (NGO) and use it for DNase-targeted natural product screening. The system with a detection limit of 0.005 U was then used to evaluate the effect of external factors on DNase I. The results show that Hg2+, As2+, Pb2+, Cd2+ and Cu2+ can inhibit DNase I activity in a concentration-dependent manner with IC50 values of 0.37 mM (Hg2+), 2.7 mM (As2+), 5 mM (Pb2+), 5.3 mM (Cd2+) and 7.8 mM (Cu2+), respectively. Meanwhile, 10 natural compounds isolated from Cyclocarya paliurus leaves were screened as DNase I inhibitors, while 5 compounds were identified as activators. Finally, the system was used to discriminate DNase activity of serum samples with and without HBV. The results showed that HBV infection significantly decreased the level of DNase I in serum samples. In summary, these data indicate that this method with the advantages of rapidity, low cost and high sensitivity is hopeful for DNase assay in biological samples as well as compound screening in vitro.
1. Introduction
DNase I is an important enzyme involved in many processes including genetic recombination, DNA repair and DNA digestion by participating DNA cleavage reactions in vivo.1–3 It is also an important enzyme tool for genomic DNA probes, DNA template digestion and high-throughput sequencing in vitro.4,5 Recently, clinical studies revealed that DNase reduced exacerbations of respiratory symptoms, inhibited tumor cell proliferation, metastasis and enhanced cell apoptosis.6–8 Furthermore, it was identified as a hopeful predictor of myocardial infarct size due to the role of the disruption of paradoxically toxic extracellular chromatin released from necrotic cells.9 Thus, developing DNase assay can provide critical data for deeply exploring the mechanism of diseases development as well as drug screening from various compound libraries. Meanwhile, the developed platform for DNase I assay can provide an alternative tool for early clinical diagnosis and therapy evaluation.
Traditional methods including gel-based electrophoresis,10 ELISA11 and colorimetric method,12 which have been widely used for DNase assay, are time-consuming, laborious and indirect in some extent. Although developed fluorescence method based on nanoparticle or molecular beacon partly overcame the limitations of conventional methods and showed promising applications,13–15 some inherited limitations such as high background and low stability still existed. In order to overcome these limitations and take DNase I assay closer to point-of-care, two-dimensional nanographene oxide (NGO) was applied to construct biosensing platform for this enzyme.16,17 As a one-atom thick two dimensional carbon material with a honeycomb structure, it has been widely used for nucleases assay in real-time.18–21 Although GO-DNA nanoassembly significantly improved the background signal and thermal stability of the fluorescence probe comparing with molecular beacon, the spatial hindrance effect of nanoassembly resulted in the unfavorable sensitivity through inhibiting recognition capability of enzyme to the substrate. In order to avoid the disturbance of the spatial hindrance effect on the enzymatic reaction and improve detection sensitivity, we employed an end-point measurement method for DNase assay and further used it to screen specific effectors from natural compounds.
2. Experimental
2.1 Materials and methods
All oligonucleotide strands (Table S1†) were purchased from Takara Biotechnology Co. Ltd (Dalian, China) and quantified using a ε260 value. DNase I was purchased from the same corporation and the stock concentration was 10.74 ng μL−1. The reactive buffer (100 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 1 mM EDTA) was used in all reactions unless otherwise indicated. All kinds of cell line were purchased from the cell library of Xiangya Central Laboratory, Central-South University. Human serum samples with HBV infection or not were obtained from the Third Xiangya Hospital, Central-South University. Graphene oxide (GO) was purchased from Nanjing XFNano Material Tech Co., Ltd. (China). The average particle size and zeta potential were determined to be 260 ± 10 nm and −28 ± 1.6 mV (Fig. S1†). All other analytical chemicals were purchased from Sigma-Aldrich Corporation (USA) without further purification.
2.2 Reaction time optimization between the substrate and NGO
GO powder was sonicated 12 h before use to obtain a water-soluble nanosized graphene oxide solution and the fluorescence probe solution was diluted with TE buffer to 10 μM. To optimize the reaction time between the probe and NGO, the nano assemblies were prepared by mixing 100 nM probe solution with 12 μg mL−1 NGO in 10 mM Tris–HCl buffer (pH 7.5, 10 mM MgCl2). This NGO addition resulted in immediate fluorescence quenching. Observed fluorescence measurements were performed at 0, 1, 5, 10, 15, 20, 25 and 30 min, respectively.
2.3 Time optimization of enzymatic reaction
To optimize the enzymatic reaction time, samples were prepared by mixing 1 μL P1 (10 μM) with same volume of DNase I (8 units) and incubated for 0, 5, 10, 15, 20, 25, 30, 35 and 40 min at 45 °C. Then, 1.2 μg NGO was added and the mixed solution was diluted with Tris–HCl buffer (pH 8.0) to 100 μL. Fluorescence measurement were performed after incubating for 10 min at room temperature.
2.4 Performance of DNase in the reactive buffer
For DNase assay, 1 μL P1 (10 μM) and various concentrations of DNase I were mixed in the buffer and incubated for 30 min at 45 °C. 1.2 μg NGO were added into solutions. After 10 min incubation, fluorescent measurements were performed with a fluorescence spectrophotometer.
2.5 Drug screening for DNase I
Compounds (1 mM in DMSO) were stored in a dry, inert environment at −20 °C and diluted to different concentrations using 20% DMSO/water. For drug screening, aliquots (1 μL) were added into reactive buffer containing 100 nM P1 and incubated at 45 °C for 10 min. Then, DNase I (2 units) was added and the sample was continued to incubate for 30 min, to which NGO of 1 μL (1.2 mg μL−1) added. After 10 min incubation, fluorescent measurements were performed using a fluorescence spectrophotometer.
2.6 DNase detection in biosamples
Tumor cells were cultured in DMEM medium and cell-free extracts were prepared as follows: 1 × 106 cells were harvested after with trypsin treatment and centrifuged at 1500g for 2 min, washed with 10 mL of cold PBS for 3 times, centrifuged and resuspended in 0.5 mL of cell lysis buffer (cell signaling) on ice for 5 min. Cells were pulse-sonicated on ice 5 times for 5 s each. Then, centrifuged extracts at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C and collected supernatants. Concentrations of cell-free extracts were quantitated by measuring the absorbance at 595 nm using Coomassie blue protein reagent (Pierce, Rockford, USA). For DNase assay of biosamples, mixture of 1 μL of P1 (10 μM) and 3 μg of cell-free extracts incubated for 30 min at 45 °C, to which of 1.2 μg NGO added. For DNase assay in serum, the mixture of 1 μL P1 probe and 1 μL serum were incubated in the reactive buffer for 30 min at 45 °C. Then, 1.2 μg NGO was added and the solution was diluted with reactive buffer to 100 μL. After 10 min incubation, fluorescence measurement was performed with a fluorescence Spectrophotometer.
000g for 20 min at 4 °C and collected supernatants. Concentrations of cell-free extracts were quantitated by measuring the absorbance at 595 nm using Coomassie blue protein reagent (Pierce, Rockford, USA). For DNase assay of biosamples, mixture of 1 μL of P1 (10 μM) and 3 μg of cell-free extracts incubated for 30 min at 45 °C, to which of 1.2 μg NGO added. For DNase assay in serum, the mixture of 1 μL P1 probe and 1 μL serum were incubated in the reactive buffer for 30 min at 45 °C. Then, 1.2 μg NGO was added and the solution was diluted with reactive buffer to 100 μL. After 10 min incubation, fluorescence measurement was performed with a fluorescence Spectrophotometer.
2.7 Fluorescence assay
Fluorescent emission spectra were performed on FL-2500 Fluorescence Spectrophotometer, Hitachi High-Technology Co., Ltd. (Tokyo, Japan). The sample cell was a 700 μL quartz cuvette. The fluorescence intensity was directly measured by spectrophotometer with excitation at 521 nm and emission range from 550 to 650 nm. Spectrophotometer slits for excitation and emission were set at 10 nm. The fitting of the experimental data was accomplished using the software Sigmaplot 12.0.
3. Results and discussion
3.1 Design of a fluorescence-based method for DNase assay
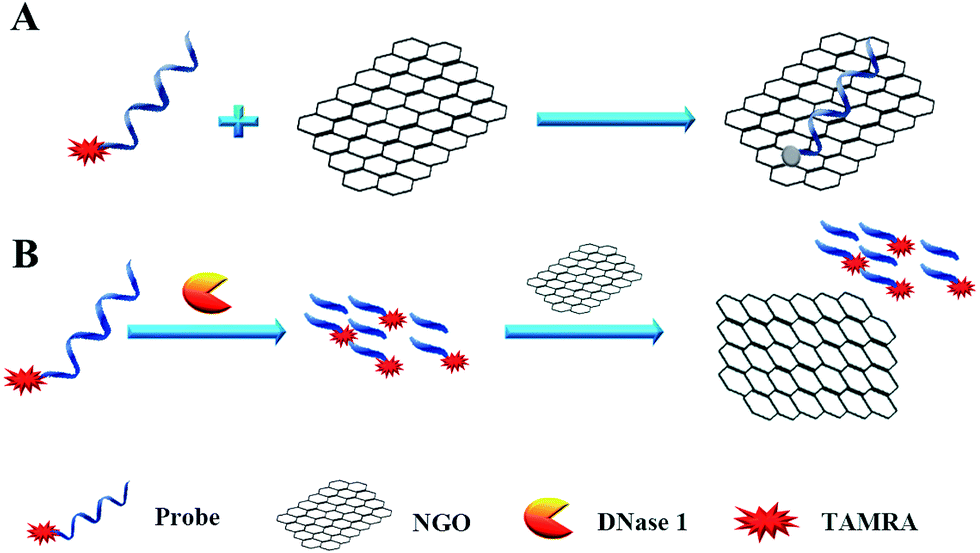
We designed a two-step detection method for DNase I assay using NGO with single-labeled fluorescence DNA as the substrate. This two-stage assay system contains three key components of the purified enzyme, a TAMRA-labeled DNA strand and NGO. The principle is that the binding of NGO with intact substrate can result in highly quenching of fluorescence signal mainly through π–π interaction between the ring of nucleobases and the hexagonal cells of the graphene if there is no enzyme present. However, DNase I can digest the long DNA stand into small fragments with weak adsorption capability on NGO. As a result, the fluorescence of the small fragments can not be quenched by NGO and they can emit strong fluorescence at 578 nm when excite at 521 nm (Fig. 1A).
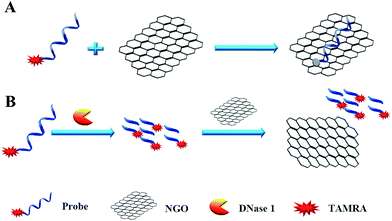 |
| | Fig. 1 Schematic diagram of the fluorescence-based DNase I assay. | |
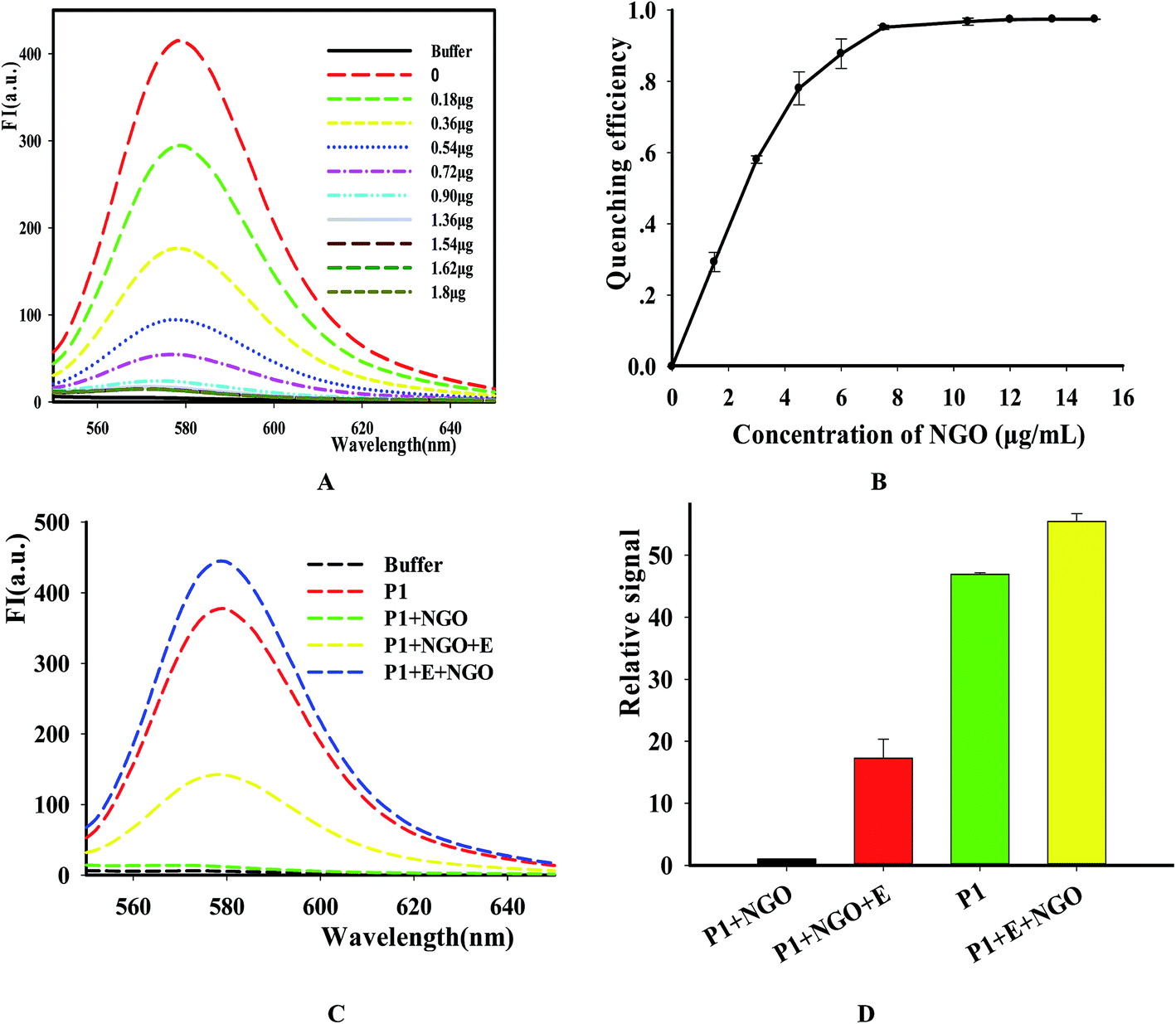
In order to simplify the manipulation process and improve sensitivity of the assay, we designed three kinds of single-stranded DNAs (Table S1†) that contain hair-pin structure or not as substrates and tested the principle of the assay using purified DNase I and fluorescence substrates. Earlier studies have shown that NGO concentration can directly influence quenching efficiency of the fluorescence probes. To test the effect of NGO concentration on the NGO–DNA interactions, the fluorescence signal of 100 nM P1 at the presence of NGO was measured and the resulting fluorescence emission spectra were shown in Fig. 2A. It can be seen that P1 possess the highest fluorescence quenching ability (99%) when 12 μg mL−1 NGO is present. This concentration is consistent with previous reports.22 For the other two probes of P2 and P3 with stem-loops, both random unpaired bases and rigid paired bases coexisted, which raised a question whether NGO can similarly quench the fluorescence signal as that of P1. We then studied the interaction of NGO with P2 and P3 using the same amounts of NGO (1.2 μg) and found that the quenching efficiency of P2 and P3 were only 70% and 60%, respectively, which are much lower than that of P1 (99%) (Fig. S2†). This dramatic fluorescence difference indicates that the existence of hairpin structure can decrease adsorption ability and NGO can efficiently differentiate these single-stranded DNAs with different spatial structures. It has been reported that the NGO/DNA interaction mainly depends on the freedom nucleobases in DNA strands and the quenching efficiency is negatively correlated with the length of complimentary bases.23,24 We further investigated the S/B ratio of the three probes at the presence of same amount of NGO (1.2 μg) and found that the ratio decreased according to the sequence of P1, P2 and P3 probes (data not shown). In addition, we monitored the fluorescence signal intensities of the reactions using “pre-mixing” and “post-mixing” strategies to test whether the DNA interacting with NGO can inhibit enzymatic cleavage protection ability through the spatial hindrance effect. The results in Fig. 2C and D indicated that adding order of NGO significantly affected the fluorescence change of P1 caused by DNase I. By comparing the S/B ratios of the two strategies, the signal of sample (P1 + E + NGO) increased about 55-fold, while the other sample (P1 + NGO + E) only increased about 17-fold (Fig. 2C and D). This result indicated that the “post-mixing” strategy is an ideal choice for manipulation of enzymatic reaction under optimized conditions without disturbance of other materials.
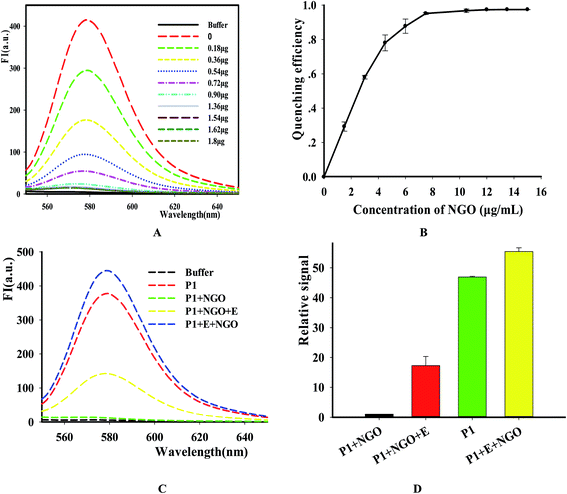 |
| | Fig. 2 The quenching efficiency of NGO on the P1. The concentrations of NGO are 0 to 16 μg mL−1, respectively. (A) Fluorescence emission spectra of TAMRA-labeled P1 upon the addition of different concentration of NGO, the indicated amount of NGO was incubated with the TAMRA-labeled substrate. After incubation for 15 min, the fluorescence was measured. (B) Fluorescence intensity versus concentration of NGO. Excitation: 521 nm. Emission: 578 nm. (C) The fluorescence intensity changes with different adding order of DNase I and NGO by using P1 as the representative. (D) The S/N detection of NGO-quenched P1 at different reaction conditions. [NGO] = 12 μg mL−1, [P1] = 100 nM. | |
3.2 Optimization of reaction time
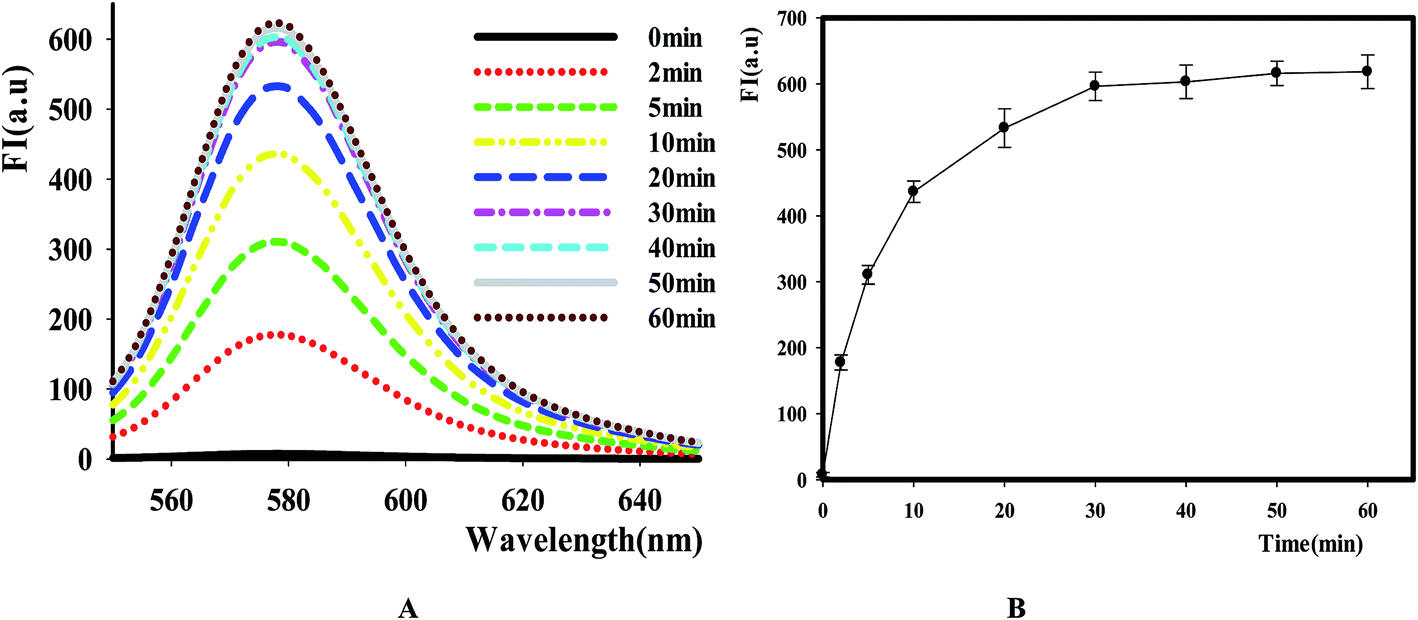
After identifying this NGO-based sensing platform for DNase assay, we then optimized the reactive time of reaction by comparing the fluorescence change at different points. The result in Fig. 3 showed that the fluorescence intensity increased as the extending of reaction time in the range of 0 to 60 min and it reached the maximum value after 30 min. Thus, this value was selected as the time point for NGO addition in the following experiments.
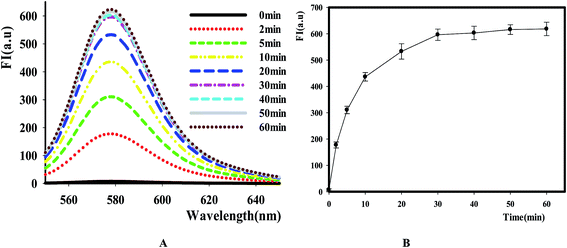 |
| | Fig. 3 The effect of time on the reaction catalyzed by DNase I. [P1] = 100 nM, E = 8 Units, [NGO] = 12 μg mL−1. (A) Fluorescence emission spectra of TAMRA-labeled P1 caused by DNase I at different time points. (B) Relative fluorescence intensity versus reactive time, Ex/Em = 521/578 nm. | |
3.3 DNase I assay
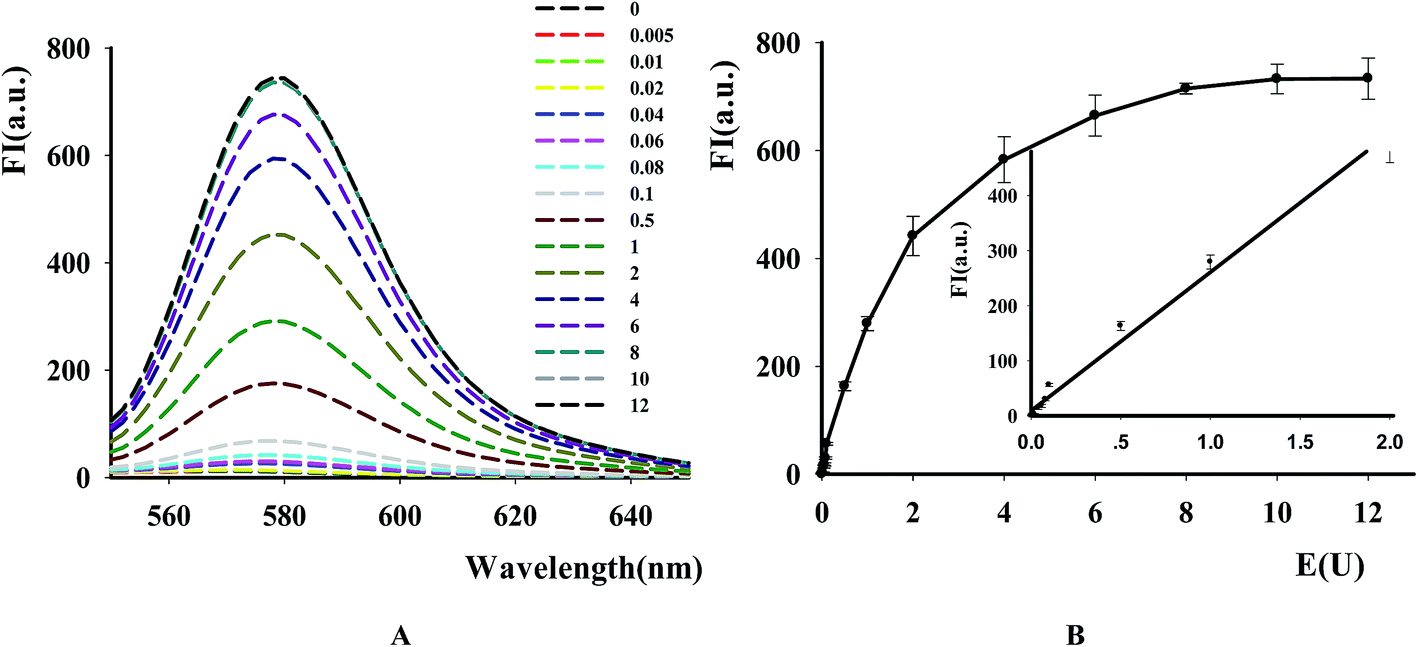
The new assay was further used to evaluate the applicability for DNase assay. From the result in Fig. 4A, it was found that the fluorescence intensity of the substrates dramatically became higher with the increase of DNase I from 0.5 × 10−2 to 10 units. The calibration curve for DNase detection in Fig. 4B showed the linear range from 0.5 × 10−2 to 2 units with linear equation y = 251.66x + 9.3582, where y is the ratio of F−F0 (where F0 and F are the fluorescence intensities of TAMRA at 578 nm in the absence and presence of DNase, respectively) and x is the DNase concentration (regression coefficient R2 = 0.99). The detection limit is estimated to be 5 × 10−3 units (3S0/S, in which S0 is the standard deviation for the blank solution, and S is the slope of the calibration curve). These results demonstrated that the NGO-based biosensor could be used for sensitive DNase assay and showed the excellent differentiate capability of NGO to DNA fragments with different lengths and high quenching efficiency on the DNA probe.
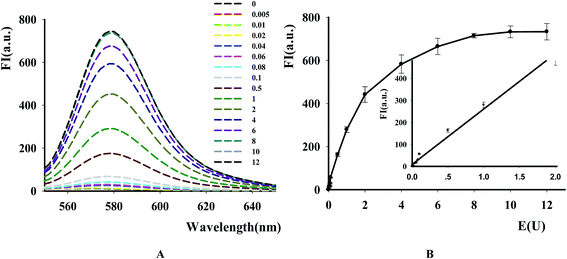 |
| | Fig. 4 The effect of DNase concentration on the fluorescence of P1. [P1] = 100 nM, [NGO] = 12 μg mL−1. (A) Fluorescence emission spectra of TAMRA-labeled P1 caused by DNase I with different concentration. (B) Fluorescence intensity versus DNase I concentration. Ex/Em = 521/578 nm. The inset in (B) shows the relationship between the fluorescence intensity and DNase concentration. | |
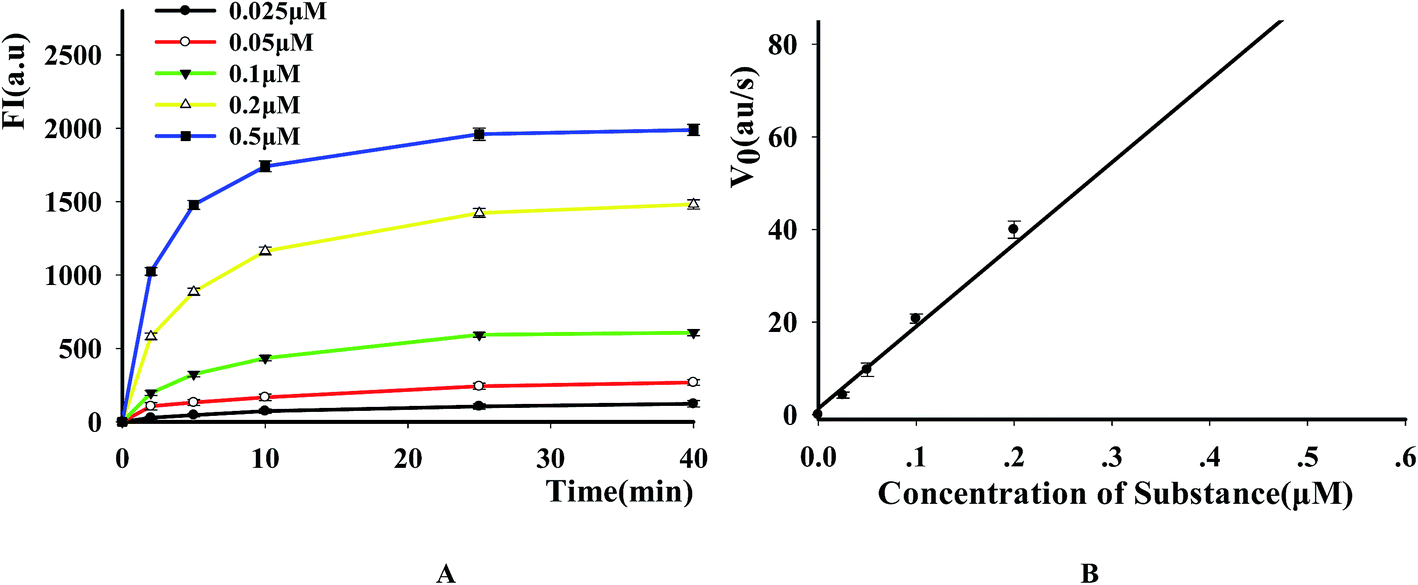
3.4 Kinetic study of DNase
To evaluate the suitability of the fluorescence-based DNase assay, we used it to carry out kinetics study of this enzyme. After incubating a fixed amount of DNase with increasing P1 concentrations, we measured the fluorescence changes as a function of time and the results indicated that the fluorescence signal increased as the increase of reactive time and substrate concentration (Fig. 5A). After obtaining the initial rates of reactions under the conditions of different substrate concentrations by linear regression, the substrate concentration was plotted versus the initial rate of reaction (Fig. 5B). From this figure, it was found that the initial velocity is linearly related with the substrate concentration in the investigated range. By using a Lineweaver–Burke plot, we determined the Km constant for DNase I for its substrate as 2.19 ± 0.07 μM. It is similar like other methods (Table S3†).
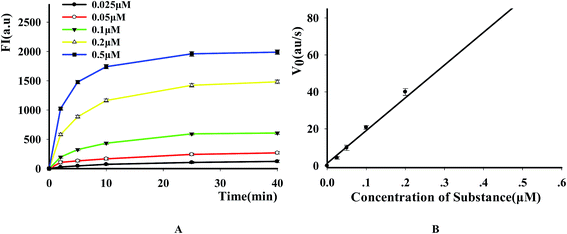 |
| | Fig. 5 Kinetic study of DNase I (A) time scan results of the reaction with different substrate concentrations. The time points are 2, 5, 10, 15, 20 and 30 min, respectively, the concentrations of 1 to 5 are 0.025, 0.05, 0.1, 0.2 and 0.5 μM, respectively. (B) The relation between the substrate concentration and the initial velocity. E = 8 U. | |
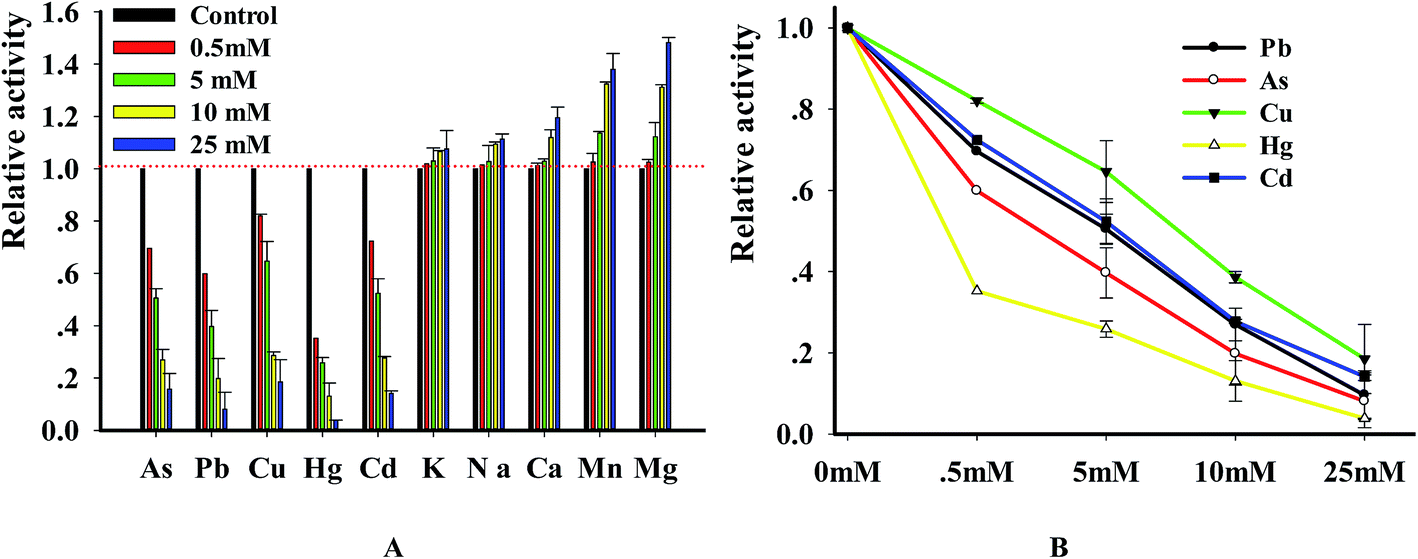
3.5 Inhibition of DNase by heavy metal ions
DNase degraded DNA in a divalent metal ion dependent manner and it prefers Mg2+ for DNA hydrolysis activity.25,26 In order to explore the relation of metal ions with enzyme, we investigated the changes of DNase activity caused by heavy metal ions. In brief, the substrate was incubated in the solution containing 10 mM MgCl2 with various metal ions. The results (Fig. 6A) indicated that the investigated ions strongly inhibited DNase activity in a concentration-dependent manner. Using the double reciprocal plot method, we got the IC50 (half- maximal inhibitory concentration) values of Pb2+, Hg2+, Cu2+, As3+ and Cd2+ with 4.95 ± 0.65, 0.39 ± 0.02, 7.82 ± 1.05, 2.7 ± 1.12 and 5.34 ± 0.46 mM, respectively. To ensure that metal ions were confidential inhibitors of DNase, we performed the reaction in the presence of 4 metal ions and analyzed the enzymatic products using PAGE method. As expected, DNase digested the substrate and produced a new band whereas the substrate remained intact in control reactions that didn't contain enzyme. However, the digested efficiency was greatly reduced in the presence of the 4 metal ions (Fig. S3†). The results, which were consistent with previous reports,27,28 indicated that the new method can be reliably used to investigate the relation of enzyme with metal ions.
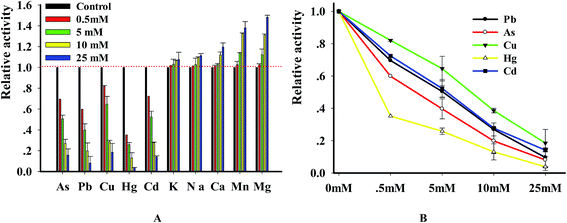 |
| | Fig. 6 (A) The graphical presentation of the effects of metal ions on the DNase activity. Relative incision activity is based on a percentage comparison between the enzyme-only control standard incision and the individual incision reaction of interest. Values shown are relative DNase digestion activities, normalized to the 0 mM, no metal control (listed here as 100), and represent the average and standard deviation of at least three independent data points. The final fluorescence at 578 nm was monitored in the presence of different factors. Assays were performed as described in the method. E = 8 U, [P1] = 100 nM. (B) Validation of inhibitory activity caused by metal ions using gel-based product analysis. | |
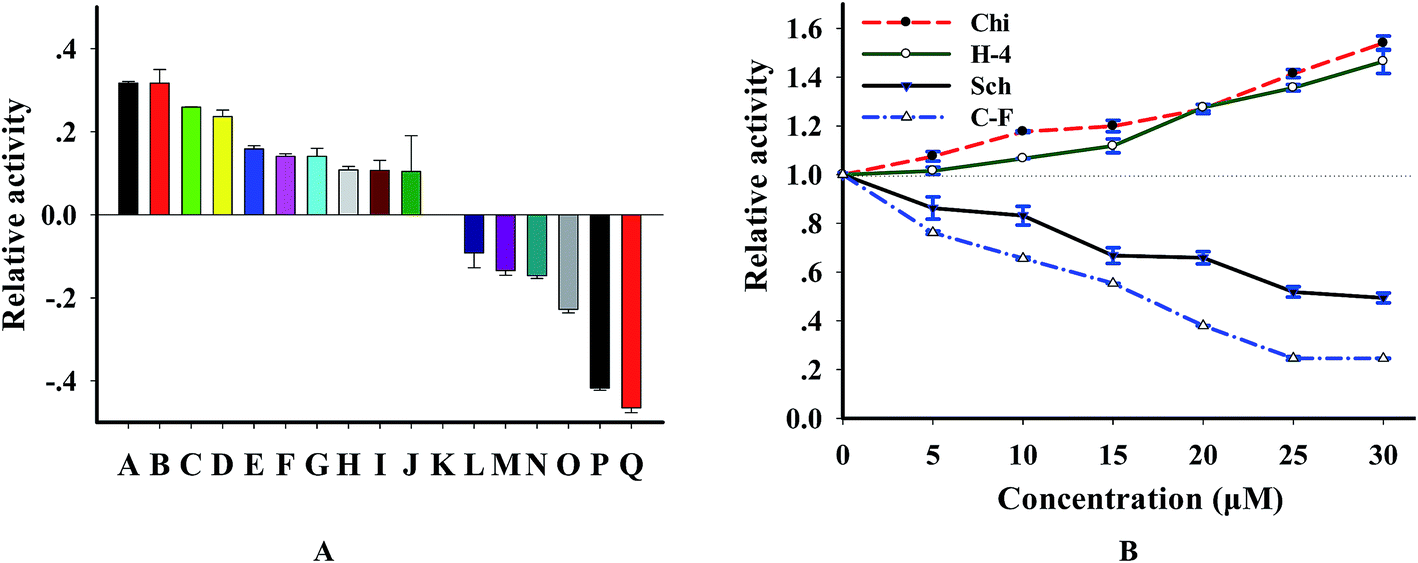
3.6 Compounds screening of DNase I
Since DNase I is tightly associated with several kinds of disease,29,30 it is becoming a useful drug for disease therapy and representing an exciting possibility for developing new therapeutics.31,32 In order to further wide the application of this method and test the potential for screening DNase-targeted drugs, we analyzed the effect of 16 natural compounds purified from Chinese herb on this enzyme. According to the results (Fig. S4† and 7A), we found that 10 compounds are activators, 6 are inhibitors and the others showed no effect when the concentration of compounds is 20 μM. By investigating the concentration effect of 4 compounds (2 inhibitors and 2 activators) on this enzyme, we found from Fig. 7B that the effect of these compounds on DNase I was concentration-dependent. We further tested the relation of enzyme activity and the concentration of 2 strong inhibitors cyclocariol F and schisanlactone E (more information is in Table S2†) and found that IC50 values (the concentration required to reduce enzyme activity by 50%) of the 2 compounds are 12.5 ± 2 μM and 30 ± 3 μM, respectively, according to the lots of maximum fluorescence intensity versus inhibitor concentration. To ensure that the identified compounds were confidential inhibitors and to exclude the possibility that these compounds were identified based on the interference with substrate, we detected the enzymatic products using PAGE method. As expected, new bands of which are shorter than that of the substrate appeared after with DNase digestion, whereas the substrate remained intact in the control reaction without DNase I. Importantly, the digested efficiency was reduced in the presence of cyclocariol F and schisanlactone E. The inhibitory effect of cyclocariol F is stronger than that of schisanlactone E. Meanwhile, significant activating effect was observed for chikusetsusaponin V and C–H-4 (Fig. S5†). Taken together, these results indicate that selective inhibition or activation of DNase using small molecules is wholly feasible. Moreover, the availability of NGO-biosensor opens up routes for the screening as well as the feasibility of identifying other natural compound libraries combing with multi-well plates.
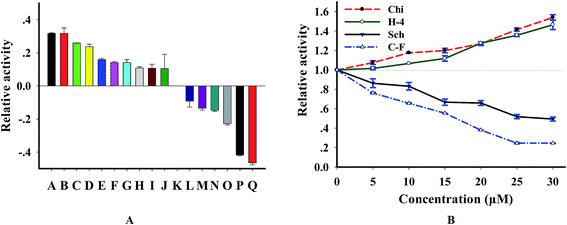 |
| | Fig. 7 (A) Effects of natural compounds on DNase I, these natural compounds were extracted from Cyclocarya paliurus leaves, the natural compounds in samples A to Q are C–H-4, chikusetsusaponin V, C–G-9, C–G-6, C–G-10, chikusetsusaponin IV, C–H-2, chikusetsusaponin IVa, C–H-3, C–H-1, control, C–G-13, C–G-11, SBB–L-1, C–G-8, schisanlactone E and cyclocariol F, respectively, [compound] = 20 μM. (B) The effect of concentration variation of chikusetsusaponin V (Chi), C–H-4 (H-4), cyclocariol F (C–F) and schisanlactone E (Sch) on DNase, [P1] = 100 nM. | |
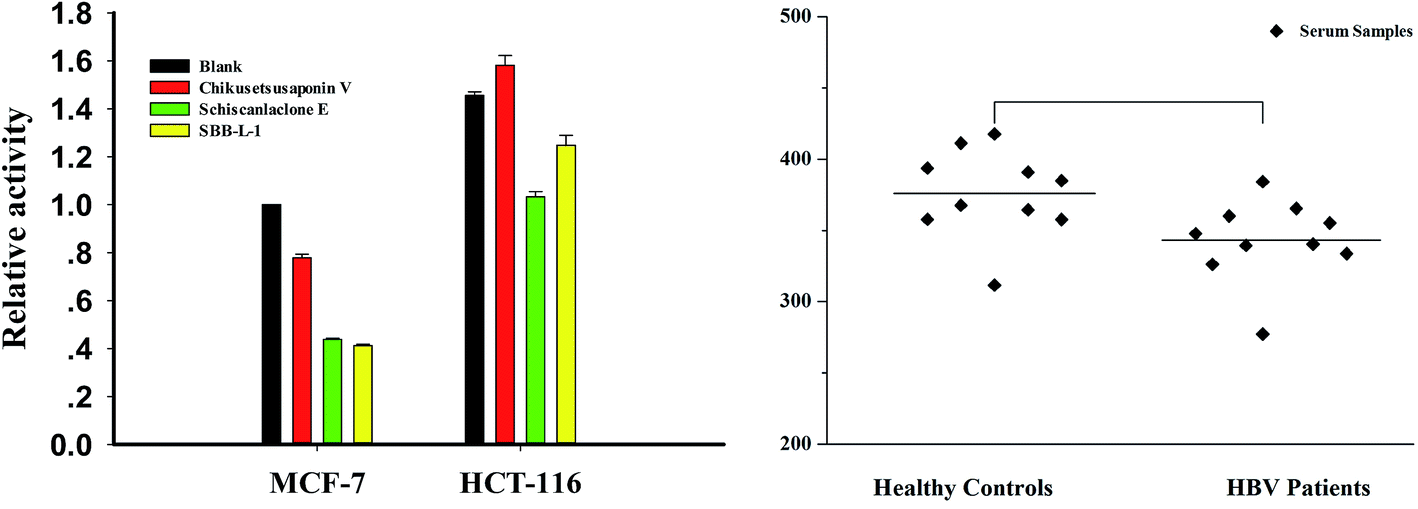
3.7 Assay of DNase in biosamples
Considering the key role in regulating important cellular processes including immuno-defense process and mammalian genome stability,33,34 developing rapid and accurate detection of DNase can provide important information in these areas. To assess the usability of the assay in complex conditions, cell-free extracts and clinic serum samples were used as assay objects. First, we incubated increasing concentrations of the MCF-7 cell-free extracts with P1 and observed a clear dose–response effect with more than 90% of maximal signal at the presence of 5 μg cell-free extracts (data not shown). Meanwhile, crystal violet (CV), a DNase I inhibitor, was used to confirm the specificity of the assay. The result indicated that more than more than 90% of maximal signal was disappeared in the presence of 50 μM CV (Fig. S6†). We then detected DNase level in different tumor cells using 3 μg cell-free extracts (a concentration for part degradation of substrates). From result, it was found that the levels of DNase of HCT-16 was 1.5 folds than that of MCF-7 cells. We also investigated the effect of several compounds, which were identified as effectors of DNase I in vitro in MCF-7 cells and HCT-116 cells and found that schisanlactone E and SBB–L-1 can markedly reduce DNase activity levels of the two cell lines, while no significant effect was observed for chikusetsusaponin V. These results demonstrate that compounds can partly affect DNase I activity in vivo (Fig. 8A). Finally, the method was used to detect DNase activity in clinic serum samples with HBV infection (n = 14) or not (n = 10). We primarily incubated increasing volume of the serum with P1 and found a clear volume-dependent effect. The highest fluorescence signal was observed when 1 μL serum was added. To accurately determine the activity of serums, we used a subsaturating amount of serum (0.5 μL). Comparing the signals obtained from samples of the two groups, we found that the DNase level slightly varied in the samples of same group, while a significant difference of the average level of DNase was observed between the two investigated groups. DNase I level in HBV group was lower than that of control group (Fig. 8B). This result implied that HBV infection could depress the immune ability of patients through inhibiting DNase I activity.35 Taking together, these results fully demonstrated that the NGO-based biosensor can be used to primarily screen DNase effector in vitro and detect DNase in complicated samples as well. The major benefits of the new method are improved experimental reproducibility, elimination of radioactivity and a substantial cost reduction when compared to conventional radio-labeled substrates.
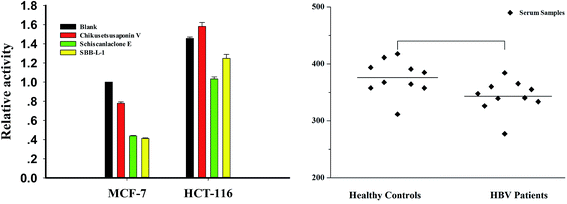 |
| | Fig. 8 DNase activity in cell-free extracts and freshly collected serum samples. Enzymatic digestion was carried out at 45 °C for 30 min; fluorescence signal was collected after NGO addition for 30 min. (A) DNase activity in cell-free extracts. (B) DNase assay in serum samples with HBV infection or not. | |
4. Conclusions
In this paper, we developed a fluorescence-based NGO-biosensor approach as a complement assay for quantitative and biochemical analysis of DNase I, an important target for therapy. Furthermore, by using the assay for screening of natural compounds, we got several DNase activators and inhibitors, which demonstrate the utility of the assay as well as the feasibility of developing selective effectors of this enzyme. Such inhibitors will be highly useful tools as chemical probes that complement existing resources available for the function study of this enzyme from different level.
Acknowledgements
This work was partially supported by the Natural Science Foundation of China (81374062, 85673179 and 31672457), the Natural Science Foundation of Hunan Province (h14JJ2049), the Science Foundation for Outstanding Young Scholars of Hunan Province (2015JJ1007) and the Fundamental Research Funds for the Central Universities of China (2015JCA03).
References
- H. Fischer, J. Scherz, S. Szabo, M. Mildner, C. Benarafa, A. Torriglia and E. Tschachler, Eckhart L DNase 2 is the main DNA-degrading enzyme of the stratum corneum, PLoS One, 2011, 6, e17581 CAS.
- P. P. Cardenas, B. Carrasco and H. Sanchez, et al., Bacillus subtilis polynucleotide phosphorylase 3′-to-5′ DNase activity is involved in DNA repair, Nucleic Acids Res., 2009, 37(12), 4157–4169 CrossRef CAS.
- T. Hendriks, M. de Hoog and M. H. Lequin, et al., DNase and atelectasis in non-cystic fibrosis pediatric patients, Critical Care, 2005, 9(4), 1 CrossRef PubMed.
- H. Sun, B. Qin and T. Liu, et al., CistromeFinder for ChIP-seq and DNase-seq data reuse, Bioinformatics, 2013, 29(10), 1352–1354 CrossRef CAS PubMed.
- C. N. N'Soukpoe-Kossi, S. Diamantoglou and H. A. Tajmir-Riahi, DNase I-DNA interaction alters DNA and protein conformations, Biochem. Cell Biol., 2008, 86(3), 244–250 CrossRef.
- L. Ge, X. Zhou and W. J. Ji, et al., Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy, Am. J. Physiol.: Heart Circ. Physiol., 2015, 308(5), H500–H509 CrossRef CAS PubMed.
- K. Samejima, H. Ogawa and A. V. Ageichik, et al., Auxin-induced Rapid Degradation of Inhibitor of Caspase-activated DNase (ICAD) Induces Apoptotic DNA Fragmentation, Caspase Activation, and Cell Death A Cell Suicide Module, J. Biol. Chem., 2014, 289(45), 31617–31623 CrossRef CAS PubMed.
- M. Oliveri, A. Daga and C. Cantoni, et al., DNase I mediates internucleosomal DNA degradation in human cells undergoing drug-induced apoptosis, Eur. J. Immunol., 2001, 31(3), 743–751 CrossRef CAS PubMed.
- B. Vogel, H. Shinagawa and U. Hofmann, et al., Acute DNase 1 treatment improves left ventricular remodeling after myocardial infarction by disruption of free chromatin, Basic Res. Cardiol., 2015, 110(2), 1–15 CrossRef CAS PubMed.
- G. H. Li, Y. Shi and Y. Chen, et al., Gelsolin regulates cardiac remodeling after myocardial infarction through DNase I-mediated apoptosis, Circ. Res., 2009, 104(7), 896–904 CrossRef CAS PubMed.
- A. Cherepanova, S. Tamkovich and D. Pyshnyi, et al., Immunochemical assay for deoxyribonuclease activity in body fluids, J. Immunol. Methods, 2007, 325(1), 96–103 CrossRef CAS PubMed.
- D. Sinicropi, D. L. Baker and W. S. Prince, et al., Colorimetric determination of DNase I activity with a DNA-methyl green substrate, Anal. Biochem., 1994, 222(2), 351–358 CrossRef CAS PubMed.
- P. Friedhoff, S. E. Matzen and G. Meiss, et al., A quantitative microtiter plate nuclease assay based on ethidium/DNA fluorescence, Anal. Biochem., 1996, 240(2), 283–288 CrossRef CAS PubMed.
- X. Xu, M. S. Han and C. A. Mirkin, A Gold-Nanoparticle-Based Real-Time Colorimetric Screening Method for Endonuclease Activity and Inhibition, Angew. Chem., 2007, 119(19), 3538–3540 CrossRef.
- P. C. Ray, A. Fortner and G. K. Darbha, Gold nanoparticle based FRET assay for the detection of DNA cleavage, J. Phys. Chem. B, 2006, 110(42), 20745–20748 CrossRef CAS PubMed.
- Z. Zhou, C. Zhu and J. Ren, et al., A graphene-based real-time fluorescent assay of deoxyribonuclease I activity and inhibition, Anal. Chim. Acta, 2012, 740, 88–92 CrossRef CAS PubMed.
- N. M. Robertson, M. S. Hizir and M. Balcioglu, et al., Monitoring the multitask mechanism of DNase I activity using graphene nanoassemblies, Bioconjugate Chem., 2015, 26(4), 735–745 CrossRef CAS PubMed.
- W. Xu, Z. Xie and C. Tong, et al., A rapid and sensitive method for kinetic study and activity assay of DNase I in vitro based on a GO-quenched hairpin probe, Anal. Bioanal. Chem., 2016, 408(14), 3801–3809 CrossRef CAS PubMed.
- J. Ge, Z. M. Huang and Q. Xi, et al., A novel graphene oxide based fluorescent nanosensing strategy with hybridization chain reaction signal amplification for highly sensitive biothiol detection, Chem. Commun., 2014, 50(80), 11879–11882 RSC.
- D. M. Zhou, Q. Xi and M. F. Liang, et al., Graphene oxide-hairpin probe nanocomposite as a homogeneous assay platform for DNA base excision repair screening, Biosens. Bioelectron., 2013, 41, 359–365 CrossRef CAS PubMed.
- J. Zhou, X. Xu and W. Liu, et al., Graphene oxide–peptide nanocomplex as a versatile fluorescence probe of protein kinase activity based on phosphorylation protection against carboxypeptidase digestion, Anal. Chem., 2013, 85(12), 5746–5754 CrossRef CAS PubMed.
- J. Gang, Graphene Oxide-based Direct Measurement of DNase I Activity with Single Stranded DNA, Bull. Korean Chem. Soc., 2014, 35(9), 2749 CrossRef CAS.
- C. H. Lu, H. H. Yang and C. L. Zhu, et al., A graphene platform for sensing biomolecules, Angew. Chem., 2009, 121(26), 4879–4881 CrossRef.
- S. He, B. Song and D. Li, et al., A graphene nanoprobe for rapid, sensitive, and multicolor fluorescent DNA analysis, Adv. Funct. Mater., 2010, 20(3), 453–459 CrossRef CAS.
- J. Wallach and P. Fonlupt, Effect of Mg2+ and Mn2+ on hydrolysis of calf thymus DNA by pancreatic deoxyribonuclease I, J. Inorg. Biochem., 1980, 13(3), 233–245 CrossRef CAS.
- V. W. Campbell and D. A. Jackson, The effect of divalent cations on the mode of action of DNase I. The initial reaction products produced from covalently closed circular DNA, J. Biol. Chem., 1980, 255(8), 3726–3735 CAS.
- J. S. Hanas and C. G. Gunn, Inhibition of transcription factor IIIA-DNA interactions by xenobiotic metal ions, Nucleic Acids Res., 1996, 24(5), 924–930 CrossRef CAS.
- L. Peng, J. Fan and C. Tong, et al., An ultrasensitive fluorescence method suitable for quantitative analysis of mung bean nuclease and inhibitor screening in vitro and vivo, Biosens. Bioelectron., 2016, 83, 169–176 CrossRef CAS PubMed.
- B. Zhu, Y. Gong and P. Chen, et al., Increased DNase I activity in diabetes might be associated with injury of pancreas, Mol. Cell. Biochem., 2014, 393(1–2), 23–32 CrossRef CAS PubMed.
- M. Gajic-Veljic, B. Bonaci-Nikolic and B. Lekic, et al., Importance of low serum DNase I activity and polyspecific anti-neutrophil cytoplasmic antibodies in propylthiouracil-induced lupus-like syndrome, Rheumatology, 2015, kev243 Search PubMed.
- S. R. Hymes, T. M. Randis and T. Y. Sun, et al., DNase inhibits Gardnerella vaginalis biofilms in vitro and in vivo, J. Infect. Dis., 2013, 207(10), 1491–1497 CrossRef CAS PubMed.
- Q. Hu, P. S. Katti and Z. Gu, Enzyme-responsive nanomaterials for controlled drug delivery, Nanoscale, 2014, 6(21), 12273–12286 RSC.
- J. Kuribara, H. Tada and Y. Kawai, et al., Levels of serum deoxyribonuclease I activity on admission in patients with acute myocardial infarction can be useful in predicting left ventricular enlargement due to remodeling, Journal of Cardiology, 2009, 53(2), 196–203 CrossRef PubMed.
- A. R. Kim, S. Song and M. Brand, et al., Nucleosome and transcription activator antagonism at human -globin locus control region DNase I hypersensitive sites, Nucleic Acids Res., 2007, 35(17), 5831–5838 CrossRef CAS PubMed.
- F. M. Valle, E. Balada and J. Ordi-Ros, et al., DNase I and systemic lupus erythematosus, Autoimmun. Rev., 2008, 7(5), 359–363 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04911k |
| ‡ These authors contributed to the work equally and should be regarded as co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Xuanming Liua,
Bin Liu
a,
Xuanming Liua,
Bin Liu