
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Three-dimensional crystal structure of novel aluminophosphate PST-5 solved using a powder charge flipping method†
Shuai Changa,
Hoi-Gu Janga,
Kwan-Young Leeb and
Sung June Cho *a
*a
aDepartment of Chemical Engineering, Chonnam National University, 77 Yongbong-Ro, Buk-gu, Gwangju 61186, Republic of Korea. E-mail: sjcho@jnu.ac.kr
bDepartment of Chemical and Biological Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
First published on 8th August 2017
Abstract
The crystal structure of the novel aluminophosphate PST-5, an as-made form of PST-6, has been determined through X-ray powder diffraction using a framework search combined with a powder charge flipping method where the partially correct structure solution obtained using the powder charge flipping method was employed for the determination of the framework through Fourier recycling in the Focus suite of programs. The result of Rietveld refinement using the initial framework structure for novel aluminophosphate PST-5, synthesized hydrothermally using low-cost diethylamine as the organic structure-directing agent, suggested a three-dimensional pore structure containing interconnected ten and eight-membered rings. PST-5 has an orthorhombic Pmmn structure in which nine T atoms exist; the lattice parameters are a = 10.28 Å, b = 36.64 Å and c = 10.89 Å, and the unit cell volume is roughly half that of PST-6.
Introduction
Zeolite, as a polycrystalline porous material, has drawn much attention because of its exceptional acid catalysis properties, characteristic sorption, and ion exchange capacity, among others.1–3 The framework structure and the corresponding unique multidimensional pore architecture of zeolite have been investigated extensively using state-of-the-art technologies including high resolution transmission electron microscopy, rotational electron diffraction, and automatic diffraction tomography.4–8 These advanced techniques have led to significant progress in the structure determination of novel porous materials that can be further extended to novel complex structures.4However, porous materials of low framework stability or containing occluded organic structure-directing agents are susceptible to electron beams that can damage or collapse the framework, inhibiting the collection of well-resolved electron diffraction or high-resolution images.9 Furthermore, the presence of amorphous phases in the polycrystalline material hampers accurate structure determination.10 Aluminophosphates are usually unstable or are transformed into other topological structures under heat treatment in the presence of water; they are indeed prone to decompose or give way when irradiated by an electron beam.11 Therefore, it is extremely difficult to readily determine the structures of aluminophosphates.
For a similar reason, the structure of PST-5, the as-made form of PST-6 that contains the organic structure-directing agent diethylamine (DEA), is still unknown because of its unstable complex structure compared to that of PST-6, the calcined form of PST-5.12 PST-6 contains nine T atoms in its unit cell, which was designated as “PSI” by the International Zeolite Association.13 This synthetic approach, using a low-cost amine, is intriguing because the hydrolysis of the aluminum precursor, and solvent evaporation prior to hydrothermal treatment, can produce various novel aluminophosphates.10,12
Recently, the novel aluminophosphate obtained from a controlled hydrothermal synthesis using DEA as the structure-directing agent was also found to have a tetragonal structure in which the lattice parameters were a = 14.44938(18) Å and c = 18.27743(18) Å.8 The corresponding novel structure was determined unambiguously using the powder charge flipping (pCF) method,14,15 and was designated as “CNU-2”. CNU-2 contains a three-dimensional pore structure with a large surface area and pore volume; approximately 350 m2 g−1 and 0.3 cm3 g−1, respectively.
In this work, the crystal structure of PST-5, which is still unknown because of its structural complexity and poor stability, was elucidated using pCF method combined with topological searching using the Focus suite of programs because the phase information from electron microscopy is limited due to the weak framework structure. The proposed crystal structure is also found to be consistent with other spectroscopic data. In addition, the present structure determination method can be extended to other porous materials that are weakly stable and where phase information from electron microscopy is limited.
Results and discussion
Synthesis of PST-5, the as-made form of PST-6
X-ray powder diffraction patterns of PST-5 and the corresponding PST-6, which were prepared using as the structure-directing agent following the literature procedure,12 are shown in Fig. 1. It was reported that the mixing of aluminum isopropoxide (AIP) and water immediately resulted in the formation of oligomeric aluminum oxide species that depended on the hydrolysis time. The AEL phase, AlPO-11,16 containing a one-dimensional ten-membered ring (10 MR), was obtained after solvent evaporation when there was no additional hydrolysis time. Further increases in AIP hydrolysis time, up to 24 h after mixing with water, led to the formation of the AWO phase,17 AlPO-21, containing a one-dimensional 8 MR. The AWO phase underwent a topotactic transformation to the ATV phase, AlPO-25, that also contains an 8 MR and maintains the same dimensional structure.18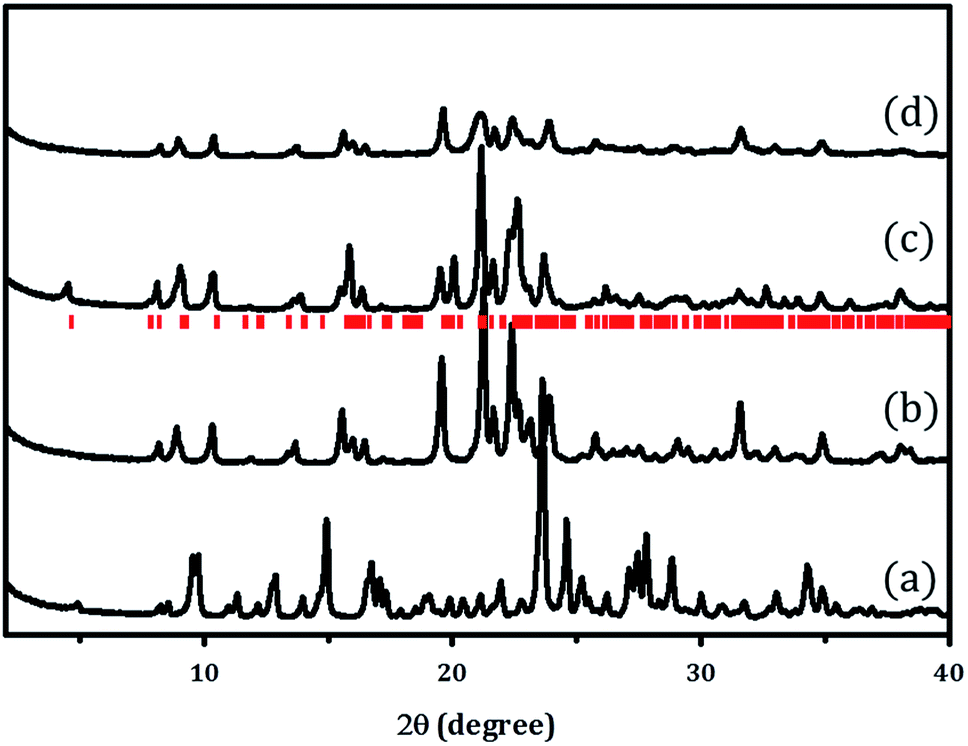 | ||
| Fig. 1 XRD patterns of (a) PST-5, (b) PST-6 (1 day), and (c) PST-6 (1 month) after calcination at 823 K under a flow of air. (d) XRD pattern of PST-6 after calcination at 423 K with ozone. The red markers indicate the PSI phase. | ||
The hydrolysis of AIP for 6 h followed by solvent evaporation readily produced PST-5 as evidenced by the XRD pattern shown Fig. 1(a). This material was further transformed into the PSI phase, PST-6, containing two parallel, highly elliptical 10 MR and 8 MR pores, after calcination at 823 K for 6 h, the XRD pattern of which is shown in Fig. 1(b). There are significant differences between the X-ray diffraction patterns of PST-5 and PST-6, which could be ascribed to the transformation of the PST-5 framework structure, in a similar manner to the AWO to ATV topotactic transformation.
PST-6 contains a framework structure consisting of two parallel, highly elliptical 10 MR and 8 MR channels that run along the c-axis.12,13 The lattice parameters of orthorhombic Cmce PST-6, were determined to be a = 8.22809 Å, b = 22.45626 Å and c = 37.89111 Å, by indexing the data in Fig. 1(b). The refinement of the X-ray diffraction pattern in Fig. 1(b) using the LeBail method provided high quality R factors; Rp = 2.76% and Rwp = 3.81% when the lattice parameters corresponding to those of PSI in the Cmce space group were employed (Fig. S1†).
Such high agreement factors and the well-matched Bragg peak positions confirm the formation of PST-6. In addition, the structure of PST-6 was examined by XRD after exposure to air at room temperature for one month. A low-angle peak appeared for the aged PST-6 and there were progressive changes in the peaks in the high-angle region (Fig. 1(c)), which can be ascribed to adsorption-induced structural changes. In order to minimize the effect of high temperature (823 K) calcination, ozone calcination was performed at 423 K to remove the occluded DEA, the XRD pattern of which shown in Fig. 1(d). Thermogravimetric and differential thermal analysis (TG-DTA) of the PST samples suggested that all occluded structure-directing agents were removed by ozone calcination at 423 K, as shown in Fig. S2.† However, the textural properties of PST-6 treated by both methods are similar, as presented in Fig. S3,† and are comparable to those reported in the literature.12 Indeed, the XRD pattern of the ozone-calcined PST-6 was similar to that of the heat-treated PST-6 at 773 K, although peak-broadening appeared implying the presence of a disordered pore structure.
The morphology of PST-5 is presented in Fig. 2. The scanning electron micrograph of PST-5 shows a planar morphology devoid of impurity phases. The transmission electron micrograph of PST-5 also reveals the same morphology; unfortunately it was not possible to observe the pores because of amorphization induced by electron-beam irradiation. However, electron diffraction did reveal the presence of a polycrystalline structure in PST-5, but it degraded rapidly because of its poor stability and the presence of occluded DEA.
 | ||
| Fig. 2 SEM and TEM images (top) and the corresponding electron diffraction patterns (bottom) for PST-5. | ||
The structure of PST-5 has also been studied by solid-state magic angle spinning (MAS) NMR spectroscopy, as shown in Fig. 3.19–21 Prior to calcination, the tetrahedral, pentagonal and octahedral aluminum peaks at 40, 11, and −2 ppm were observed.12 The pentagonal and octahedral peaks disappeared upon calcination, suggesting that the PST-5 framework is tetrahedrally coordinated. The 31P MAS NMR spectrum of PST-5 exhibited no less than seven peaks ranging from −13 ppm to −35 ppm. Upon calcination, these peaks merged into two peaks at −27 and −32 ppm, which correspond to the tetrahedral framework of the phosphorous atoms.
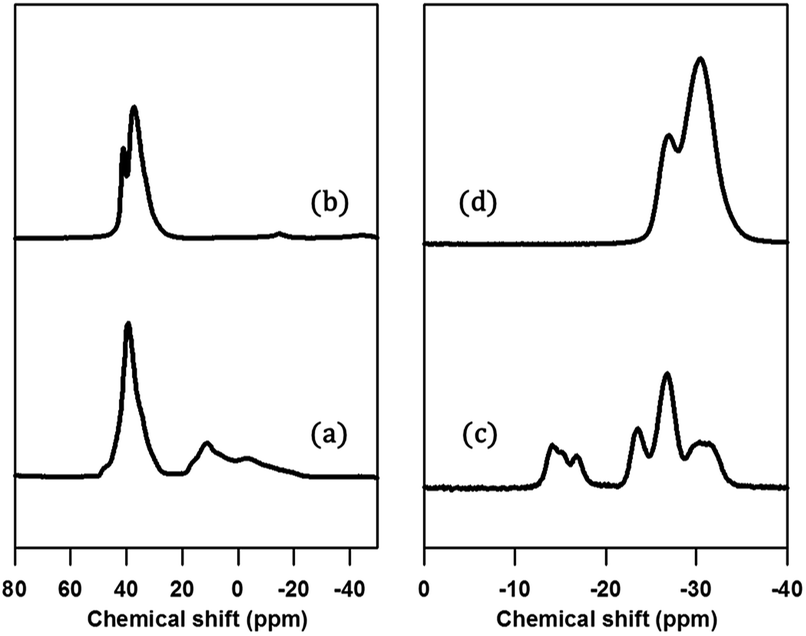 | ||
| Fig. 3 (a, b) 29Al and (c, d) 31P MAS NMR spectra of PST-5; (a, c) before, and (b, d) after calcination at 823 K under a flow of air. | ||
Chemical shifts between around −13 and −25 ppm suggest the presence of P(OAl)x(OH)y or possibly even a layered phase. Prior to deconvolution, inspection of the 2nd derivatives of the NMR peaks indicated nine deconvolved P species. Deconvolution of the 31P NMR spectrum in Fig. 4 suggested the presence of nine species; four peaks near −15 ppm appear to correspond to hydroxyl group bridges, while the remaining five peaks can be considered to belong to framework species. These 29Al and 31P MAS NMR spectral results for PST-5 and PST-6 are consistent with those reported earlier.12
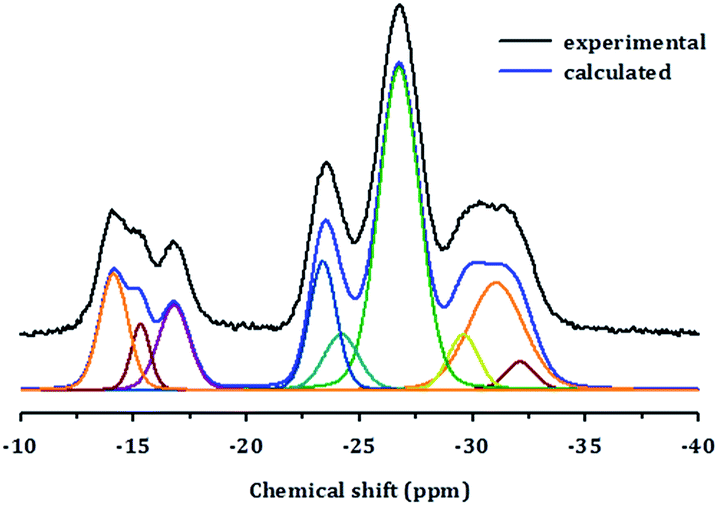 | ||
| Fig. 4 Deconvolution of the 31P MAS NMR spectrum of PST-5. | ||
Structure determination of PST-5, the as-made form of PST-6
In order to determine the crystal structure of PST-5, powder diffraction data were collected using synchrotron radiation at the BL-9B station, Pohang Accelerator Laboratory. The indexing was performed with the DICVOL06 program to obtain high M(20) and F(20) values of 51.9 and 163.2, respectively, indicating that the orthorhombic crystal system cell parameters, of which the lattice parameters are a = 36.55301 Å, b = 10.87706 Å, and c = 10.27363 Å, are reasonably correct. The most probable highest space group was found to be Pmnm from systematic absences. Later the space group was transformed into Pmmn, a standard setting (origin choice 2). Intensity extraction was performed up to d = 1.0 Å using the LeBail method to further search for the structure solution.22 The number of separated reflections was only 338 among 3918 total reflections, which implies that 92% of the reflections overlapped, within 0.3 × fwhm (full width at half maximum). Indeed, such high statistical overlapping hampered the structure determination.6Initial attempt to obtain the framework structure solution using Focus6,9 was failed over the equipartitioned X-ray intensity. In the meantime, the electron-density map can be obtained readily using the pCF method employing histogram matching method.22–24 However, the electron-density map was also not interpretable to give the complete framework structure because of significant loss or lack of phase information even though the pore and the framework region could be identified. Therefore, following the scheme as shown in Fig. 5, the obtained framework structure was considered to be partially correct due to the presence of the well-resolved XRD peaks at low angles that are similar to those from single-crystal diffraction. Therefore, the partially correct framework structure can be used as a model for the decomposition of the overlap peaks. Such model-biased repartitioning of the XRD intensities thus provided the best estimate of each reflection for the Focus framework search program.6,9,22 During 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 trial runs, 38 possible frameworks were generated and 66% of the solutions were the same framework structure, which was believed to be correct.
000 trial runs, 38 possible frameworks were generated and 66% of the solutions were the same framework structure, which was believed to be correct.
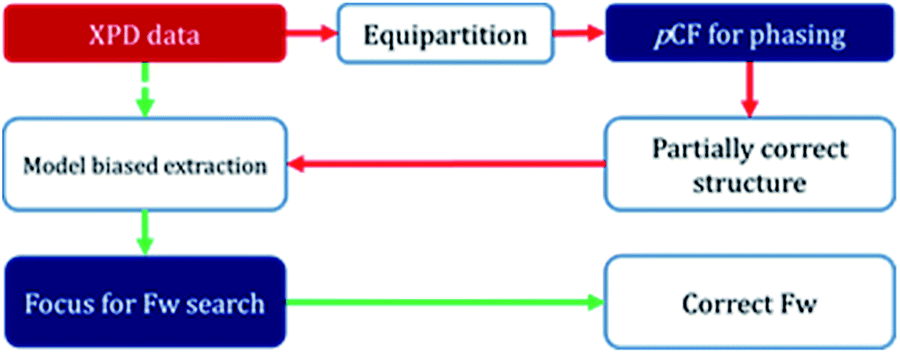 | ||
| Fig. 5 Schematic diagram for framework search combing pCF method and Focus program. | ||
Fig. S4† displays the best possible framework structures obtained from the Focus program. It is interesting that the structure obtained for PST-5 is strikingly similar to that of PST-6, with the exception that the unit cell volume is almost half that of PST-6. The projection of the obtained PST-5 framework structure along the a-axis is the same as that of PST-6. The electron-density map was obtained again using the pCF method with the corresponding framework as the model structure in order to see if the penta or hexa coordinated T atom was present. After 1500 trials, the ten-highest probable electron-density maps, with low Rsym of about 21%, were merged to give the most probable electron-density map. The electron-density map obtained in this manner was almost the same as that from the framework search in Focus with no penta or hexa coordinated T atom. The partially correct structure solution from the pCF method, which also contains partially correct phase information, provides the best estimate of XRD intensities through model-biased repartitioning for the Focus program. The current strategy in Fig. 5 suggests that the correct structure solution can be obtained by combining the pCF method with the Focus framework search program in cases where phase information is limited.
Fig. 6 displays the final Rietveld refinement of the XRD pattern of PST-5 with the reasonable agreement factors detailed in Tables 1–4. There are nine unique T atoms in the unit cell, while approximately 13.4 DEA molecules are also present; this corresponds to a 17.8 wt% weight loss due to decomposition during calcination, which is partly consistent with the DTA measurement results as shown in Fig. S2.† The initial structure of the DEA molecules in the pores were determined using the MM2 method implemented in ChemBio3D.25 The structure of DEA was subject to the determination of location with the parallel-tempering algorithm implemented in the Free Objects for Crystallography program.26 During the final Rietveld refinement, each DEA was initially considered to be rigid, and subsequently the corresponding thermal motion was described using the TLS (translation-libration-screw) method with distance restraints.27 Thereby, the current Rietveld refinement provided an Rp of 8.55% and an Rwp of 11.63%, with a reasonable average T–O bond distance of 1.67 Å, and an average O–T–O angle of 110°. In addition, the C–N and C–C bond distances were consistent with single C–C and C–N bonds. The corresponding angles were also in the 120 ± 10° range. Such a DEA location in PST-5 suggests that it played an important role as the structure-directing agent during the formation of the porous aluminophosphate structure.
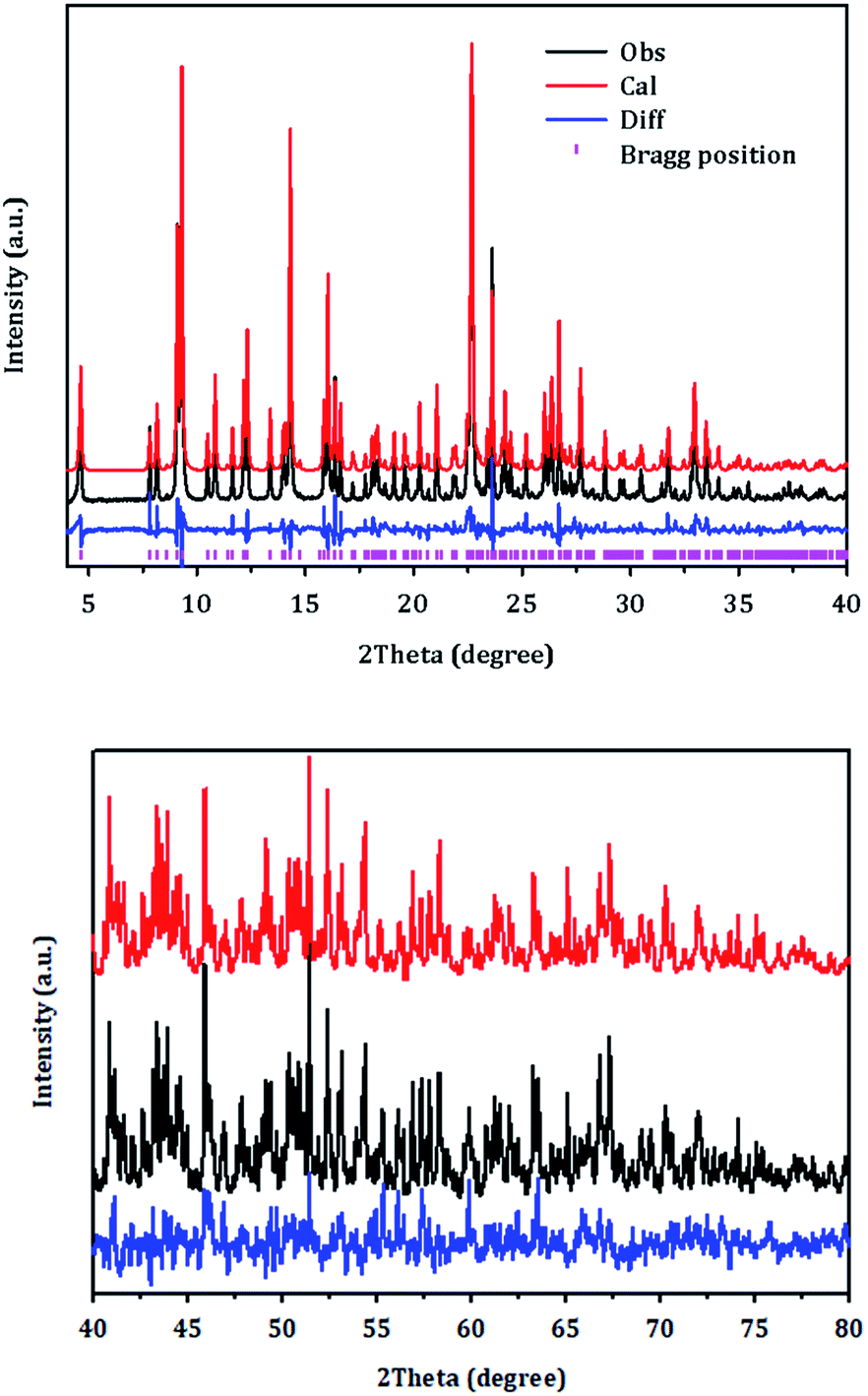 | ||
| Fig. 6 Rietveld refinement plot for PST-5. Tick marks indicate Bragg positions. For clarity, the plot has been magnified five times between 40 and 80°. | ||
| Material | PST-5 |
| Unit cell composition | |(DEA)13.4(H2O)10.9|T72O144 |
| Symmetry | Orthorhombic |
| Space group | Pmmn |
| a, Å | 10.2796(7) |
| b, Å | 36.643(3) |
| c, Å | 10.8918(8) |
| Cell volume, Å3 | 4102.7(5) |
| Wavelength, Å | 1.4862 |
| Radiation type | Synchrotron radiation, PAL |
| No. observation | 3911 |
| No. parameters | 117 |
| No. restraints | 47 |
| No. constraints | 1 |
| Max change/s.u. | 0.0480 |
| Largest peak | 0.96 |
| Deepest hole | −0.99 |
| Rp, % | 8.55 |
| Rwp, % | 11.63 |
| RF, % | 9.81 |
| GOF | 5.12 |
| Atom | x | y | z | Uiso, 102× Å2 | Mult. | Occ. |
|---|---|---|---|---|---|---|
| a The thermal displacement factor was calculated from TLS method. | ||||||
| O1 | 0.25 | 0.595(3) | 0.678(10) | 0.046(6) | 4 | 1 |
| O2 | 0.568(8) | 0.129(2) | 0.474(6) | 0.046(6) | 8 | 1 |
| O3 | −0.022(6) | 0.0776(19) | 0.344(7) | 0.046(6) | 8 | 1 |
| O4 | 0.620(7) | 0.1408(18) | 0.219(6) | 0.046(6) | 8 | 1 |
| O5 | 0.25 | 0.654(3) | 0.475(9) | 0.046(6) | 4 | 1 |
| O6 | 0.087(8) | 0.6203(17) | 0.309(6) | 0.046(6) | 8 | 1 |
| O7 | 0.045(6) | 0.6870(19) | 0.359(7) | 0.046(6) | 8 | 1 |
| O8 | 0.25 | 0.194(3) | 0.610(10) | 0.046(6) | 4 | 1 |
| O9 | 0.100(11) | 0.25 | 0.642(9) | 0.046(6) | 4 | 1 |
| O10 | 0.129(7) | 0.1992(17) | 0.823(5) | 0.046(6) | 8 | 1 |
| O11 | −0.049(7) | 0.1818(18) | 0.035(6) | 0.046(6) | 8 | 1 |
| O12 | 0.480(6) | 0.1170(18) | 0.089(7) | 0.046(6) | 8 | 1 |
| O13 | 0.25 | 0.664(3) | 0.923(10) | 0.046(6) | 4 | 1 |
| O14 | 0.074(7) | 0.5586(17) | 0.335(6) | 0.046(6) | 8 | 1 |
| O15 | 0.044(6) | 0.0146(18) | 0.280(6) | 0.046(6) | 8 | 1 |
| O16 | 0.25 | 0.516(3) | 0.455(9) | 0.046(6) | 4 | 1 |
| O17 | 0 | 0 | 0.5 | 0.046(6) | 4 | 1 |
| O18 | 0.464(6) | 0.5992(19) | 0.096(6) | 0.046(6) | 8 | 1 |
| O19 | 0.25 | 0.582(3) | 0.137(9) | 0.046(6) | 4 | 1 |
| O20 | 0.25 | 0.071(3) | 0.268(9) | 0.046(6) | 4 | 1 |
| O21 | 0.140(7) | 0.0686(18) | 0.111(6) | 0.046(6) | 8 | 1 |
| O22 | 0.25 | 0.080(3) | −0.028(10) | 0.046(6) | 4 | 1 |
| O23 | 0.25 | 0.203(3) | 0.036(10) | 0.046(6) | 4 | 1 |
| O24 | 0.093(11) | 0.25 | 0.078(8) | 0.046(6) | 4 | 1 |
| T1 | 0.616(4) | 0.1136(11) | 0.339(4) | 0.050(3) | 8 | 1 |
| T2 | 0.112(4) | 0.6527(11) | 0.411(4) | 0.050(3) | 8 | 1 |
| T3 | 0.105(4) | 0.2045(9) | 0.677(4) | 0.050(3) | 8 | 1 |
| T4 | 0.608(4) | 0.1446(10) | 0.077(4) | 0.050(3) | 8 | 1 |
| T5 | 0.098(4) | 0.5143(11) | 0.397(3) | 0.050(3) | 8 | 1 |
| T6 | 0.121(4) | 0.5825(11) | 0.213(3) | 0.050(3) | 8 | 1 |
| T7 | 0.092(4) | 0.0606(10) | 0.245(3) | 0.050(3) | 8 | 1 |
| T8 | 0.092(4) | 0.0914(11) | −0.013(4) | 0.050(3) | 8 | 1 |
| T9 | 0.089(4) | 0.2046(10) | −0.015(3) | 0.050(3) | 8 | 1 |
| O25 | 0.25 | 0.756(7) | 0.088(8) | 0.08(4) | 4 | 1 |
| O26 | 0.25 | 0.527(6) | −0.10(2) | 0.08(4) | 4 | 0.4931 |
| O27 | 0.25 | 0.135(4) | 0.023(14) | 0.08(4) | 4 | 0.7802 |
| O28 | 0.43(2) | 0.75 | 0.14(2) | 0.08(4) | 4 | 0.4496 |
|
||||||
| Rigid body with TLS method for diethylamine-aa | ||||||
| C1 | 0.622(10) | 0.2667(9) | 0.336(3) | 0.52(14) | 8 | 0.67(2) |
| C3 | 0.473(10) | 0.2625(9) | 0.346(3) | 0.52(14) | 8 | 0.67(2) |
| C6 | 0.269(10) | 0.3173(9) | 0.385(3) | 0.52(14) | 8 | 0.67(2) |
| C8 | 0.322(10) | 0.3518(9) | 0.450(3) | 0.52(14) | 8 | 0.67(2) |
| N1 | 0.356(10) | 0.2865(9) | 0.359(3) | 0.52(14) | 8 | 0.67(2) |
|
||||||
| Rigid body with TLS method for diethylamine-ba | ||||||
| C2 | 0.266(8) | 0.4961(15) | 0.654(4) | 0.17(4) | 8 | 1 |
| C4 | 0.211(8) | 0.4584(15) | 0.642(4) | 0.17(4) | 8 | 1 |
| C5 | 0.314(8) | 0.3987(15) | 0.612(4) | 0.17(4) | 8 | 1 |
| C7 | 0.463(8) | 0.3937(15) | 0.574(4) | 0.17(4) | 8 | 1 |
| N2 | 0.300(8) | 0.4323(15) | 0.697(4) | 0.17(4) | 8 | 1 |
| Bond distance | Bond distance | ||
|---|---|---|---|
| T1–O1 | 1.56(7) | T6–O18 | 1.67(7) |
| T1–O2 | 1.65(8) | T6–O19 | 1.56(6) |
| T1–O3 | 1.64(8) | T7–O3 | 1.71(8) |
| T1–O4 | 1.64(8) | T7–O15 | 1.80(8) |
| T2–O2 | 1.60(8) | T7–O20 | 1.68(5) |
| T2–O5 | 1.58(6) | T7–O21 | 1.57(7) |
| T2–O6 | 1.66(8) | T8–O12 | 1.64(8) |
| T2–O7 | 1.53(8) | T8–O18 | 1.62(8) |
| T3–O7 | 1.72(8) | T8–O21 | 1.65(8) |
| T3–O8 | 1.70(7) | T8–O22 | 1.68(5) |
| T3–O9 | 1.71(4) | T9–O10 | 1.83(6) |
| T3–O10 | 1.62(7) | T9–O11 | 1.74(8) |
| T4–O4 | 1.56(8) | T9–O23 | 1.76(5) |
| T4–O11 | 1.55(8) | T9–O24 | 1.94(6) |
| T4–O12 | 1.67(8) | C1–C3 | 1.55(14) |
| T4–O13 | 1.62(6) | C3–N1 | 1.44(12) |
| T5–O14 | 1.80(8) | N1–C6 | 1.49(10) |
| T5–O15 | 1.75(8) | C6–C8 | 1.54(7) |
| T5–O16 | 1.69(5) | C2–C4 | 1.51(11) |
| T5–O17 | 1.59(4) | C4–N2 | 1.45(10) |
| T6–O6 | 1.78(8) | N2–C5 | 1.53(9) |
| T6–O14 | 1.65(8) | C5–C7 | 1.60(11) |
| Angle | Angle | ||
|---|---|---|---|
| O1–T1–O2 | 122(5) | O16–T5–O17 | 110(4) |
| O1–T1–O3 | 100(5) | O6–T6–O14 | 82(4) |
| O1–T1–O4 | 98(5) | O6–T6–O18 | 93(4) |
| O2–T1–O3 | 94(4) | O6–T6–O19 | 119(5) |
| O2–T1–O4 | 121(4) | O14–T6–O18 | 131(4) |
| O3–T1–O4 | 122(4) | O14–T6–O19 | 133(5) |
| O2–T2–O5 | 85(5) | O18–T6–O19 | 92(4) |
| O2–T2–O6 | 95(4) | O3–T7–O15 | 91(4) |
| O2–T2–O7 | 128(5) | O3–T7–O20 | 119(5) |
| O5–T2–O6 | 117(5) | O3–T7–O21 | 137(4) |
| O5–T2–O7 | 123(5) | O15–T7–O20 | 116(5) |
| O6–T2–O7 | 106(4) | O15–T7–O21 | 117(4) |
| O7–T3–O8 | 127(5) | O12–T8–O18 | 85(4) |
| O7–T3–O9 | 107(5) | O12–T8–O21 | 82(4) |
| O7–T3–O10 | 108(4) | O12–T8–O22 | 130(5) |
| O8–T3–O9 | 99(5) | O18–T8–O21 | 142(4) |
| O8–T3–O10 | 105(5) | O18–T8–O22 | 140(5) |
| O9–T3–O10 | 110(5) | O21–T8–O22 | 72(5) |
| O4–T4–O11 | 114(4) | O10–T9–O11 | 115(4) |
| O4–T4–O12 | 86(4) | O10–T9–O23 | 96(4) |
| O4–T4–O13 | 87(5) | O10–T9–O24 | 127(4) |
| O11–T4–O12 | 105(4) | O11–T9–O23 | 130(5) |
| O11–T4–O13 | 89(5) | O11–T9–O24 | 105(4) |
| O12–T4–O13 | 166(5) | O23–T9–O24 | 82(5) |
| O14–T5–O15 | 104(4) | (O–T–O)av | 110(4) |
| O14–T5–O16 | 103(5) | C1–C3–N1 | 138(3) |
| O14–T5–O17 | 118(3) | N1–C6–C8 | 120(8) |
| O15–T5–O16 | 126(5) | C2–C4–N2 | 109(10) |
| O15–T5–O17 | 97(3) | C4–N2–C6 | 109(6) |
Fig. 7 displays PST-5 in the natural tile representation. There are five different kinds of tile used for the construction of the PST-5 structure: {33.42.5.62.72}, {42.62.82}3, {42.63.8}2 {43.6.82} and {43.63}2. The projections of PST-5 along the [100], [010] and [001] directions show the presence of intersecting eight and ten-membered rings, implying the formation of a three-dimensional structure. A double four-membered ring (D4R) was found in the {42.62.82}3 tile, which may later lead the structural collapse of the material upon the calcination. It was of particular interest to know whether or not a highly elliptical 10 MR was present. However, such a highly elliptical 10 MR was found to be a simple projection in the [100] direction. In fact, all the T atoms in the {33.42.5.62.72} and {42.62.82} tile are not in the same plane, as shown in Fig. 7 and 8.
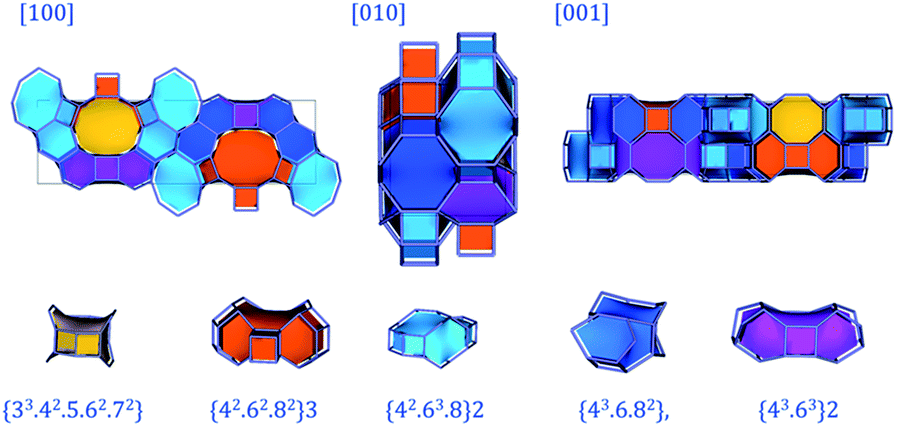 | ||
| Fig. 7 Natural tile representations of PST-5 obtained from the topological analysis. | ||
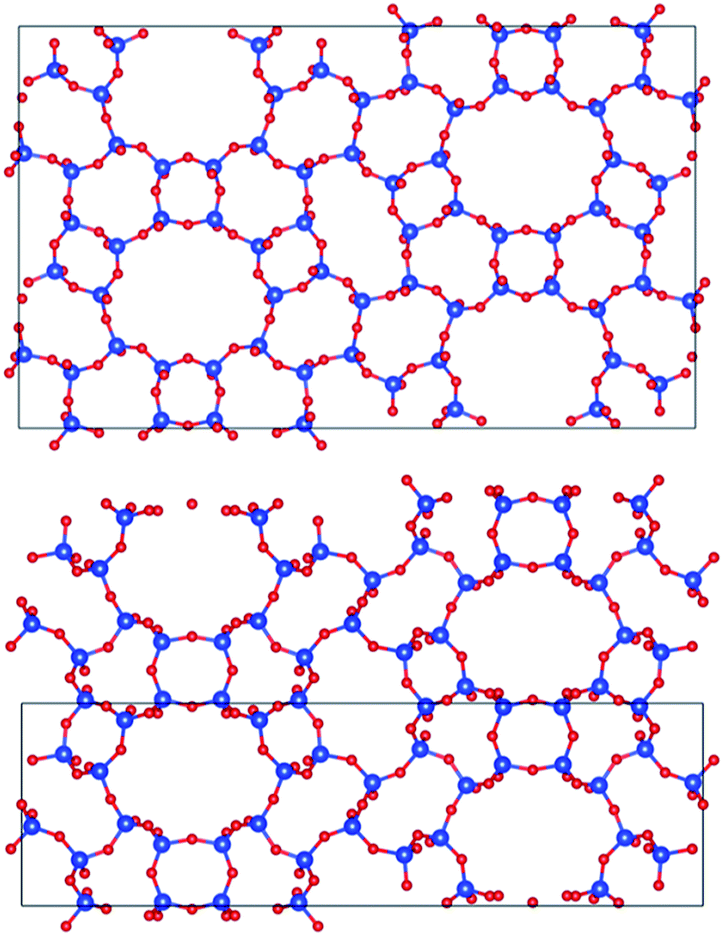 | ||
| Fig. 8 Projections of PST-5 (lower panel), and PST-6 (upper panel) in the [100] direction. | ||
In order to check framework stability, the obtained structure of PST-5 was also subjected to lattice-energy minimization calculations using the General Utility Lattice Program (GULP) with the zeolite shell model as the potential model.28–30 The obtained total lattice energy was determined to be −2.135 eV Å−3 which is relatively high compared to that of PST-6, −2.646 eV Å−3. The presence of the D4R significantly may increase the total lattice energy, leading to the transformation or transition of the structure. The results of the lattice-energy calculations are consistent with the proposed structural transformation of PST-5 to PST-6.
Fig. 9 visualizes the connectivity within PST-5. There is an interconnection between the 8 MR and 10 MR, suggesting the formation of a three-dimensional structure. Furthermore, one of the DEA molecules lies between the 8 MR and 10 MR sections, as shown in Fig. S5.† Such a DEA location supports the formation of a three-dimensional structure in PST-5.
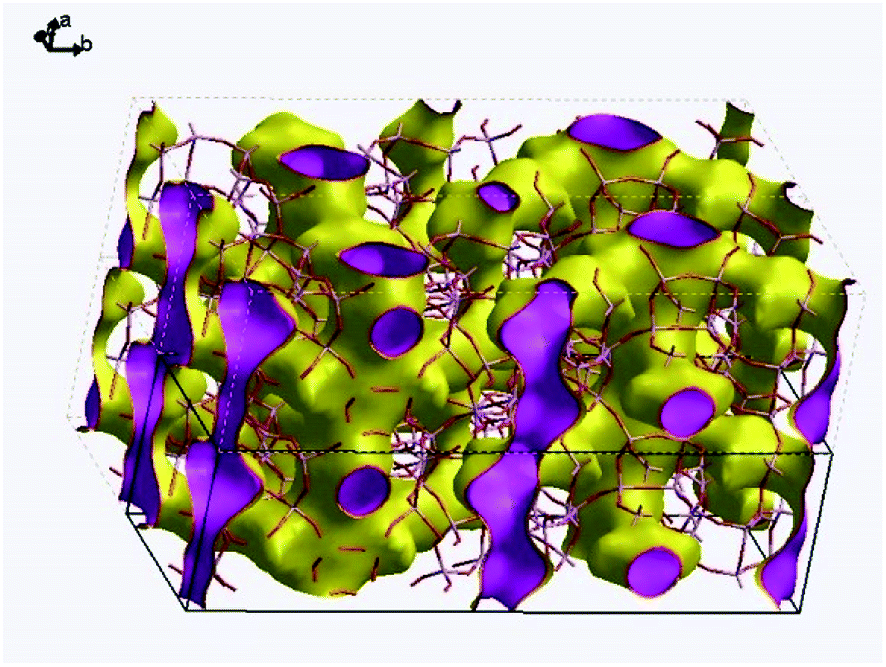 | ||
| Fig. 9 Pore connectivity within PST-5. The surface represents zero electron density. | ||
Conclusions
The crystal structure of PST-5 has been clarified using the pCF method in combination with the Focus framework search program. PST-5 has an orthorhombic Pmmn structure in which the lattice parameters are a = 10.28 Å, b = 36.64 Å and c = 10.89 Å, which are similar to those of PST-6, with the exception the one of the lattice parameters is approximately half that of PST-6. The obtained structure contains 9 T atoms and has three-dimensional pore connectivity, which is comparable to the one-dimensional structure of PST-6. Energy calculations for the PST-5 and PST-6 structures suggest that the transition from the 3D to the 1D pore structure is favorable. The structure determination methodology used in this work can also be extended to other weakly stable porous materials where phase information is limited. In addition, the partially correct structure solution was found to enhance the search for the true structure solution. Furthermore, the location and thermal motion of the structure-directing agent can be determined, despite structural overlap, highlighting the role that the structure-directing agent plays in the formation of the unique novel porous structure.Experimental section
Synthesis of PST-5
Aluminophosphates were synthesized as described below.10,12 H3PO4 (3.84 g, 85%, Daejung) was dissolved in doubly distilled water (5.63 g) and the solution was stirred for 1 h. Aluminum isopropoxide (AIP, 6.95 g, 98%, Aldrich) was slowly added dropwise to doubly distilled water (5.64 g) with vigorous stirring to achieve hydrolysis. The phosphoric acid solution was then combined with the hydrolyzed aluminum hydroxide solution with vigorous stirring and the mixture was stirred for further 1 h. Diethylamine (DEA, 2.45 g, 99.5%, Aldrich) was added dropwise to the mixture with vigorous stirring. The resulting mixture had a pH of 8.5 to 9.0. No less than 90% of isopropanol in solvent was evaporated at room temperature. The resulting gel was transferred to an autoclave and hydrothermally condensed at 473 K for 5 d under static conditions. The nominal gel composition was 2DEA:1Al2O3:1P2O5:40H2O.
After cooling room temperature, the solid product was recovered by filtration and washed with copious amounts of doubly distilled water. The solid was dried in a vacuum oven for 12 h at room temperature. The obtained aluminophosphate was treated to remove occluded organic molecules under a flow of air while the temperature was increased to 823 K at a rate of 2 K min−1 and held at 823 K for 6 h. Alternatively, ozone was employed in order to remove DEA at 423 K.
Physical characterization of PST-5
For the determination of phase purity and sample crystallinity, powder X-ray diffraction spectra were recorded using a D/MAX Ultima III instrument (Rigaku, Japan) with Cu Kα radiation, or an EMPYREAN diffractometer (PANanalytical). Data were collected with a fixed divergence slit (0.50°) and Soller slits (incident and diffracted = 0.04 rad). Nitrogen adsorption–desorption isotherms were measured at 77 K with an ASAP2020 instrument (Micromeritics, USA). Prior to nitrogen adsorption measurement, the sample was evacuated at 573 K for 2 h in order to remove adsorbed impurities.Multinuclear MAS NMR measurements were performed mainly on a Unity Solid Inova 400 MHz spectrometer (Agilent Technologies, U.S.A) at the Korea Basic Science Institute. For 27Al MAS NMR, the sample was spun at a rate of 12 kHz. The 27Al MAS spectra were recorded at 104.2797 MHz with a π/6 rad pulse length of 1.0 μs, a recycle delay of 2 s; about 2048–4096 pulse transients were acquired a AlCl3(aq) as a reference. The 31P MAS NMR spectra were recorded at a spinning rate of 12.0 kHz and were measured on the same spectrometer at the 31P frequency of 162.0046 MHz. The spectra were acquired after about 100 pulse transients, which were repeated with a π/6 rad pulse length of 2.0 μs and a recycle delay of 20 s. The 31P chemical shifts are referenced to H3PO4. The 13C MAS NMR spectra, at a spinning rate of 4.5 kHz, were measured at the 13C frequency of 100.6302 MHz with a π/2 rad pulse length of 3.0 μs and a recycle delay of 12 s. Typically, 10000 pulse transients were accumulated. Scanning electron micrographs were obtained using an S-4700 instrument (Hitachi, Japan) operating at 25 kV. Elemental analyses were performed with an Oxford Instruments ISIS EDS X-ray microanalysis system attached to an EMAX instrument. Thermal gravimetric and differential thermal analyses (TG-DTA) were carried out on DTG-60H instrument (Shimadzu, Japan) at a ramping rate of 10 K min−1 under an air stream.
Structure determination of PST-5
For detailed structural analysis of PST-5, synchrotron diffraction data were collected on beamline 9B at the Pohang Accelerator Laboratory (Pohang, Korea) using highly collimated monochromatic synchrotron radiation (λ = 1.4862 Å). The detector arm of the vertical scan diffractometer consisted of seven sets of Soller slits, flat Ge(111) crystal analyzers, anti-scatter baffles, and scintillation detectors, with each set separated by 20°. Data were obtained on the sample at room temperature in flat-plate mode, with a step size of 0.01° for a scan time of 3 s per step, and overlaps of 2° to the next detector bank over the 2θ range of 5–145°.Diffraction patterns obtained were indexed using the DICVOL06 program implemented in the FullProf program suite.10 The initial structure of DEA in the framework was obtained by the MM2 method implemented in ChemBio3D.25 Subsequently the parallel tempering algorithm implemented in the Free Objects for Crystallography program was applied to determine the framework structure containing DEA.26 Then, profile refinement of the structure model, comprising the framework and the included organic molecules was performed using the Rietveld method in the JANA2006 package.22 During Rietveld refinement, a pseudo-Voigt function and microscopic broadening, together with a manually interpolated background, were used to describe the peak shapes. The framework was modeled as completely siliceous. Isotropic displacement parameters for all T atoms were constrained to be equal in order to minimize the number of parameters, as were those of all O atoms, and the TLS method was used to describe the thermal motion of DEA molecule. The framework T (Al or P)–O distances were soft-restrained to 1.62 Å with the standard uncertainty of 0.005 Å. A combination of the refined occupancies of the structure-directing agents and their site multiplicities suggested the presence of 13.4 DEA and 10.9 water molecules per unit cell.
Energy optimization of PST-5
The obtained structure was subject to lattice energy minimization calculations using the General Utility Lattice Program (GULP) (version 4.3).28–30 All atomic coordinates and cell parameters are optimized to zero force using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization method. Calculations were performed at constant pressure using a zeolite shell model as the potential model in which a Buckingham function was used to describe the short-range interactions and a three-body (bond bending) term was included to accurately model O–T–O angles. A shell model was also used to simulate the polarizability of the oxygen atoms. The Newton–Raphson optimizer was employed during energy minimization, with maximum function and gradient tolerances of 0.0001 and 0.001 eV Å−1, respectively; the symmetry was constrained and the cell parameters varied. A gradient-norm convergence criterion of 0.001 eV Å−1 was used for all optimizations. All calculations were performed on a Dell XPS 8700 desktop computer with 8 cores based on the Intel Core i7-4770 CPU processor (3.4 GHz) with 24 GB RAM.Acknowledgements
This research was supported by the Basic Science Research Program and the Global Frontier Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (MSIP) (NRF-2016R1A5A1009592 and NRF-2015M3A6B1065266). The authors thank the Pohang Accelerator Laboratory (PLS) for the collection of X-ray powder diffraction data.Notes and references
- X. Meng and F.-S. Xiao, Chem. Rev., 2014, 114, 1521–1543 CrossRef CAS PubMed.
- J. Cejka, A. Corma and S. Zones, Zeolites and Catalysis: Synthesis Reaction and Applications, Wiley, 2010 Search PubMed.
- J. Li, J. Yu and R. Xu, Proc. R. Soc. A, 2012, 468, 1955–1967 CrossRef CAS.
- J. Su, E. Kapaca, L. Liu, V. Georgieva, W. Wan, J. Sun, V. Valtchev, S. Hovmöller and S. Zou, Microporous Mesoporous Mater., 2014, 189, 115–125 CrossRef CAS.
- Y. Lorgouilloux, M. Dodin, E. Mugnaioli, C. Marichal, P. Caullet, N. Bats, U. Kolb and J.-L. Paillaud, RSC Adv., 2014, 4, 19440–19449 RSC.
- S. Smeets, L. B. McCusker, C. Baerlocher, E. Mugnaioli and U. Kolb, J. Appl. Crystallogr., 2013, 46, 1017–1023 CrossRef CAS.
- J. Sun, C. Bonneau, A. Cantin, A. Corma, M. J. Diaz-Cabanas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154 CrossRef CAS PubMed.
- W. Hua, H. Chen, Z. B. Yu, X. Zou, J. Lin and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 5868 CrossRef CAS PubMed.
- L. B. McCusker and C. Baerlocher, Chem. Commun., 2009, 1439–1451 RSC.
- S. Chang, G. C. Jo, J. H. Kim and S. J. Cho, Adv. Porous Mater., 2016, 4, 179–188 CrossRef and reference therein.
- S. Xu, Z. Zhao, M. Y. Hu, X. Han, J. Z. Hu and X. Bao, Microporous Mesoporous Mater., 2016, 223, 241–246 CrossRef CAS.
- J. K. Lee, A. Turrina, L. Zhu, S. Seo, D. Zhang, P. A. Cox, P. A. Wright, S. Qiu and S. B. Hong, Angew. Chem., Int. Ed., 2014, 53, 7480–7483 CrossRef CAS PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/ Search PubMed.
- C. Baerlocher, D. Xie, L. B. McCusker, S. J. Hwang, I. Y. Chan, K. Ong, A. W. Burton and S. I. Zones, Nat. Mater., 2008, 7, 631–635 CrossRef CAS PubMed.
- C. Baerlocher, L. B. McCusker and L. Palatinus, Z. Kristallogr., 2007, 222, 47–53 CrossRef CAS.
- J. M. Bennett, J. W. Richardson Jr, J. J. Pluth and J. V. Smith, Zeolites, 1987, 7, 160–162 CrossRef CAS.
- J. M. Bennett, J. M. Cohen, G. Artioli, J. J. Pluth and J. V. Smith, Inorg. Chem., 1985, 24, 188–193 CrossRef CAS.
- J. Li, J. Yu, K. Wang, G. Zhu and R. Xu, Inorg. Chem., 2001, 40, 5812–5817 CrossRef CAS PubMed.
- D. Muller, E. Jahn and G. Ladwig, Chem. Phys. Lett., 1984, 109, 332–336 CrossRef.
- C. S. Blackwell and R. L. Patton, J. Phys. Chem., 1984, 88, 6135–6139 CrossRef CAS.
- J. Zhuang, D. Ma, G. Yang, Z. Yan, X. Liu, X. Liu, X. Han, X. Bao, P. Xie and Z. Liu, J. Catal., 2004, 228, 234–242 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345–420 CAS.
- C. Baerlocher, F. Gramm, L. Massüger, L. B. McCusker, Z. He, S. Hovmöller and X. Zou, Science, 2007, 315, 1113–1116 CrossRef CAS PubMed.
- D. Xie, L. B. McCusker and C. Baerlocher, J. Am. Chem. Soc., 2011, 133, 20604–20610 CrossRef CAS PubMed.
- M. J. Dudek and J. W. Ponder, J. Comput. Chem., 1995, 16, 791–816 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- A. H. Van Benschoten, P. V. Afonine, T. C. Terwilliger, M. E. Wall, C. J. Jackson, N. K. Sauter, P. D. Adams, A. Urzhumtsev and J. S. Fraser, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1657–1667 CAS.
- J. D. Gale, J. Chem. Soc., Faraday Trans., 1997, 93, 629–637 RSC.
- J. D. Gale, J. Phys. Chem. B, 1998, 102, 5423–5431 CrossRef CAS.
- J. D. Gale, Z. Kristallogr., 2005, 220, 552–554 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental details and additional information on thermogravimetric analysis, Rietveld refinement and N2 sorption results. CCDC 1563410 and CSD 433368. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05100j |
| This journal is © The Royal Society of Chemistry 2017 |