
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSchiff base derived Fe3+-selective fluorescence turn-off chemsensors based on triphenylamine and indole: synthesis, properties and application in living cells†
Chengqiang Pana,
Kai Wanga,
Shaomin Jia,
Huaqian Wang*a,
Zongzhi Lia,
Huahong Heb and
Yanping Huo *ac
*ac
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China. E-mail: yphuo@gdut.edu.cn; huaqianwang@126.com; Fax: +86 20 39322235; Tel: +86 20 39322236 Tel: +86 13798135622 Tel: +86 13560355016
bGuangzhou Institute for Drug Control, Guangzhou 510160, China
cGuangdong Engineering Research Center for Scientific Research and Biochemical Detection Reagent, Guangzhou 510006, China
First published on 19th July 2017
Abstract
Two Schiff base derivatives (S1 and S2) containing triphenylamine and indole fluorophores groups have been synthesized via a one-pot reaction and utilized as fluorescence turn-off sensors towards Fe3+ ions in THF. S1 and S2 demonstrated fluorescence turn-off sensing towards Fe3+ ions, via photoinduced electron transfer (PET). The 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometries of the sensor complexes ([S1, S2] + Fe3+) were calculated from Job plots based on UV-vis absorption titrations. The detection limits (LODs) of [S1, S2] + Fe3+ sensor responses were calculated by their standard deviation, linear fitting and from their fluorescence binding isotherms. More importantly, [S1, S2] + Fe3+ sensors were found to be active in aqueous media within a wide range of pH values. In addition, biological imaging and membrane permeability demonstrated that S1 could act as a turn-off fluorescence chemosensor for Fe3+ in living cells.
:1 stoichiometries of the sensor complexes ([S1, S2] + Fe3+) were calculated from Job plots based on UV-vis absorption titrations. The detection limits (LODs) of [S1, S2] + Fe3+ sensor responses were calculated by their standard deviation, linear fitting and from their fluorescence binding isotherms. More importantly, [S1, S2] + Fe3+ sensors were found to be active in aqueous media within a wide range of pH values. In addition, biological imaging and membrane permeability demonstrated that S1 could act as a turn-off fluorescence chemosensor for Fe3+ in living cells.
1. Introduction
The design and synthesis of high selectivity and sensitivity chemical sensors to detect metal ions in the ecological environment and in biology has attracted a great deal of attention.1–9 Current chemosensors present a large number of appealing advantages, including high sensitivity and selectivity, generally non-destructive characteristics, low cost, facile operation, a fast response time, and suitability as a diagnostic tool for biological purposes.10–12As one of the essential trace elements in biological systems, Fe3+ plays a crucial role in living organisms and metabolism.13,14 For example, it is an essential element for the formation of the hemoglobin of vertebrate red cells and plays an vital role in the storage and transport of oxygen to tissues.15,16 However, iron level should be balanced in human being. Excessive Fe3+ in the human body has been found to be related to an increased incidence of certain cancers and the dysfunction of certain organs, with symptoms such as diarrhea, vomiting, stomach pain, and heart/liver damage.17,18 On the other hand, iron deficiency can lead to anemia.19 Hence a convenient and rapid method for detecting the concentration of Fe3+ in biological samples is of great interest in biological and environmental concerns. Recently, some turn-off chemosensors for Fe(III)-selective detection were reported and a few of them have been successfully used in biological applications.20 However, some defects of these chemosensors such as synthetic difficulties, ligand cytotoxicity, cross-sensitivity toward other metal cations, poor water solubility, a narrow pH detection span, a low fluorescence quantum yield in aqueous media, and long response times, have been found in actual practice.21–28 Therefore, we tend to develop the specific sensory for Fe3+ detections in aqueous media with low cytotoxicity, a high fluorescence quantum yield and a broad pH detection span via simple synthetic pathways.
Currently, the fluorogenic materials with aggregation-induced emission (AIE) attributes have become a hot research topic since the debut of AIE concept in 2001,29 and have been found to serve as chemosensors, bioprobes, stimuli-responsive nanomaterials and active layers in the construction of efficient organic light-emitting diodes.30 However, many of these fluorogenic molecules also have synthetic difficulties, so we try to develop molecules that could exhibit the AIE effects with less synthetic difficulties.
With these considerations, Schiff bases are the suitable candidates having simple synthetic steps and also applied to many fluorescent sensors, as well as in AIE applications.31,32 However, to develop Schiff base sensors with AIE properties, the presence of strong fluorophores are required.33 Triphenylamine and indole derivatives were evidenced as excellent fluorophores and widely used in the developments of fluorescence (FL) sensors because of their excellent photoluminescence properties and chemical stabilities. Therefore, we developed the triphenylamine- and indole- based Schiff bases and evaluated their sensor and AIE properties (Fig. 1).
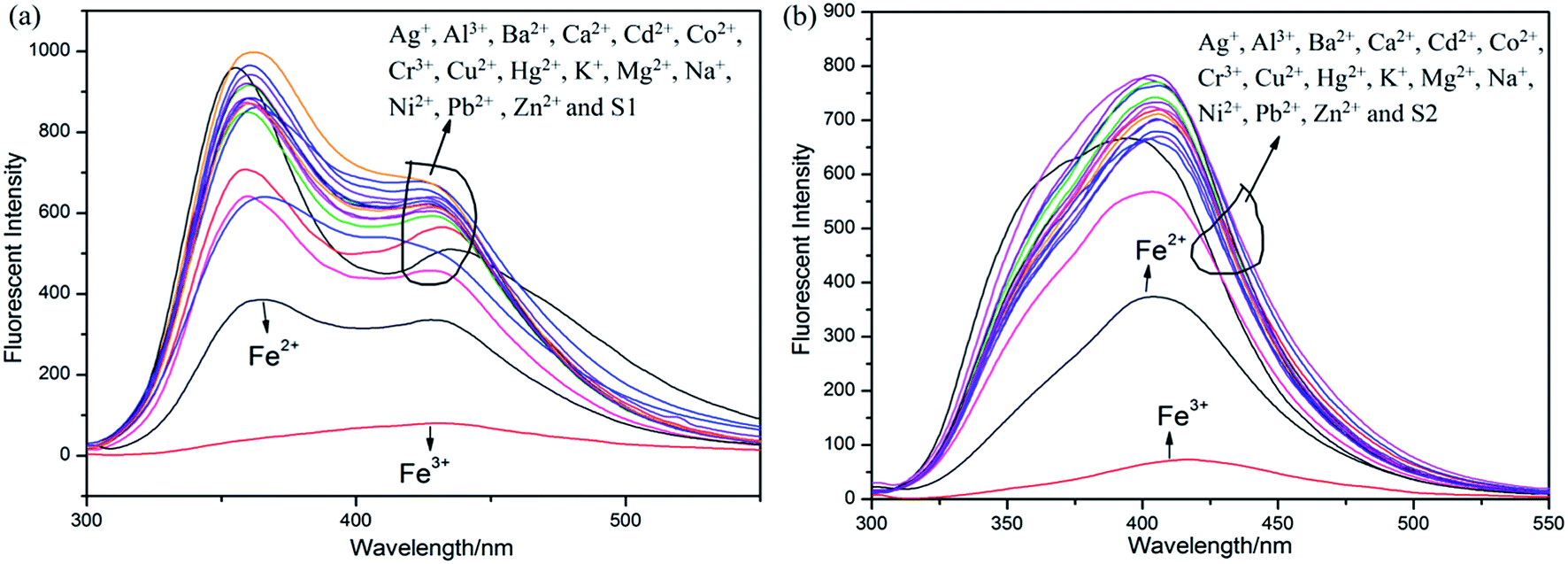 | ||
| Fig. 1 Sensor responses of (a) S1 and (b) S2 (1 × 10−5 M) in THF (λex = 300 nm) towards different nitrate salts in THF. | ||
Herein, we report triphenylamine- and indole- based Schiff bases derivatives (S1 and S2) with AIE properties for the first time as Fe3+ turn-off sensors via PET and S1 was also utilized for the detection of Fe3+ in living cells.
2. Experimental
2.1 Materials and methods
All of the chemicals are commercially available, and used without further purification. Elemental analyses were performed with an EA1110 CHNS-0 CE elemental analyzer. All fluorescence measurements were carried out on a LS 50B Luminescence Spectrometer (Perkin Elmer, Inc., USA). All UV-vis absorption spectra were recorded on a Lambda 20 UV-vis Spectrometer (Perkin Elmer, Inc., USA). 1H and 13C NMR experiments were carried out on a MERCURYplus 400 spectrometer operating at a resonance frequency of 400 MHz. Electrospray ionization mass spectra (ESI-MS) were recorded on a Finnigan LCQ mass spectrometer. All the materials for synthesis were purchased from commercial suppliers and used without further purification. The metal ion solutions were prepared from FeCl2·4H2O, Ni(NO3)2·6H2O, Co(NO3)2·6H2O, Cu(NO3)2·3H2O, Mg(NO3)2·6H2O, Zn(NO3)2·6H2O, Ca(NO3)2·4H2O, Fe(NO3)3·9H2O, Al(NO3)3·9H2O, KNO3, NaNO3, Cd(NO3)2·4H2O, Cr(NO3)3·9H2O, Hg(NO3)2·H2O, Ba(NO3)2 and Pb(NO3)2, respectively.2.2 Synthesis of compound S1
2-Aminophenol (440.0 mg, 4.03 mmol) was added to a solution of 4-(diphenylamino)benzaldehyde (1096.05 mg, 4.01 mmol) dissolved in 30 ml of methanol. The resulting mixture was refluxed for 12 h. After completion of the reaction (confirmed by thin layer chromatographic analysis), the reaction mixture was filtered off and a light brown oily residue was obtained upon removal of the solvent from the filtrate under vacuum. This oily residue on treatment with ethanol gave an light brownish solid precipitate, which was collected by filtration and dried to give S1 as a pure product (1095 mg, 75%). 1H NMR (400 MHz, DMSO-d6, TMS) δ (ppm): 9.52 (s, 1H), 8.26 (d, J = 8.6 Hz, 1H), 8.05 (d, J = 16.2 Hz, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.61 (d, J = 8.5 Hz, 2H), 7.34 (dd, J = 16.5, 9.3 Hz, 6H), 7.10 (t, J = 9.9 Hz, 6H), 7.00 (d, J = 8.5 Hz, 2H). 13C NMR (500 MHz, CDCl3, TMS) δ (ppm): 158.73, 151.52, 150.52, 146.93, 138.72, 130.69, 130.25, 127.31, 125.67, 124.72, 121.22, 119.93, 119.30, 116.24. ESI-MS m/z: 365.1 ([M + H]+). Elemental analysis: found C: 82.12, H: 5.21, N: 7.28; calculated for (C25H20N2O) C: 82.42, H: 5.49, N: 7.69 (%).2.3 Synthesis of compound S2
2-Aminophenol (445.25 mg, 4.08 mmol) was added to a solution of 1-methyl-2-phenyl-3-formylindole (952.92 mg, 4.05 mmol) dissolved in 30 ml of methanol. The resulting mixture was refluxed for 10 h. After completion of the reaction (confirmed by thin layer chromatographic analysis), the reaction mixture was filtered off and a light brown oily residue was obtained upon removal of the solvent from the filtrate under vacuum. This oily residue on treatment with ethanol gave an light brownish solid precipitate, which was collected by filtration and dried to give S2 as a pure product (911.38 mg, 69%). 1H NMR (400 MHz, DMSO-d6, TMS) δ (ppm): 9.73 (s, 1H), 8.57 (s, 2H), 7.61–7.52 (m, 3H), 7.51–7.44 (m, 2H), 7.44–7.32 (m, 3H), 7.07 (t, J = 7.2 Hz, 1H), 6.99 (dd, J = 11.2, 8.0 Hz, 2H), 6.79 (t, J = 7.6 Hz, 1H), 3.66 (s, 3H). 13C NMR (400 MHz, CDCl3, TMS) δ (ppm): 152.75, 150.47, 146.54, 137.12, 136.83, 129.91, 128.71, 128.45, 127.68, 125.81, 124.41, 122.68, 121.41, 118.85, 114.42, 113.13, 112.08, 108.80, 30.05. ESI-MS m/z: 327.4 ([M + H]+). Elemental analysis: found C: 80.54, H: 5.36, N: 8.28; calculated for (C22H18N2O) C: 80.98, H: 5.52, N: 8.59 (%).3. Results and discussion
3.1 Synthesis of S1–S2
As shown in Scheme 1, S1 and S2 were synthesized via a one pot aldehyde and amine condensation in methanol with 75 and 69% yields, respectively.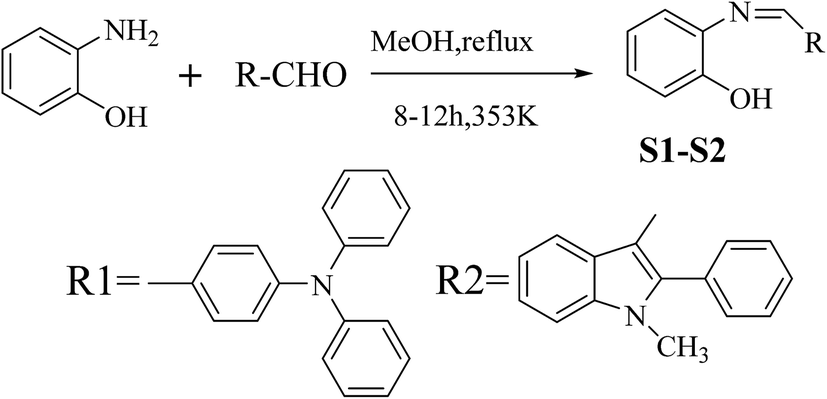 | ||
| Scheme 1 Synthesis of S1–S2. | ||
3.2 Spectroscopic properties
Initially, S1 and S2 (20 μM) in THF were investigated towards 60 μM (3 equiv.) of metal ions (Ag+, Al3+, Ba2+, Ca2+, Cd2+, Co2+, Cr3+, Cu2+, Fe2+, Fe3+, Hg2+, K+, Mg2+, Na+, Ni2+, Pb2+ and Zn2+) in H2O. As noticed in Fig. 1, S1 and S2 show better selectivities to Fe3+ ions, respectively, upon treatment with 3 equiv. of metal ions, respectively. In addition, in order to establish the specific selectivities of S1 and S2 to Fe3+, respectively, we performed the single and dual metal competitive analysis, as shown in Fig. 2. In a single metal system (black bars), all the metal (Ag+, Al3+, Ba2+, Ca2+, Cd2+, Co2+, Cr3+, Cu2+, Fe2+, Fe3+, Hg2+, K+, Mg2+, Na+, Ni2+, Pb2+ and Zn2+ in H2O) concentrations were kept as 60 μM. However, for the dual-metal (red bars) studies, two equal amounts of aqueous solutions of Fe3+ and other metal ions (60 μM + 60 μM) were combined. Furthermore, in the single metal analysis, the Fe3+ effect at 60 μM was investigated. In addition to the selectivity of S1 and S2 towards Fe3+ ion, Fe2+ ion also showed little selectivity (Fig. 1).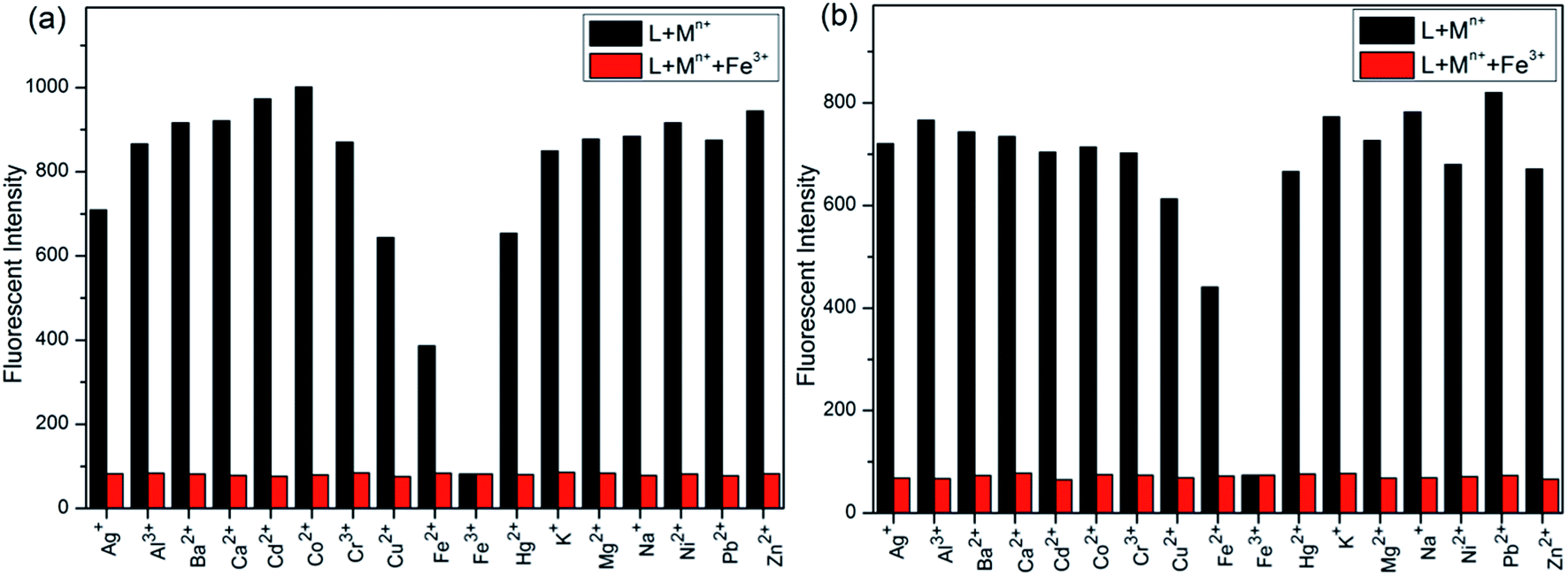 | ||
| Fig. 2 Relative fluorescence intensities of (a) S1 and (b) S2 in THF (λex = 300 nm) in THF in the presence of competing metal ions. Black bar; S1 and S2 in THF with stated metal ions in H2O, respectively. Red bar; S1 and S2 in THF with Fe3+ + stated metal ions in H2O. | ||
Moreover, the specific selectivities of S1 and S2 by single and dual metal studies also conformed their respective selectivities toward Fe3+ ion. The effect of Fe3+ concentration on the emission of S1 and S2 was investigated in detail. Fig. 3 illustrates the fluorescence spectral changes of S1 and S2 (20 μM) as a function of Fe3+ concentration (0–4.0 equiv.) in THF at room temperature. Importantly, as the concentration of the Fe3+ ions increased, a progressive decrease in the fluorescence emission intensity at 470 nm and 405 nm was observed, respectively.
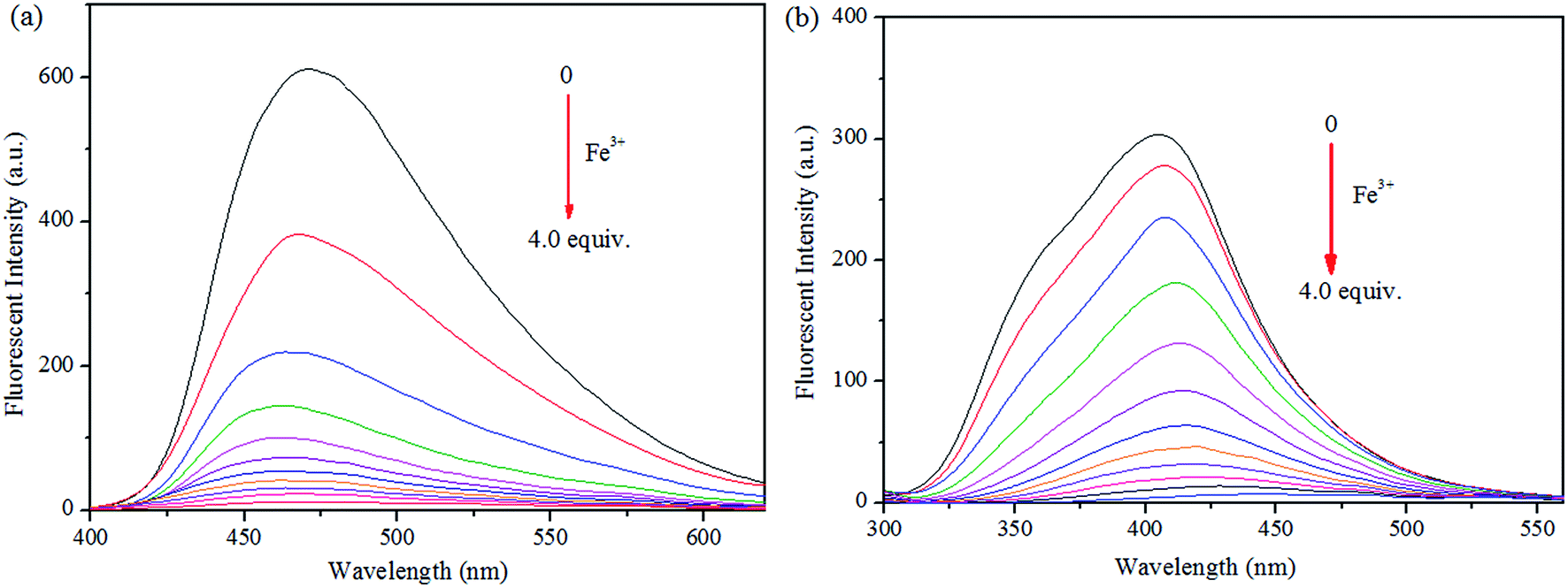 | ||
| Fig. 3 Fluorescence spectra of (a) S1 and (b) S2 and in THF (λex = 300 nm) with 0–4 equiv. of Fe3+ in H2O. | ||
For better understanding of the chromofluorophore, DFT study was done for S1 and S2. All theoretical calculations were carried out using the Gaussian 09 program. The B3LYP exchange correlation functional under LANL2DZ basis set was performed to calculate HOMO and LUMO levels. The geometries were optimized, and electron distribution in the MOs was calculated (Fig. 4). The HOMO–LUMO orbital distribution showed that the electron cloud of S1 was spread from the phenylamine moiety to the phenol moiety. The major contribution for HOMO and LUMO of S2 was well-distributed. Indeed, it has been demonstrated that fluorescence quenching of these compounds may occur by the photoinduced electron transfer (PET) from intraligand charge transfer.
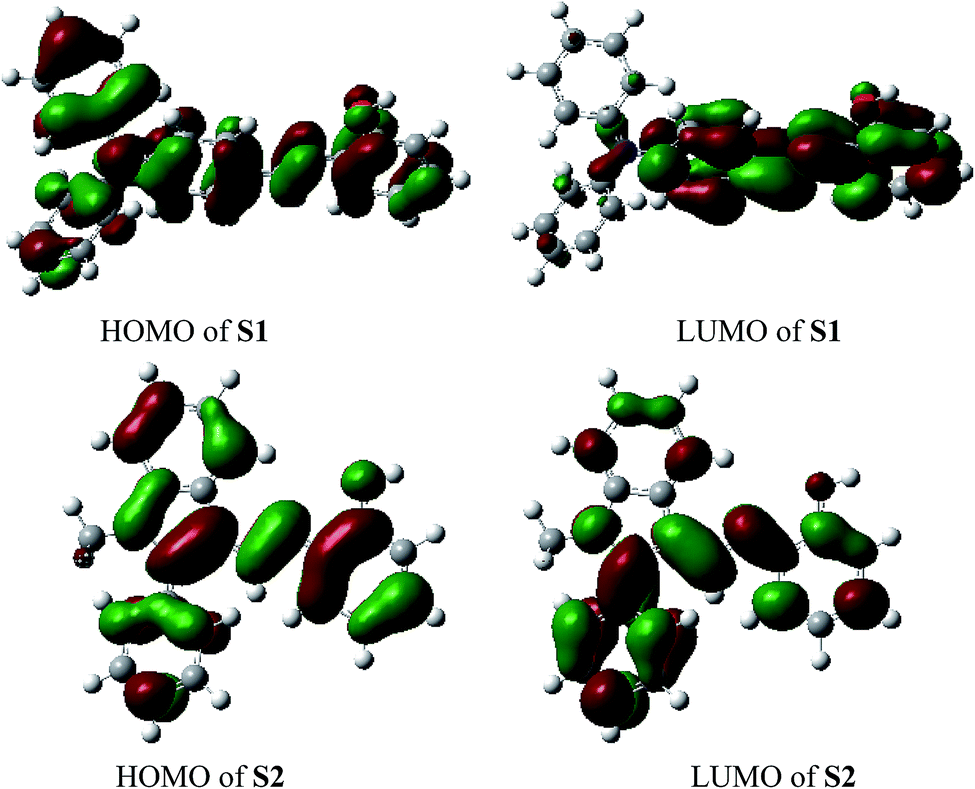 | ||
| Fig. 4 Molecular orbitals of HOMO and LUMO for S1–S2. | ||
To ensure the binding sites of sensor responses of, the stoichiometries of S1 or S2 + Fe3+ were calculated through Job's plots as shown in Fig. 5. The stoichiometries of S1 or S2 + Fe3+ were established by Job's plots between the mole fraction (XM) and absorption maximum changes at 435 nm, respectively. Upon the addition of 0–60 μM of Fe3+ (with an equal span of 6 μM). The Job's plots were plotted between XM and absorption changes at 435 nm, where they went through maxima at molar fractions of ca. 0.323 and 0.385, respectively, as shown in Fig. 5, representing their 2:1 stoichiometric. From the ESI, the ESI mass spectrum of S1 or S2 and Fe3+ was also studied to find further support for this binding behavior. The complexes of S1 or S2 and Fe3+ show the peak at m/z 782 and 706, (calculated m/z 782 and 706 for S1 or S2 and Fe3+), respectively, illustrating the structure of the complexes of S1 or S2 and Fe3+, confirming the 2:1 stoichiometric ratio between the host and guest. Therefore, the possible sensing mechanism based on the excimer formation was proposed as noted in Fig. 6. The fluorescence changes observed in the ligand solutions of S1 and S2 upon binding to Fe3+ are due to deprotonation of phenolic –OH which allows the charge transfer from ligand to metal ion. The charge transfer from ligand to metal is also responsible for fluorescence quenching behavior of ligands upon metal complexation.33
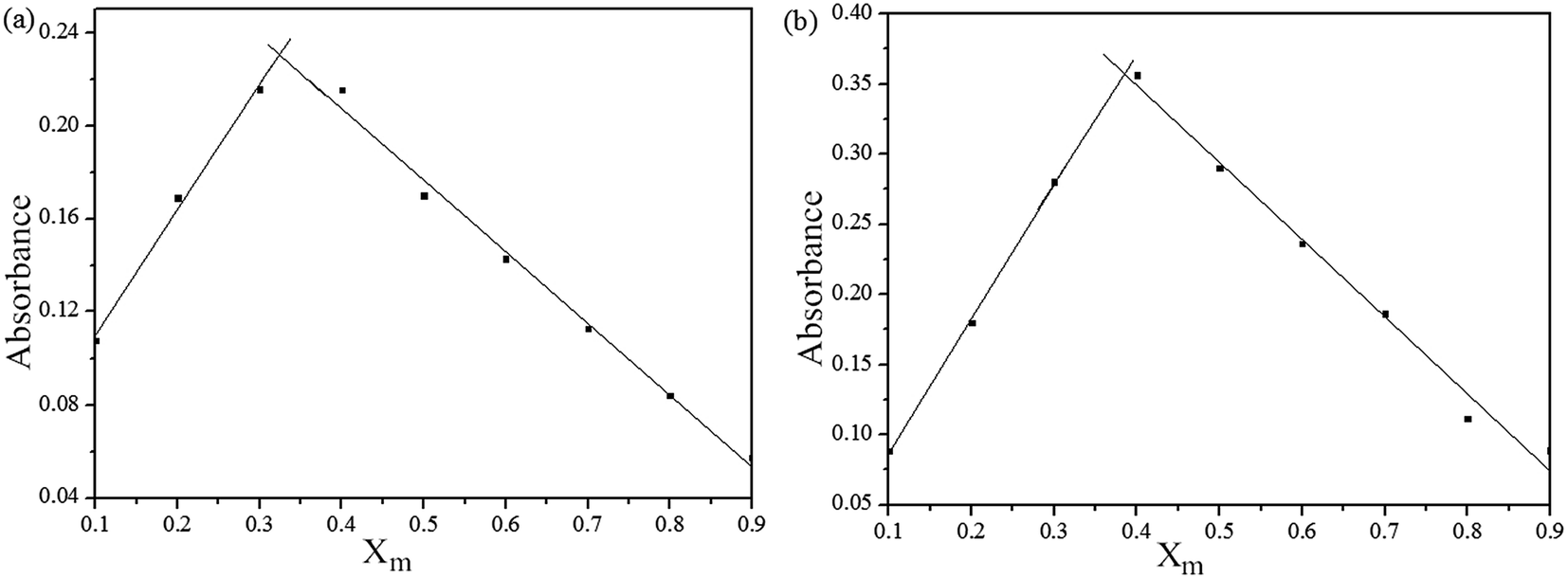 | ||
| Fig. 5 Absorbance spectral changes of (a) S1 and (b) S2 in THF, titrated with of Fe3+ ions in H2O and (b) stoichiometry calculations of (a) S1 and (b) S2 based on absorbance changes; Xm = [Fe3+]/[Fe3+] + [S]; where Xm = mole fraction, [Fe3+] and [S] are concentrations of Fe3+ and S1 or S2. | ||
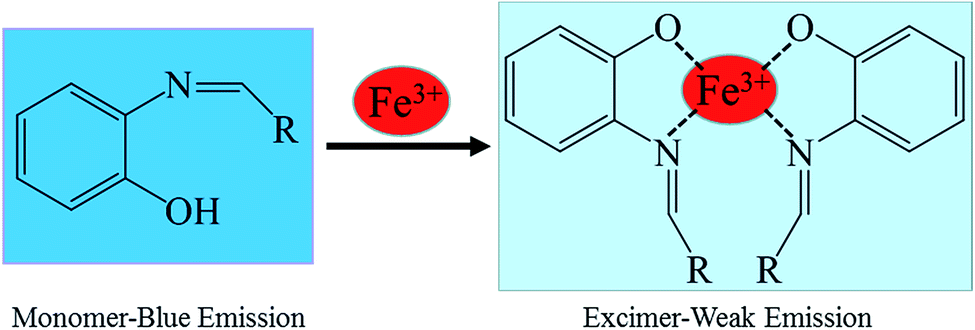 | ||
| Fig. 6 Possible proposed binding mechanism of S1 or S2, towards Fe3+ ions. | ||
In order to prove the selectivities of S1 and S2 towards Fe3+, respectively, the calculations of detection limits (LODs) were performed through standard deviations and linear fittings as shown in Fig. 7. By plotting the relative fluorescence intensity (I0/I) changes as a function of concentration the detection limits of S1 or S2 + Fe3+ were calculated as 4.51 × 10−5 and 3.37 × 10−6 M, respectively. The detection limit (LODs) was determined from the following equation: LODs = 3σ/S where σ is the standard deviation of the blank solution; S is the slope of the calibration curve.
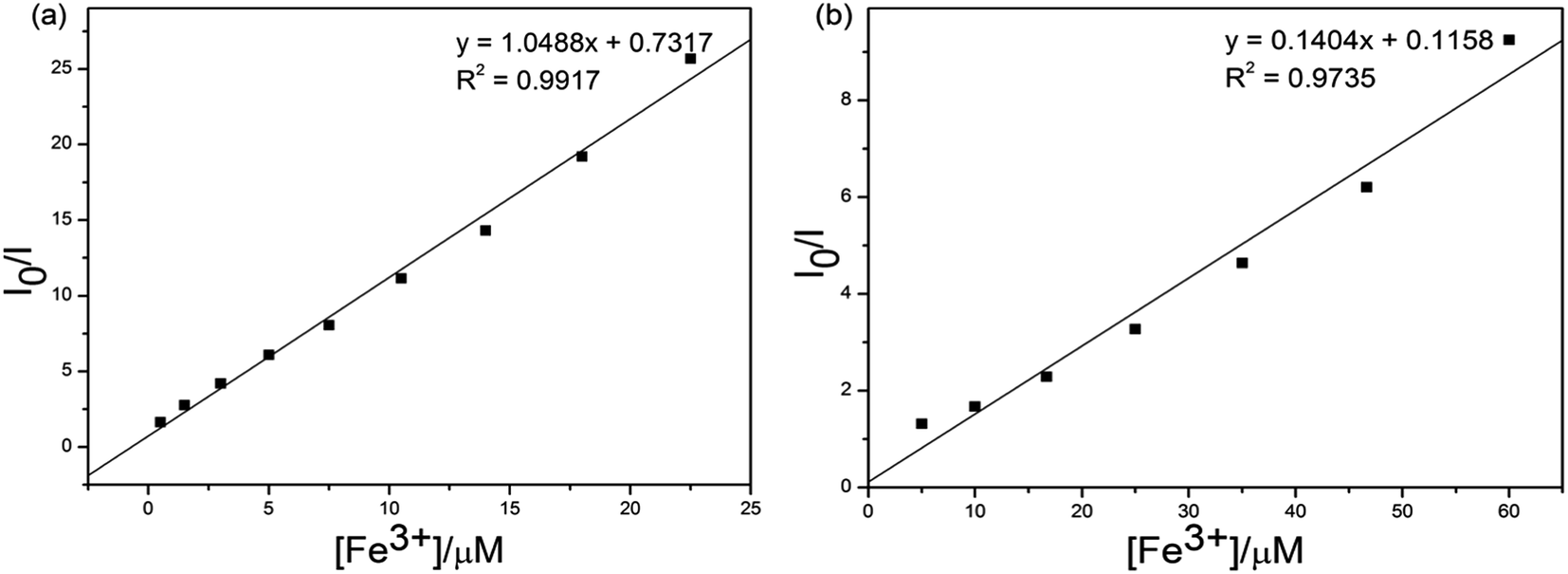 | ||
| Fig. 7 Standard deviations and linear fitting for detection limit calculations of (a) S1 and (b) S2 + Fe3+. | ||
Considering the influence of pH to the sensors. The S1 or S2 and S1 or S2 + Fe3+ sensors responses were verified between pHs 0 and 14, maintained by the respective buffers (100 μM). In contrast to separate titrations of pHs (0–14) solutions (100 μM) to S1 and S2. The experiment results show that the sensors of S1 or S2 were stable in wide ranges of pHs 3–12 and 4–12, respectively (see Fig. 8). Meanwhile the S1 or S2 + Fe3+ sensors were active in wide ranges of pHs 1–14 in both cases, as shown in Fig. 8. The experiment results show that S1 or S2 + Fe3+ were stable in wide ranges of pHs 3–12 and 4–12 respectively. In pH 3–12, due to ESIPT involving the phenolic protons and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N isomerization. The CN isomerization is the predominant decay process of the excited state for S1 and S2. Upon addition of Fe3+, the coordination of S1 and S2 with the metal ion inhibits the CN isomerization and prevents ESIPT.34 But fluorescence dramatically change from pH 12 are most likely caused by the deprotonation of the hydroxy group of S1 and S2.35 The above observation confirmed that the sensors can be utilized in acidic and basic and not affected by pH change.
N isomerization. The CN isomerization is the predominant decay process of the excited state for S1 and S2. Upon addition of Fe3+, the coordination of S1 and S2 with the metal ion inhibits the CN isomerization and prevents ESIPT.34 But fluorescence dramatically change from pH 12 are most likely caused by the deprotonation of the hydroxy group of S1 and S2.35 The above observation confirmed that the sensors can be utilized in acidic and basic and not affected by pH change.
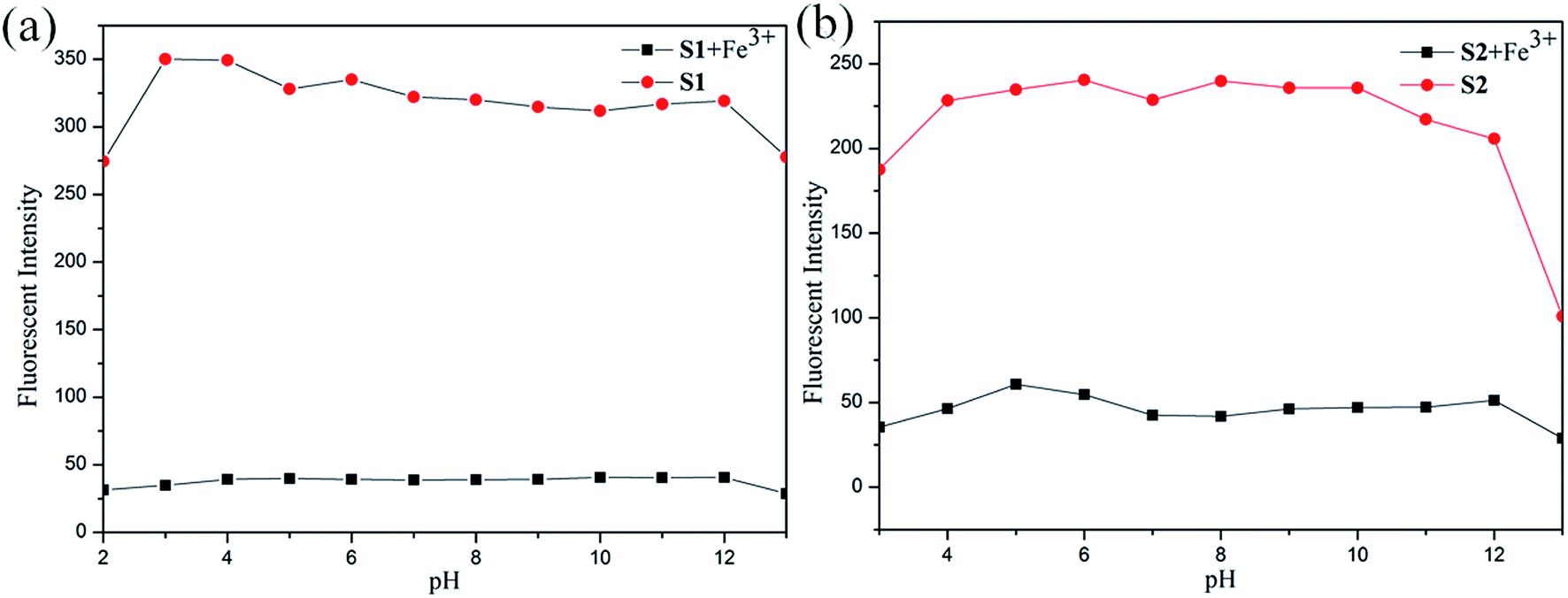 | ||
| Fig. 8 Fluorescence intensity of (a) S1 and (b) S2 with different pH conditions. | ||
Since many sensor responses are affected by the presence of counter ions, we performed the sensor titrations of S1 and S2 towards Fe3+, respectively, with different counterion salts. As evidenced in Fig. 9, the Fe3+ sensor response was found to be decreased in the presence of counter ions (Br−, NO3−, Cl−, F−, ClO4−, OH− and SO42−). Hence, it was concluded that both the Fe3+ sensor responses of S1 and S2 were not affected incredibly in the presence of different counter ions.
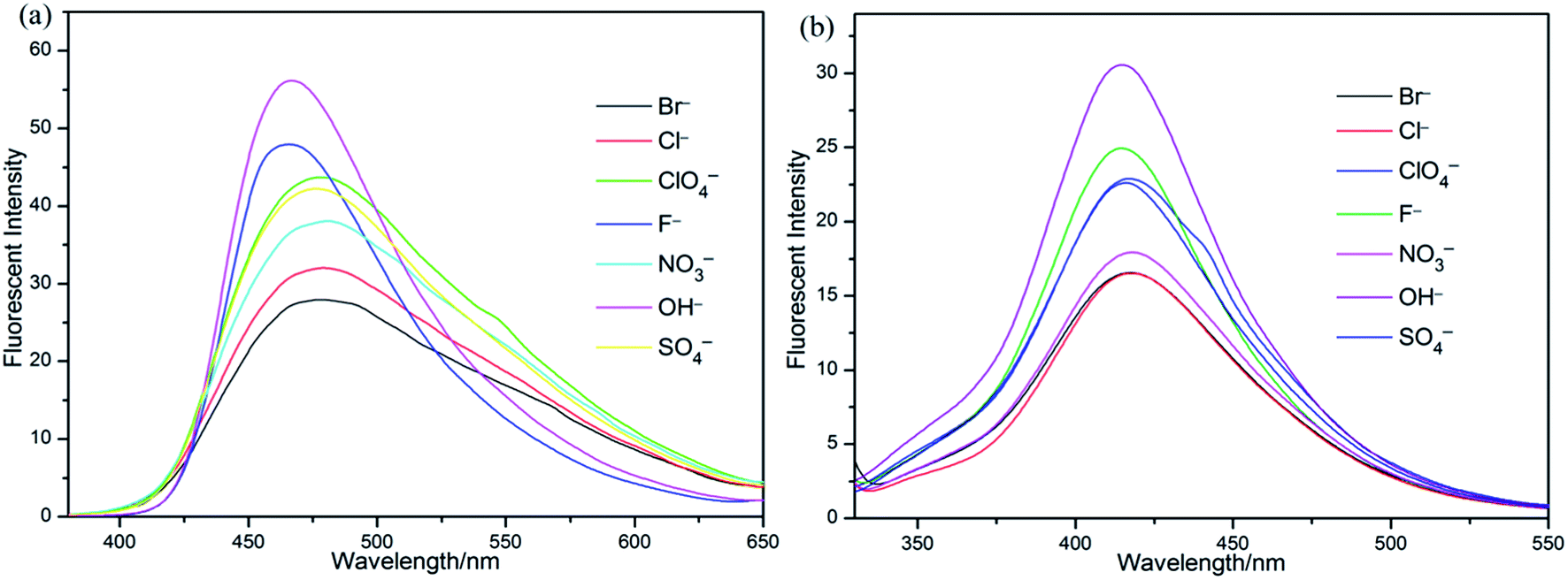 | ||
| Fig. 9 Sensor responses of (a) S1 and (b) S2 towards Fe3+ in presence of different counter ions. | ||
More interestingly, while preparing the stock solutions of S1 and S2 in THF by varying the water concentrations for sensor titrations, the fluorescence spectra were enhanced with spectral shifts due to their aggregated nature. Hence, we proceeded the aggregation induced enhanced emission (AIEE) analysis further. As shown in Fig. 10, the PL intensities were enhanced by increasing the concentrations of H2O (up to 90% or 80%), at the same time, the PL emissions were shifted from 460 to 479 nm of S1 and 430 to 424 nm of S2 in both extremes. The fluorescence changes arose from AIE were visualized by the photographs of S1 (0 and 80%) and S2 (0 and 90%), respectively.
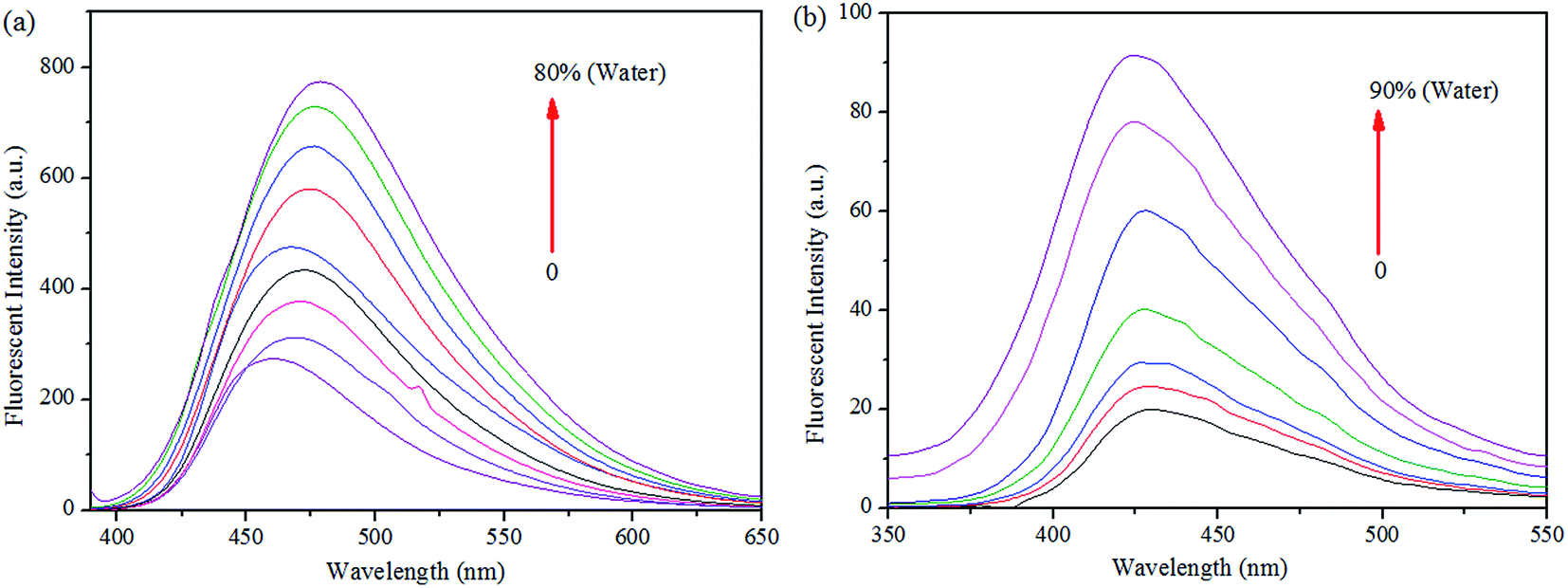 | ||
| Fig. 10 Fluorescence spectra of (a) S1 and (b) S2 in THF, upon increasing the concentration of water. | ||
As reported by previous literature,36 the AIEs mechanism of both S1 and S2 possibly arose from the restriction of intramolecular rotation (RIR). Since, the single bond rotation is mainly responsible for the dominant nonradiative decay. A single bond that links a benzene ring of S1 and S2 can be rotated. The RIR effect might be the cause for the AIEE nature of S1 and S2. The PET process and suppression of charge transfer (CT) or intramolecular charge transfer (ICT) are the other mechanistic approaches for AIE. However, our observations on the PL spectra of AIEEs for S1 and S2 suggested that both the red and blue shifts by aggregation were probably originated from the suppression of twisted intramolecular charge transfer (TICT). The above justification was also well supported by the similar reports available in the field of AIEs.37
To demonstrate the potential of S1 to detect Fe3+ in living matrices, fluorescence imaging experiments were carried out in living cells (Fig. 11). First, we selected human prostatic carcinoma cell line (PC-3) to investigate the permeability of S1. PC-3 cells were obtained from the Cell Resources Center of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Human prostate cancer cell PC-3 was derived from a 62 year-old grade IV prostate cancer in white male patients with bone marrow metastases. The patient with prostate cancer signed informed consent before any study procedures. The cells were cultured in RPMI 1640 (Gibco) and supplemented with 10% fetal bovine serum (FBS; Gibco) at 37 °C in a humidified atmosphere of 5% CO2. For all experiments, cells were harvested from subconfluent cultures by the use of trypsin and were resuspended in fresh complete medium before plating. PC-3 cells were incubated with S1 (10 μM) for 1 h at 37 °C, and washed with PBS to remove the remaining compound S1. The results are shown in Fig. 11. One can clearly observe significant imaging changes of the medium upon addition of Fe3+ for 30 min (10 equiv.) at 37 °C. PC-3 cells incubated with S1 initially display a strong fluorescence image (Fig. 11b), but the fluorescence image immediately becomes faint even quenching in the presence of Fe3+ (Fig. 11c). Thus, the chemosensor S1 can be a suitable fluorescence chemosensing probe for Fe3+ detection in biological systems.
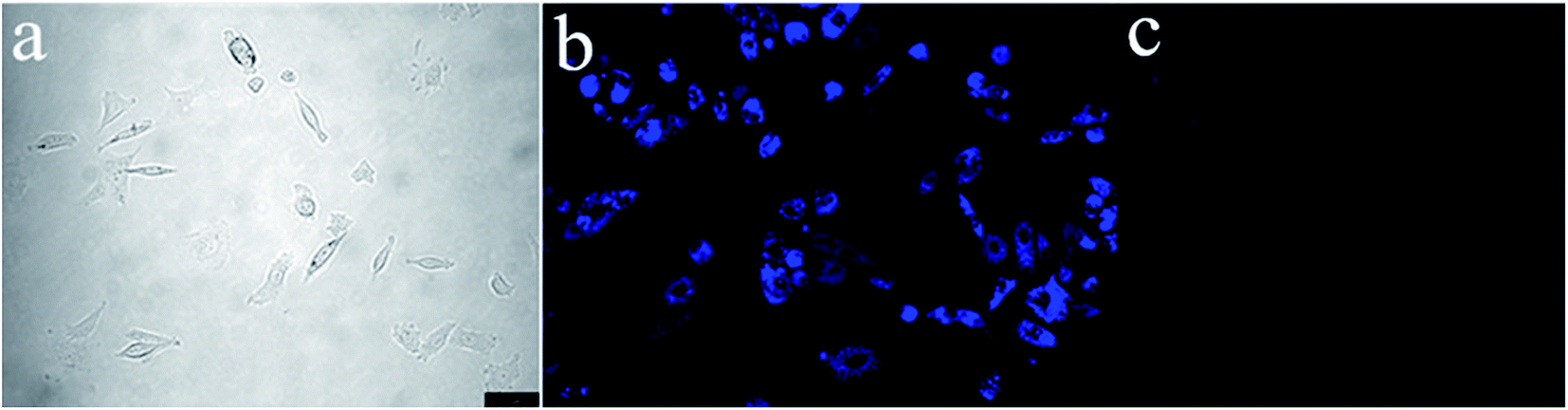 | ||
| Fig. 11 Fluorescence microscopy images of Fe3+ in PC-3 cells (Olympus IX71, 40× objective lens). (a) Bright-field transmission image of PC-3 cells. (b) Fluorescence image of PC-3 cells incubated with S1 (10 μM). (c) Further incubated with addition of various concentrations of Fe3+ (10 equiv.). | ||
At the same time, we also studied the fluorescence imaging of S2 in PC-3 cells. But the results showed that PC-3 cells incubated with S2 initially almost no fluorescence angiography (Fig. S2†), which suggests that the AIE phenomenon of S2 was unobvious compared with the S1. In the medium solution, both S1 and S2 have an AIE effect. As shown in Fig. 10, the AIE phenomenon of S1 was significantly better than S2. The fluorescence intensity of S2 was weak in the medium solution. So there was no fluorescence of S2 in PC-3 cells.
4. Conclusion
In summary, two Schiff based turn-off chemosensors with AIE were designed for detection of Fe3+, which were investigated with much higher selectivity over other competitive metal ions. Adequate characterization and studies were carried out to confirm the chemosensors S1 and S2 and their sensing behaviors toward Fe3+ ion. The 2:1 stoichiometry of sensors to Fe3+ were calculated from Job plots based on UV-vis absorption titrations. Furthermore, by standard deviations and linear fittings the detection limits (LODs) of S1 or S2 + Fe3+ were calculated as 4.51 × 10−5 and 3.37 × 10−6, respectively. And their recognition behavior can function well over a wide range of pH, and not affected incredibly in the presence of different counter ions, making them suitable for detection of Fe3+ ion in industrial and environmental fields. As well as, fluorescence imaging experiments of S1 in living cells clearly observe good cell permeability and significant imaging changes of the aqueous solution medium upon addition of Fe3+. Therefore, the turn-off chemosensor S1 can be a suitable fluorescence chemosensing probe for Fe3+ detection in biological systems. This is of great significance for medical research. Similar to the sensor properties, S1 and S2 in THF showed greater AIEE properties with shifted PL emission peaks.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (61671162 and 21372051) and Technology Plan of Guangdong Province (2016A010103031). This work is also supported by Guangdong Province Universities and Colleges Young Pearl River Scholar Funded Scheme (2016).References
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed.
- J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed.
- S. K. Kim, D. H. Lee, J. I. Hong and J. Y. Yoon, Acc. Chem. Res., 2009, 42, 23–31 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, Acc. Chem. Res., 2009, 42, 193–203 CrossRef CAS PubMed.
- R. Martínez-Mánez and F. Sancenón, Chem. Rev., 2003, 103, 4419–4476 CrossRef PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed.
- C. R. Lohani and K. H. Lee, Sens. Actuators, B, 2010, 143, 649–654 CrossRef CAS.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2012, 256, 170–192 CrossRef CAS.
- A. T. Wright and E. V. Anslyn, Chem. Soc. Rev., 2006, 35, 14–28 RSC.
- J. Liu and Y. Lu, J. Fluoresc., 2004, 14, 343–354 CrossRef CAS PubMed.
- J. H. Lee, Z. Wang, J. Liu and Y. Lu, J. Am. Chem. Soc., 2008, 130, 14217–14226 CrossRef CAS PubMed.
- O. K. Fix and K. V. Kowdley, Minerva Med., 2008, 99, 605–617 CAS.
- P. Aisen, M. Wessling-Resnick and E. A. Leibold, Curr. Opin. Chem. Biol., 1999, 3, 200–206 CrossRef CAS PubMed.
- X. B. Zhang, G. Cheng, W. J. Zhang, G. L. Shen and R. Q. Yu, Talanta, 2007, 71, 171–177 CrossRef CAS PubMed.
- K. Vijay, C. Nandib and S. D. Samant, RSC Adv., 2016, 6, 49724–49727 RSC.
- (a) E. D. Weinberg, Eur. J. Cancer Prev., 1996, 5, 19–36 CAS; (b) D. Galaris, V. Skiada and A. Barbouti, Cancer Lett., 2008, 266, 21–29 CrossRef CAS PubMed.
- F. O. Omara and B. R. Blakley, J. Nutr., 1993, 123, 1649–1655 CAS.
- L. H. Allen, J. Nutr., 2002, 132, 813S–819S CAS.
- (a) L. Huang, F. P. Hou, J. Cheng, P. X. Xi, F. J. Chen, D. Bai and Z. Z. Zeng, Org. Biomol. Chem., 2012, 10, 9634–9638 RSC; (b) S. K. Sahoo, D. Sharma, R. K. Bera, G. Crisponi and J. F. Callan, Chem. Soc. Rev., 2012, 41, 7195–7227 RSC; (c) B. K. Kanungo, M. Baral, R. K. Bera and S. K. Sahoo, Monatsh. Chem., 2010, 141, 157–168 CrossRef CAS.
- H. S. Jung, P. S. Kwon, J. W. Lee, J. I. Kim, C. S. Hong, J. W. Kim, S. H. Yan, J. Y. Lee, J. H. Lee, T. H. Joo and J. S. Kim, J. Am. Chem. Soc., 2009, 131, 2008–2012 CrossRef CAS PubMed.
- D. En, Y. Guo, B. Chen, B. Dong and M. Peng, RSC Adv., 2013, 4, 248–253 RSC.
- K. Mehdi, B. Alireza and M. Ghodsi, J. Fluoresc., 2015, 25, 1297–1302 CrossRef PubMed.
- R. Kagit, M. Yildirim, O. Ozay, S. Yesilot and H. Ozay, Inorg. Chem., 2014, 53, 2144–2151 CrossRef CAS PubMed.
- M. Kumar, R. Kumar, V. Bhalla, P. R. Sharma and T. Kaur, Dalton Trans., 2012, 41, 408–412 RSC.
- L. Fu, J. Mei, J. Zhang, Y. Liu and F. Jiang, Luminescence, 2013, 28, 602–606 CrossRef CAS PubMed.
- X. Zhang, G. Cheng, W. Zhang, G. Shen and R. Yu, Talanta, 2007, 71, 171–177 CrossRef CAS PubMed.
- J. Nandre, S. Patil, P. Patil, S. Sahoo and C. Redshaw, J. Fluoresc., 2014, 24, 1563–1570 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- (a) M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC; (b) Z. Liu, W. Xue, Z. Cai, G. Zhang and D. Zhang, J. Mater. Chem., 2011, 21, 14487–14491 RSC.
- (a) V. Bhalla, R. Tejpal, R. Kumar and A. Sethi, Inorg. Chem., 2009, 48, 11677–11684 CrossRef CAS PubMed; (b) D. Maity, A. K. Manna, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, Chem.–Eur. J., 2011, 17, 11152–11161 CrossRef CAS PubMed; (c) S. Sumalekshmy and C. J. Fahrni, Chem. Mater., 2011, 23, 483–500 CrossRef CAS PubMed; (d) D. Maity and T. Govindaraju, Inorg. Chem., 2011, 50, 11282–11284 CrossRef CAS PubMed; (e) J. F. Jang, Y. Zhou, J. Yoon and J. S. Kim, Chem. Soc. Rev., 2011, 40, 3416–3429 RSC; (f) P. Song, X. Chen, Y. Xiang, L. Huang, Z. Zhou, R. Wei and A. Tong, J. Mater. Chem., 2011, 21, 13470–13475 RSC.
- M. Shellaiah, Y. H. Wu, A. Singh, M. V. R. Raju and H. C. Lin, J. Mater. Chem. A, 2013, 1, 1310–1318 CAS.
- N. Narayanaswamy and T. Govindaraju, Sens. Actuators, B, 2012, 161, 304–310 CrossRef CAS.
- L. Yang, W. Zhu, M. Fang, Q. Zhang and C. Li, Spectrochim. Acta, Part A, 2013, 109, 186–192 CrossRef CAS PubMed.
- M. J. C. Marenco, C. Fowley, B. W. Hyland, G. R. Hamilton, D. Galindo-Riaño and J. F. Callan, Tetrahedron Lett., 2012, 53, 670–673 CrossRef CAS.
- (a) S. Karupannan and J. C. Chambron, Chem.–Asian J., 2011, 6, 964–984 CrossRef PubMed; (b) L. Fabbrizzi, M. Lichelli, P. Pallavicini, D. Sacci and E. Taglietti, Analyst, 1996, 121, 1763–1768 RSC.
- (a) M. Cai, Z. Gao, X. Zhou, X. Wang, S. Chen, Y. Zhao, Y. Qian, N. Shi, B. Mi, L. Xie and W. Huang, Phys. Chem. Chem. Phys., 2012, 14, 5289–5296 RSC; (b) Y. Hong, L. Meng, S. Chen, C. W. T. Leung, L. T. Da, M. Faisal, D. A. Silva, J. Liu, J. Wing, Y. Lam, X. Huang and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 1680–1689 CrossRef CAS PubMed; (c) A. Qina, J. W. Y. Lamb and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182–209 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05064j |
| This journal is © The Royal Society of Chemistry 2017 |