
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative dehydrogenation of n-butane to butenes on Mo-doped VMgO catalysts
Xue Liu ab,
Linhai Duan*a,
Weishen Yangb and
Xuefeng Zhu*b
ab,
Linhai Duan*a,
Weishen Yangb and
Xuefeng Zhu*b
aCollege of Chemistry, Chemical Engineering and Environmental Engineering, Liaoning Shihua University, Fushun, 113001, P. R. China. E-mail: lhduan@126.com; Tel: +86-024-5686-0757
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, P. R. China. E-mail: zhuxf@dicp.ac.cn; Tel: +86-411-8437-9182
First published on 7th July 2017
Abstract
VMgO catalysts with different molybdenum doping amounts were prepared by an impregnation method. The structure, specific surface area, and basic and redox properties of these catalysts were determined by XRD, BET, CO2-TPD, and H2-TPR. The XRD results revealed that all catalysts contained an orthovanadate phase (Mg3(VO4)2), while no metavanadate and pyrovanadate phases were detected. BET surface area analysis showed that the Mo-doped catalysts possessed lower surface areas than the undoped one. The reducing and basicity properties of the catalysts were characterized by H2-TPR and CO2-TPD measurements, which demonstrated that Mo-doping improved the redox temperature and reduced the number of basic sites. The performances of these catalysts were investigated at different C4H10/O2 molar ratios, temperatures, and contact times. The Mo-doping not only improved the selectivity of butenes but also inhibited the deep oxidation reactions, although cracking reactions occurred with high levels of Mo doping. When the Mo/V atomic ratio was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, n-butane conversion of 34.5% and total butene selectivity of 79.3% were achieved at 630 °C. To the best of our knowledge, the oxidative dehydrogenation performance of the synthesized Mo-doped VMgO catalysts described in this work represents a remarkable improvement compared to previous reports.
:100, n-butane conversion of 34.5% and total butene selectivity of 79.3% were achieved at 630 °C. To the best of our knowledge, the oxidative dehydrogenation performance of the synthesized Mo-doped VMgO catalysts described in this work represents a remarkable improvement compared to previous reports.
1 Introduction
Butenes, including 1-butene, 1,3-butadiene, 2-butene, and isobutene, are the third most important petrochemical intermediate products among the olefins.1 They are widely used in various chemical processes for the production of synthetic resins, plastics, rubber, automotive fuel components, and other valuable products.2–6 In view of the increasing demand for butenes, the oxidative dehydrogenation (ODH) of n-butane is regarded as a low-cost and promising process for the production of butenes.7The types of side reactions that would happen during the process of n-butane ODH, such as deep oxidation reactions, cracking reactions and isomerization reactions, are shown in Scheme 1. The ODH of n-butane is very complex and difficult to control. Therefore, a catalyst with high performance is required for the ODH of n-butane.8,9 In recent studies, much effort has been devoted to the development of more efficient catalysts in order to achieve both high activity and selectivity.10–13 The critical issue related to the ODH of butane is that deep oxidation reactions can occur, leading to the low selectivity of butenes. Costine et al. conducted an extensive data mining study on C3 and C4 selective oxidation reactions occurring in the gas phase over solid catalysts, and suggested that there is a clear limitation in terms of selectivity and conversion, beyond which experimental progresses are difficult to achieve. In their study, the selectivity of n-butane to butenes could only reach 60%, when the conversion was 30%.14
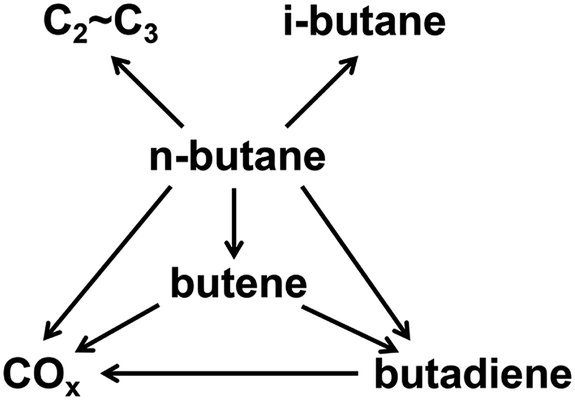 | ||
| Scheme 1 Reactions of the ODH of n-butane. | ||
The effects of the supports, e.g. TiO2, CeO2, ZrO2, Al2O3, and MgO, have been widely investigated for V-based catalysts.8,15–25 To the best of our knowledge, MgO is the best among these supports, and VMgO catalysts have been extensively studied as highly efficient catalysts for the ODH of n-butane.8,15,20–24 It has been reported that Mg3(VO4)2 is the active phase for the ODH of n-butane, while other vanadate phases (such as metavanadate and pyrovanadate) are unfavorable for the production of butenes. However, most studies disclosed that the as-prepared catalysts contained minor quantities of other vanadate phases.22,26 Therefore, the preparation method is critical for obtaining a good VMgO catalyst.
The catalytic performance of the VMgO catalysts can be slightly improved by adding transition metals, such as Ni, Mo, or Zr. Liu et al. examined the influence of the addition of Ni to the VMgO catalysts in the ODH of n-butane. The authors found that a moderate doping amount of Ni (n(Ni)/n(V) = 0.3) could promote the formation of the Mg3(VO4)2 phase and increase the selectivity of butenes.26 Dejoz et al. prepared Mo-doped VMgO catalysts with a Mo:V atomic ratio in the range 0–1.4 to determine the influence of the addition of Mo on the ODH of n-butane. The results demonstrated that the Mo–VMgO-based catalysts showed an enhanced selectivity of the ODH products, and especially to butadiene. However, the selectivity of cracking products increased with increasing Mo-doping amount.15 Lee et al. studied Mg(VO4)2/MgO–ZrO2 catalysts with different Mg:Zr ratios. The MgO–ZrO2 supports were prepared by a gel-oxalate coprecipitation method, while the Mg3(VO4)2/MgO–ZrO2 catalysts were obtained by a wet impregnation method. The results of this study indicated that Mg3(VO4)2/MgO–ZrO2 (Mg:Zr ratio = 4:1) exhibited the best catalytic performance for the ODH of n-butane, providing a total butenes (including 1-butene, 2-butene, and 1,3-butadiene) yield of ∼23%.27 Rischard et al. prepared Mo-doped VMgO catalysts, and performed the ODH reaction at different O2/n-butane molar ratios in a two-zone fluidized bed reactor. When the reaction was conducted at 580 °C and the O2/n-butane molar ratio was 1.9, the selectivity of 1,3-C4H6 reached 50.7% with an n-butane conversion of 64.5%. Particularly, the authors reported that the catalysts could be regenerated in a single vessel.28
In this work, a series of 10 wt% V2O5/MgO catalysts devoid of metavanadate and pyrovanadate phases were prepared via an impregnation method. Oxalic acid was used to dissolve ammonium metavanadate and ammonium molybdate in order to allow a good dispersion of the V and Mo species on the catalyst surface. The influence of the Mo:V atomic ratio on the physical and chemical properties was determined by various analytical techniques and the catalytic performance was investigated in a fixed bed reactor. We found that the addition of a small amount of Mo can significantly change the redox and basic properties of the corresponding catalyst, improving the total butenes selectivity.
2 Experimental
2.1 Catalyst preparation
All catalysts were prepared by an impregnation method. The different Mo:V atomic ratios used were 0, 3:100, and 5:100. The MgO support was pretreated in a muffle furnace at 900 °C for 3 h. Appropriate amounts of oxalic acid, ammonium metavanadate, and ammonium heptamolybdate were dissolved in deionized water. The mixed solution was added into a beaker containing the MgO powder and the stirring was continued for 2 h at room temperature. The resultant slurry was dried at 120 °C overnight, and then calcined at 700 °C for 10 h. The resultant powders were pressed into pellets, and then crushed and sieved to 40–60 mesh.
2.2 Catalyst characterization
The surface areas of the catalysts were measured using a BET apparatus (OMNISORP 100CX). The crystalline phases of the catalysts were investigated using a XRD diffractometer (Rigaku, D/MAX-RB) with Cu Kα radiation operated at 40 kV and 100 mA. The infrared spectra were recorded on a Nicolet 6700 Fourier-transform infrared spectroscopy-attenuated total reflectance (FTIR-ATR, Thermo Scientific Co.) spectrometer. H2 temperature programmed reduction (H2-TPR) was performed to elucidate the reducibility of the catalysts using a ChemStar chemisorption analyzer. CO2 temperature programmed desorption (CO2-TPD) was performed to determine the amount of surface basic sites with a ChemStar chemisorption analyzer.2.3 Catalyst performance test
The ODH of n-butane was performed in a fixed bed quartz reactor (10 mm i.d.) under atmospheric pressure. For each catalytic test, 0.50 g of catalyst (40–60 mesh) was used. A thermocouple tube with an o.d. of 5 mm was placed at the center of the quartz reactor. The catalytic reaction conditions were as follows unless otherwise stated: the total space velocity (GHSV) was kept at 58800 h−1; the concentration of n-butane at the feed inlet was 2 vol%; the molar ratio of n-butane to oxygen was 1:1 or 1.5:1; nitrogen was used as balance gas. The contact time was adjusted by changing the total flow rate, while maintaining constant the composition of the feed gas mixture. The products of the reaction were analyzed by an online gas chromatograph equipped with a thermal conductivity detector (TCD). The conversion of n-butane, selectivity to total butenes (including 1-C4H8, 2-t-C4H8, 2-c-C4H8, and 1,3-C4H6), selectivity to monobutenes (including 1-C4H8, 2-t-C4H8, and 2-c-C4H8) and selectivities to other products were calculated based on the following equations, respectively:
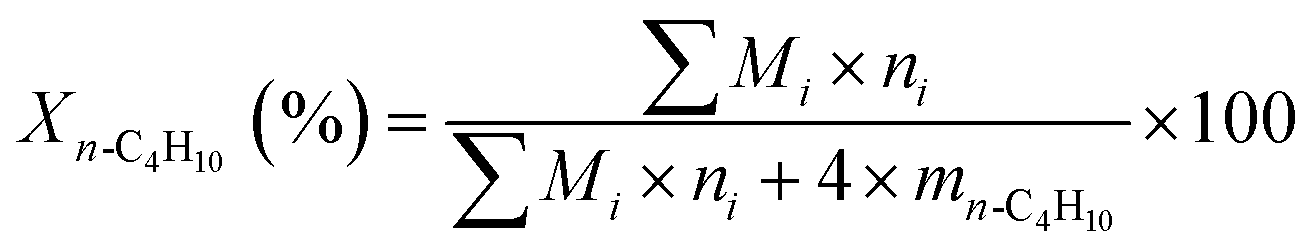 | (1) |
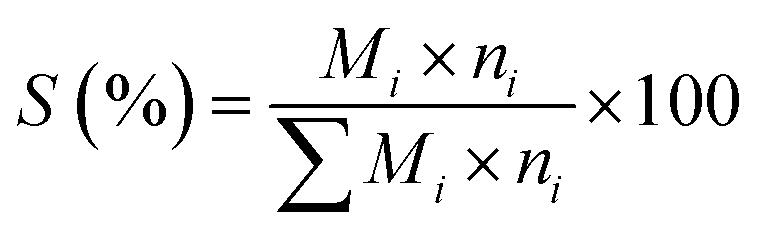 | (2) |
3 Results and discussion
3.1 Catalyst characterization
The BET surface areas of all the samples before and after the reaction are listed in Table 1. A slight decrease in the surface area can be noticed as the doping amount of molybdenum increases. The specific surface area of all the catalysts decreased to about 20 m2 g−1 after the reaction. This decrease may be ascribed to the sintering of the catalysts under the reaction conditions.29| Samples | V2O5 content (wt%) | Mo/V atomic ratio | SBET/(m2 g−1) | |
|---|---|---|---|---|
| Before reaction | After reaction | |||
| VMgO | 10 | 0 | 44.2 | 22.0 |
| 3Mo–VMgO | 10 | 3:100 |
33.8 | 21.0 |
| 5Mo–VMgO | 10 | 5:100 |
26.6 | 17.1 |
Fig. 1 shows the XRD patterns of the as-prepared catalysts. The two crystalline phases can be assigned to MgO (JCPDS no. 45-0946) and Mg3(VO4)2 (JCPDS no. 19-0778) respectively.15,30 No metavanadate and pyrovanadate phases were detected, and no Mo-based crystal phase was found due to the extremely low Mo content and its high dispersion in the catalysts.31
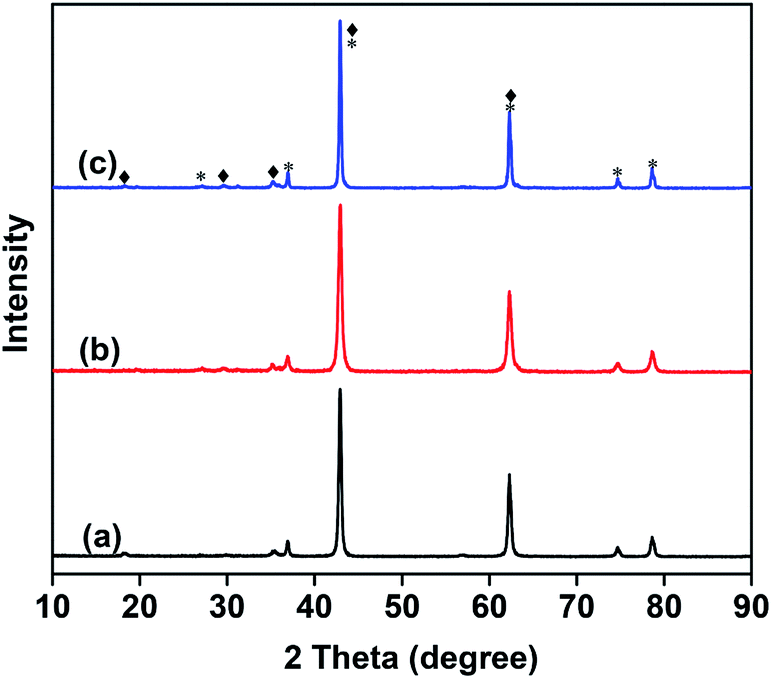 | ||
| Fig. 1 XRD patterns 10 wt% V2O5/MgO catalysts with different Mo doping amounts. (a) VMgO, (b) 3Mo–VMgO, (c) 5Mo–VMgO. (*) MgO, JCPDS no. 45-0946; (♦) Mg3(VO4)2, JCPDS no. 19-0778. | ||
Fig. 2 shows the FTIR spectra of the as-prepared catalysts. All samples exhibited strong absorption bands at 800–900 cm−1, which correspond to the stretching vibration of VO43−.32 As shown in Fig. 2, the stretching vibration of VO43− becomes stronger with the increase in Mo content. The FTIR results are consistent with those of XRD, further confirming the absence of metavanadate and pyrovanadate phases except for the Mg3(VO4)2 phase.
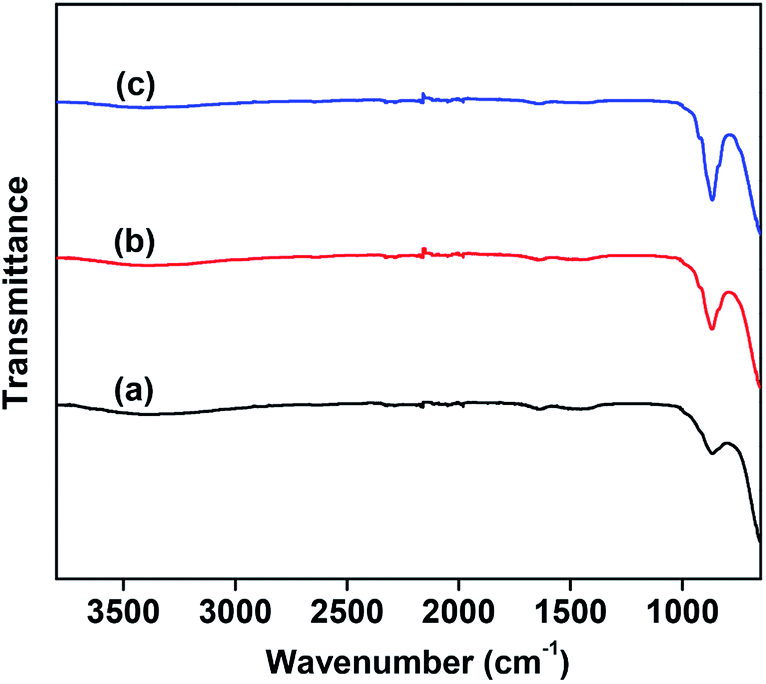 | ||
| Fig. 2 FTIR spectra of the 10 wt% V2O5/MgO catalysts with different Mo doping amounts. (a) VMgO, (b) 3Mo–VMgO, (c) 5Mo–VMgO. | ||
The number of basic sites and the basic strength that significantly affect the CO2 desorption were measured by means of CO2-TPD. The results are shown in Fig. 3. For comparison, the CO2-TPD curve of the MgO support was also provided, as shown in Fig. 3(a). The VMgO catalyst without Mo doping exhibited a similar desorption peak area to the peak of the MgO support. This suggests that the VMgO catalyst has a similar number of basic sites to that of the MgO support. The increase in the Mo-doping amount strongly reduced the number of basic sites, but did not obviously change the basic strength since the CO2 desorption peak at about 300 °C did not shift to high or low temperatures after the Mo-doping. With the increase in the Mo-doping amount, the CO2 desorption peak area decreased significantly according to the order 5Mo–VMgO < 3Mo–VMgO < VMgO.
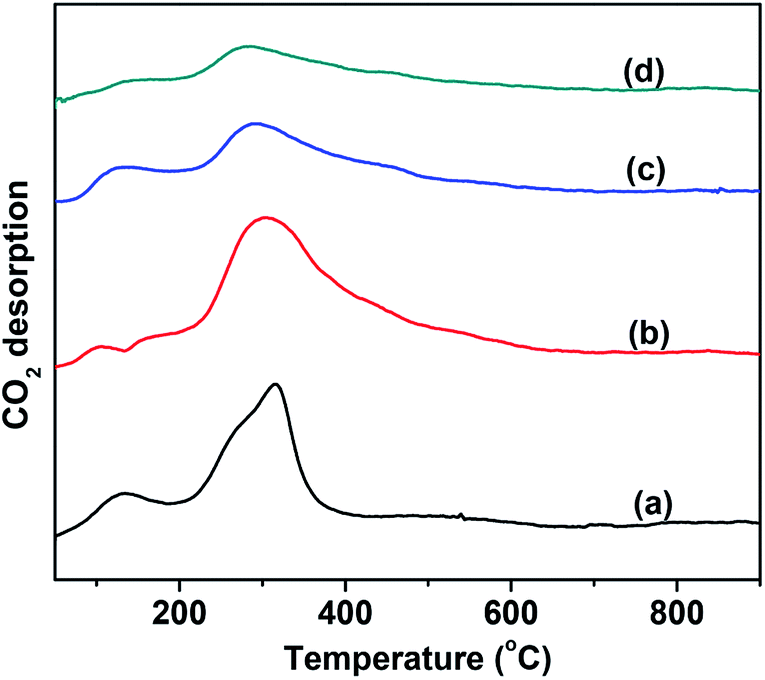 | ||
| Fig. 3 CO2-TPD profiles of the 10 wt% V2O5/MgO catalysts with different Mo-doping amounts. (a) MgO, (b) VMgO, (c) 3Mo–VMgO, (d) 5Mo–VMgO. | ||
The catalytic behavior of VMgO and the Mo-doped VMgO catalysts during the ODH of n-butane strongly depends on the redox properties of the V-species. Therefore, the redox properties of the catalysts were characterized by H2-TPR, and the results are shown in Fig. 4. It can be seen that the content of reducible lattice oxygen slightly increases with the increase in Mo-doping amount, while the reduction peak shifts to higher temperatures. This indicates that the V–O bonding energy increases with the addition of Mo.
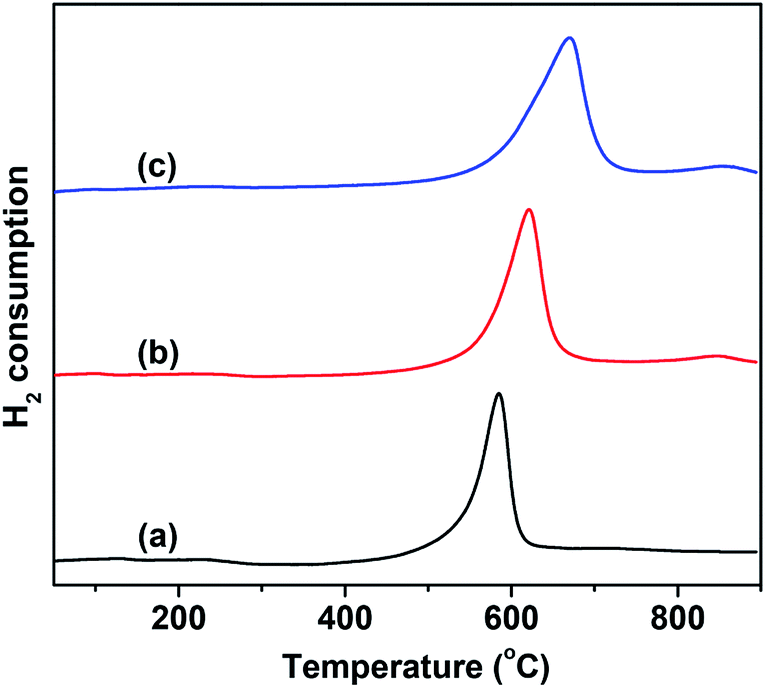 | ||
| Fig. 4 H2-TPR profiles of the 10 wt% V2O5/MgO catalysts with different Mo-doping amounts. (a) VMgO, (b) 3Mo–VMgO, (c) 5Mo–VMgO. | ||
It is generally believed that the ODH of alkanes requires moderately active lattice oxygen,28,33–35 since more highly active lattice oxygen can easily lead to the deep oxidation of alkanes, thus resulting in a low selectivity, while much less active lattice oxygen is responsible for low conversion. The H2 consumption peak appearing at high temperatures suggests that the lattice oxygen of the catalyst is less active upon Mo-doping, as shown in Fig. 4. As the Mo/V ratio increased from 0 to 5:100, the H2 consumption peak gradually shifted from 580 °C to 630 °C. Therefore, a lower selectivity to COx is expected to be achieved on the 5Mo–VMgO catalyst due to the lower oxygen lattice reactivity of the Mg3(VO4)2 phase, as measured by H2-TPR.
3.2 Catalytic performance
:1. Monobutenes, butadiene, and COx were the main carbon-containing products. Some small amounts of other carbon-containing products like C2–C3 olefins were also detected as by-products of the ODH of n-butane.
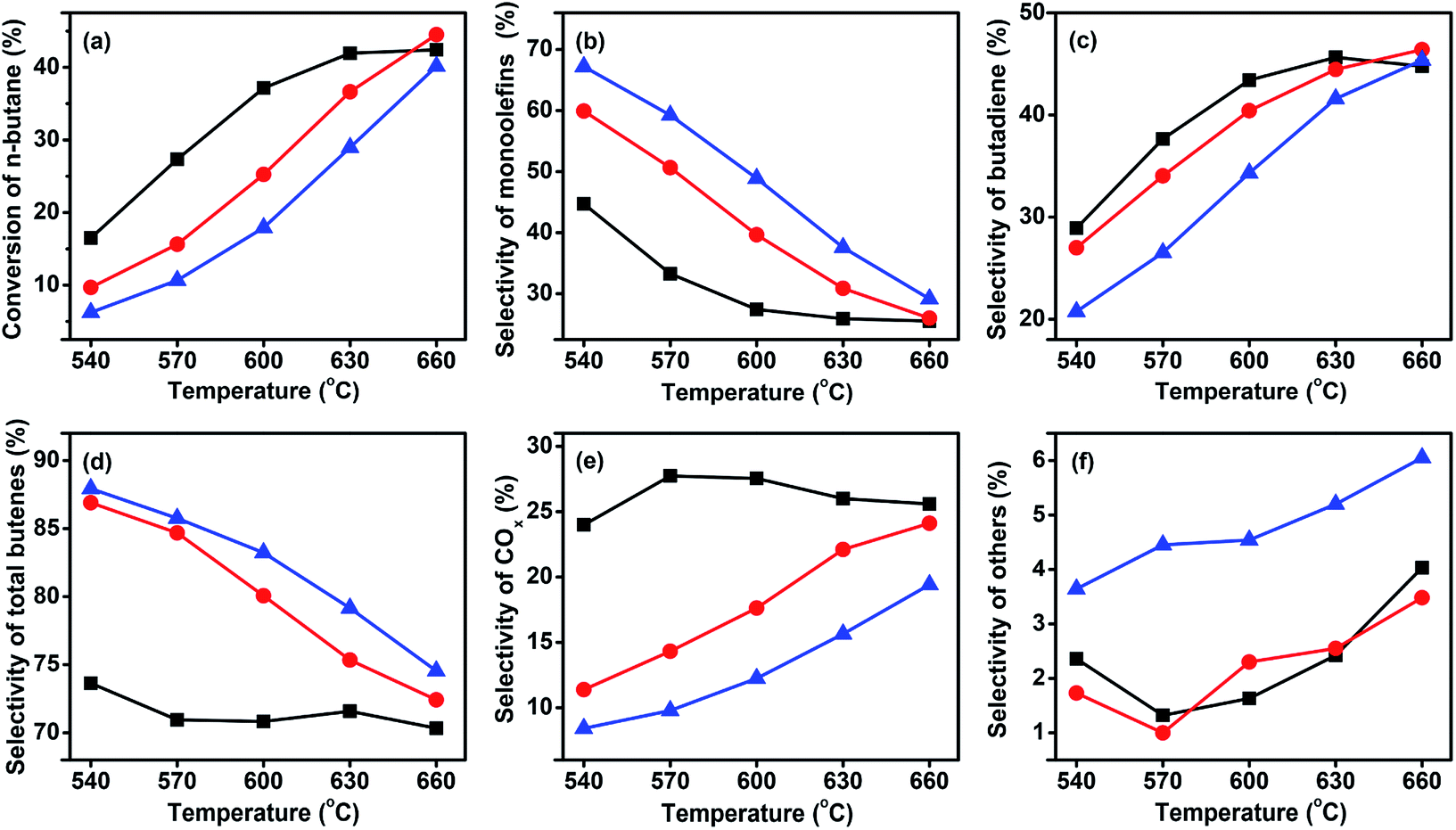 | ||
Fig. 5 Catalytic performance of the three catalysts at GHSV of 58800 h−1 and a C4H10/O2 molar ratio of 1:1. ( ) VMgO, ( ) VMgO, ( ) 3Mo–VMgO, ( ) 3Mo–VMgO, ( ) 5Mo–VMgO. ) 5Mo–VMgO. | ||
As expected, with the rising of the temperature the conversion increased, while the selectivity decreased. Furthermore, the selectivity of monobutenes gradually decreased, whereas that of butadiene increased upon increasing temperatures, suggesting that the monobutenes were gradually converted into butadiene during the ODH of n-butane. This phenomenon has also been reported by other researchers.9,36–38 The selectivity to total butenes gradually decreased with the rising of temperature. The selectivities to other products, such as C2–C3, increased with the addition of Mo, indicating that more Mo-doping leads to cracking reactions. In the temperature range of 540–630 °C, the Mo-free sample, VMgO, acted as the most active catalyst. This result was consistent with the H2-TPR results. At 630 °C, the n-butane conversion over the VMgO catalyst reached 42.8% at a C4H10/O2 molar ratio of 1:1. This value was higher than those obtained using 3Mo–VMgO (37.0%) and 5Mo–VMgO (30.1%) under the same conditions. Thus, the addition of Mo can decrease the activity of the catalyst, accompanied by an improvement of the selectivity to total butenes and inhibition of the deep oxidation reactions. The total butenes selectivity over the VMgO catalyst at 630 °C reached 71.6%, while that on the 3Mo–VMgO and 5Mo–VMgO catalysts was 75.4 and 79.2%, respectively. For the selectivity to butadiene, the value obtained using the VMgO catalyst was similar to that of 3Mo–VMgO and close to 45% at 630 °C, while that of 5Mo–VMgO was 41.6%. All these catalysts exhibited better performances than those reported in the literature.8,15,26,30,35
As discussed above, while the conversion of n-butane decreased, the selectivity to total butenes increased with the increase of the Mo-doping amount. This change can be attributed to the fact that the addition of Mo might deactivate part of the high active sites on the catalyst surface and suppress the deep oxidation reactions to produce COx. Therefore, a small amount of Mo oxide can enhance the ODH performance of butane, in agreement with other literature reports.31
For selective oxidation reactions, the selectivity is usually more important than the conversion; in the presence of a certain catalyst, a higher conversion often means lower selectivity, higher consumption of raw materials, more heat release, and higher risk of thermal runaway, thus the conversion is usually regulated at low levels. The ODH of butane is strongly exothermic, and during our study it was found difficult to control the catalyst bed temperature at a C4H10/O2 ratio lower than 1. Thus, the three catalysts were tested at a higher C4H10/O2 ratio. Fig. 6 shows the performance of the three catalysts at a C4H10/O2 molar ratio of 1.5:1. In comparison to the reaction at a C4H10/O2 molar ratio of 1:1, an increase of total butenes selectivity was accompanied by a slight decrease of butane conversion. For example, for the 3Mo–VMgO catalyst at 630 °C and C4H10/O2 molar ratio of 1:1, the total butenes selectivity and the butane conversion were 75.4 and 37.0%, respectively, while those at a 1.5:1 C4H10/O2 molar ratio were 79.3 and 34.5%, respectively.
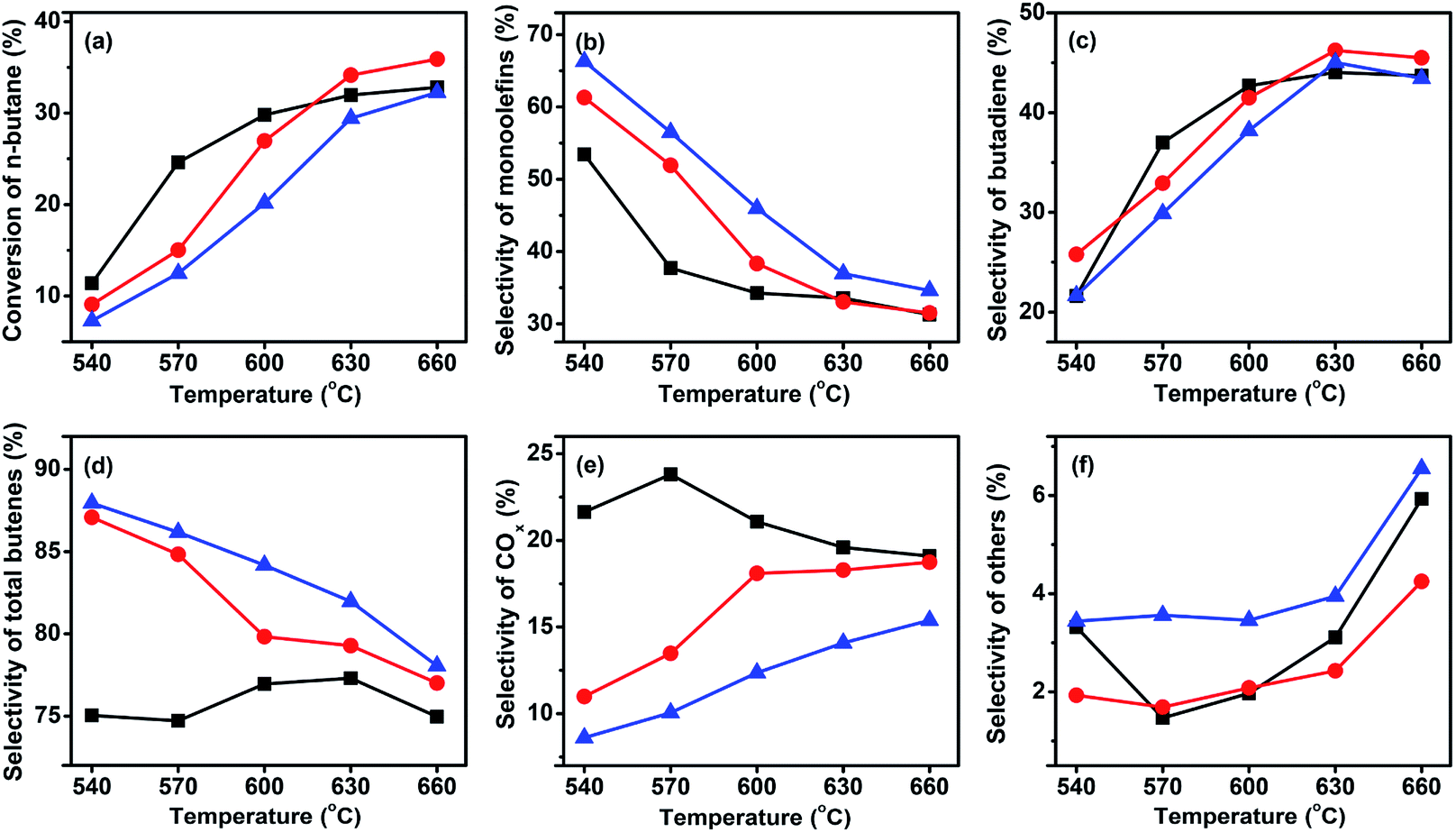 | ||
Fig. 6 Catalytic performance of the three catalysts at GHSV of 58800 h−1 and a C4H10/O2 molar ratio of 1.5:1. ( ) VMgO, ( ) VMgO, ( ) 3Mo–VMgO, ( ) 3Mo–VMgO, ( ) 5Mo–VMgO. ) 5Mo–VMgO. | ||
The catalytic activity of vanadium-based catalysts is closely related to the activity of the lattice oxygen, and it is based on the redox cycle V5+ ↔ V4+.15,39 For the reaction, the lattice oxygen is consumed by oxidation reactions, while being supplied by oxygen gas. The results of the H2-TPR analysis indicate that the addition of Mo to the VMgO catalyst can increase the reduction temperature of V5+ to V4+, therefore the catalytic activity is reduced but accompanied by an improvement of the selectivity to total butenes. A small amount of Mo-doping can significantly improve the selectivity to total butenes, however further additions of Mo can lead to an increase of cracking reactions. As shown in Fig. 5 and 6, when the Mo/V ratio was 3:100, no effect on the cracking reactions was observed, while a significant increase in the selectivity to total butenes was noticed. However, as the Mo/V ratio slightly increased to 5:100, the selectivity to cracking reactions was more than doubled. The results of the CO2-TPD experiments indicate that the Mo-doping reduced the number of basic sites on the catalysts, thus higher levels of Mo-doping might promote the cracking reactions. Dejoz et al. found that the Lewis acid sites increased with the addition of Mo to the VMgO catalyst, and reported a similar finding in their study, i.e., that the selectivity of the cracking reactions increased with Mo-doping increases.15 Therefore, the optimal Mo/V ratio for the Mo–VMgO catalysts is 3:100 by considering the n-butane conversion and total butenes selectivity.
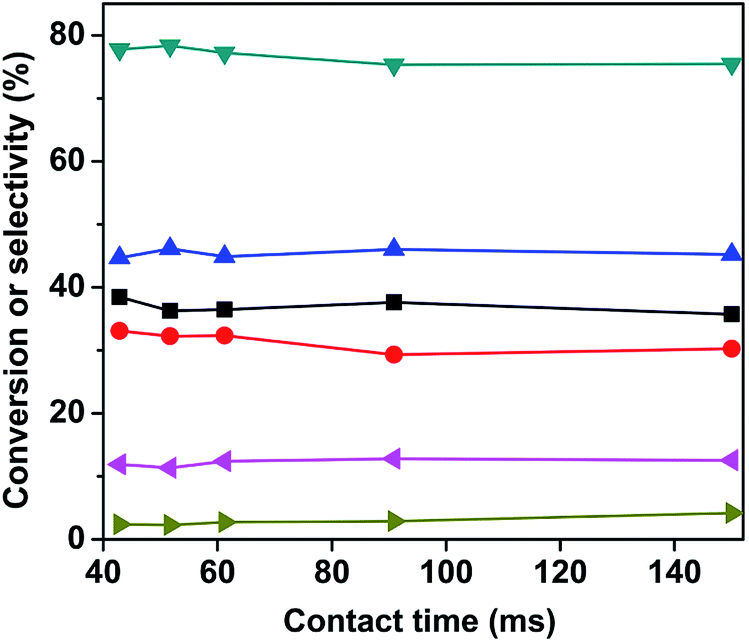 | ||
Fig. 7 Effect of the contact time on the catalytic performance. Reaction conditions: 630 °C, C4H10/O2 molar ratio = 1.5:1. ( ) n-butaneconversion, ( ) n-butaneconversion, ( ) total butenes selectivity, ( ) total butenes selectivity, ( ) butadiene selectivity, ( ) butadiene selectivity, ( ) monoolefin selectivity, ( ) monoolefin selectivity, ( ) COx selectivity, ( ) COx selectivity, ( ) other products selectivity. ) other products selectivity. | ||
:1 and 630 °C, and the results are shown in Fig. 8. As it can be seen, the conversion of n-butane was maintained around 34%, the total butenes selectivity remained close to 80%, and the butadiene selectivity was around 45% within the first 50 h. After 50 h, the n-butane conversion and butadiene selectivity decreased slightly, however the total butenes selectivity gradually increased. This change in conversion and selectivity was related to the catalyst sintering, which resulted in the reduction of the specific surface area (as shown in Table 1) and number of active sites.
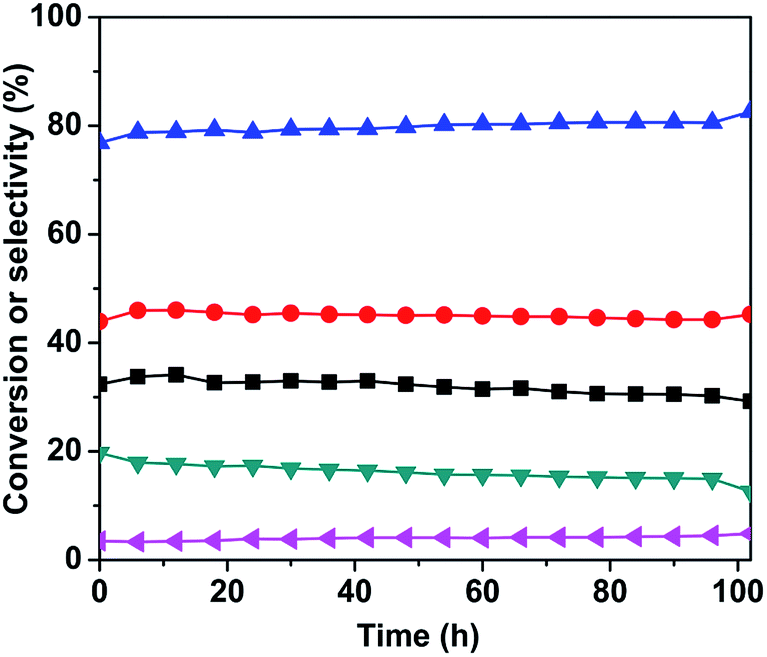 | ||
Fig. 8 Stability of the 3Mo–VMgO catalyst. Reaction conditions: at GHSV of 58800 h−1, 630 °C, C4H10/O2 molar ratio = 1.5:1. ( ) n-butaneconversion, ( ) n-butaneconversion, ( ) total butenes selectivity, ( ) total butenes selectivity, ( ) butadiene selectivity, ( ) butadiene selectivity, ( ) COx selectivity, ( ) COx selectivity, ( ) other products selectivity. ) other products selectivity. | ||
4 Conclusions
Mo-Doped VMgO catalysts containing Mg3(VO4)2 as the only vanadate phase were prepared by an impregnation method. BET surface area tests revealed that the Mo-doped catalysts possessed a lower surface area as comparing to the undoped sample. H2-TPR and CO2-TPD results indicated that the Mo-doping improved the redox temperature, while decreasing the number of basic sites. On the basis of the n-butane ODH catalytic tests, it was found that the addition of a small amount of Mo effectively improved the selectivity of butenes and inhibited the deep oxidation reactions. However, further addition of Mo decreased the catalyst activity, leading to an increase in the selectivity of cracking reactions. Therefore, the optimal Mo/V ratio for this series of catalysts was 3:100 by considering the n-butane conversion and total butenes selectivity. Changing the contact time produced only a slight increase in the selectivities to monoolefins and total butenes. The 3Mo–VMgO catalyst maintained a stable performance during the initial 50 h, but it then underwent a gradual deactivation due to sintering, which induced a reduction of the active sites.
Acknowledgements
The authors gratefully acknowledge financial support from the Fundamental Research Program for Clean Energy and National Natural Science Foundation of China (21476101), Youth Innovation Promotion Association of the Chinese Academy of Sciences, the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB17020400) and Dalian Institute of Chemical Physics (DICP DMTO201503).References
- Y. B. Xu, J. Y. Lu and J. D. Wang, Progr. Chem., 2007, 19, 1481–1487 CAS.
- E. Finocchio, G. Ramis, G. Busca, V. Lorenzelli and R. J. Willey, Catal. Today, 1996, 28, 381–389 CrossRef CAS.
- S. D. Jackson and S. Rugmini, J. Catal., 2007, 251, 59–68 CrossRef CAS.
- P. R. Pujado, B. V. Vora and C. Make, Hydrocarbon Process., 1990, 69, 65–72 CAS.
- S. S. Choi and Y. K. Kim, J. Ind. Eng. Chem., 2011, 17, 394–396 CrossRef CAS.
- L. H. Duan, X. H. Gao, X. H. Meng, H. T. Zhang, Q. Wang, Y. C. Qin, X. T. Zhang and L. J. Song, J. Phys. Chem. C, 2012, 116, 25748–25756 CAS.
- G. Raju, B. M. Reddy, B. Abhishek, Y.-H. Mo and S.-E. Park, Appl. Catal., A, 2012, 423, 168–175 CrossRef.
- J. M. López Nieto, A. Dejoz, M. I. Vázquez, W. O'Leary and J. Cunningham, Catal. Today, 1998, 40, 215–228 CrossRef.
- J. M. López Nieto, P. Concepción, A. Dejoz, H. Knözinger, F. Melo and M. I. Vázquez, J. Catal., 2000, 189, 147–157 CrossRef.
- O. Rubio, J. Herguido and M. Menéndez, Chem. Eng. Sci., 2003, 58, 4619–4627 CrossRef CAS.
- R. Vidal-Michel and K. L. Hohn, J. Catal., 2004, 221, 127–136 CrossRef CAS.
- N. Kijima, M. Toba and Y. Yoshimura, Catal. Lett., 2009, 127, 63–69 CrossRef CAS.
- S. Ge, C. Liu, S. Zhang and Z. Li, Chem. Eng. J., 2003, 94, 121–126 CrossRef CAS.
- A. Costine and B. K. Hodnett, Appl. Catal., A, 2005, 290, 9–16 CrossRef CAS.
- A. Dejoz, J. M. López Nieto, F. Márquez and M. I. Vázquez, Appl. Catal., A, 1999, 180, 83–94 CrossRef CAS.
- N. Madaan, R. Haufe, N. R. Shiju and G. Rothenberg, Top. Catal., 2014, 57, 1400–1406 CrossRef CAS.
- N. Madaan, N. R. Shiju and G. Rothenberg, Catal. Sci. Technol., 2016, 6, 125–133 Search PubMed.
- Z. Strassberger, E. V. Ramos-Fernandez, A. Boonstra, R. Jorna, S. Tanase and G. Rothenberg, Dalton Trans., 2013, 42, 5546–5553 RSC.
- B. Xu, X. F. Zhu, Z. W. Cao, L. N. Yang and W. S. Yang, Chin. J. Catal., 2015, 36, 1060–1067 CrossRef CAS.
- O. Rubio, J. Herguido and M. Menéndez, Chem. Eng. Sci., 2003, 58, 4619–4627 CrossRef CAS.
- R. Vidal-Michel and K. L. Hohn, J. Catal., 2004, 221, 127–136 CrossRef CAS.
- N. Kijima, M. Toba and Y. Yoshimura, Catal. Lett., 2009, 127, 63–69 CrossRef CAS.
- S. Ge, C. Liu, S. Zhang and Z. Li, Chem. Eng. J., 2003, 94, 121–126 CrossRef CAS.
- Y. Wang, S. H. Xie, B. Yue, S. J. Feng and H. Y. He, Chin. J. Catal., 2010, 31, 1054–1060 CAS.
- Y. F. Liu, X. P. Wang, F. P. Tian and C. Y. Yia, Chin. J. Catal., 2004, 25, 721–776 CAS.
- R. Liu, X. P. Wang, C. Y. Jia and W. Shi, Chin. J. Catal., 2005, 26, 650–654 CrossRef CAS.
- J. K. Lee, H. Lee, U. G. Hong, J. Lee, Y.-J. Cho, Y. Yoo, H.-S. Jang and I. K. Song, J. Ind. Eng. Chem., 2012, 18, 1096–1101 CrossRef CAS.
- J. Rischard, C. Antinori, L. Maier and O. Deutschmann, Appl. Catal., A, 2016, 511, 23–30 CrossRef CAS.
- A. Barrera, K. Muramatsu, T. Viveros, S. Gomez, J. A. Montoya, P. del Angel, G. Pérez and J. Campa Molina, Appl. Clay Sci., 2009, 42, 415–421 CrossRef CAS.
- N. Kijima, M. Toba and Y. Yoshimura, Catal. Lett., 2009, 127, 63–69 CrossRef CAS.
- C. Caro, K. Thirunavukkarasu, M. Anikumar, N. R. Shiju and G. Rothenberg, Adv. Synth. Catal., 2012, 354, 1327–1336 CrossRef CAS PubMed.
- X. Y. Fu, S. Y. Niu, H. W. Zhang and Q. Xin, Spectrosc. Spectral Anal., 2006, 26, 27–29 CAS.
- C. Téllez, M. Abon, J. A. Dalmon, C. Mirodatos and J. Santamaría, J. Catal., 2000, 195, 113–124 CrossRef.
- M. E. Harlin, V. M. Niemi and A. O. I. Krause, J. Catal., 2000, 195, 67–78 CrossRef CAS.
- H. Lee, J. K. Lee, U. G. Hong, Y. Yoo, Y.-J. Cho, J. Lee, H.-S. Jang, J. C. Jung and I. K. Song, J. Ind. Eng. Chem., 2012, 18, 808–813 CrossRef CAS.
- A. A. Lemonidou, G. J. Tjatjopoulos and I. A. Vasalos, Catal. Today, 1998, 45, 65–71 CrossRef CAS.
- B. Solsona, F. Ivars, P. Concepción and J. M. López Nieto, J. Catal., 2007, 250, 128–138 CrossRef CAS.
- N. C. Ramani, D. L. Sullivan and J. G. Ekerdt, J. Catal., 1998, 173, 105–114 CrossRef CAS.
- T. Blasco and J. M. López Nieto, Appl. Catal., A, 1997, 157, 117–142 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |