
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 5,5-difluoro-5-phosphono-pent-2-en-1-yl nucleosides as potential antiviral agents†
F. Chevriera,
Z. Chamasa,
T. Lequeuxb,
E. Pfundb,
G. Andreic,
R. Snoeckc,
V. Roya and
L. A. Agrofoglio *a
*a
aUniversité d'Orléans et CNRS, ICOA, UMR 7311, F-45067, Orléans, France. E-mail: luigi.agrofoglio@univ-orleans.fr
bLaboratoire de Chimie Moléculaire et Thio-organique, ENSICAEN, UNICAEN, UMR, CNRS 6507, FR 3038, Caen, France
cREGA Institute for Medical Research, KU Leuven, Leuven, Belgium
First published on 23rd June 2017
Abstract
A series of hitherto unknown acyclic 5,5-difluoro-5-phosphono-pent-2-en-1-yl-pyrimidines (9a, b, 13a, b), -purines (16a, b) and -(1,2,4)-triazolo-3-carboxamide (19) were successfully synthesized from (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene in a stereoselective manner. All the synthetized compounds were assayed for antiviral activity against various viruses, but were found to be neither active nor toxic.
Introduction
Viruses are infectious agents that can replicate their genome within host cells. Many antiviral drugs are nucleoside or nucleotide analogs. Acyclic nucleoside phosphonates (ANPs)1 are a class of nucleotide analogs, originally developed by A. Holy's group,2 which exhibit a broad spectrum of antiviral activities. ANPs possess a common structure, a nucleobase attached to an aliphatic side chain containing a phosphonate moiety (C–P), and have an increased metabolic stability and resistance to chemical and biological degradation.3 Their activities are reliant on their diphosphorylation by NDP and NTP kinases, and further incorporation into the viral DNA where they can act as chain terminators. Three ANPs are in current clinical use for the treatment of serious viral infections, adefovir (PMEA), tenofovir [(R)-PMPA] and cidofovir [(S)-CDV] against hepatits B virus (HBV), human immunodeficiency virus (HIV) and cytomegalovirus (CMV), respectively, (Fig. 1).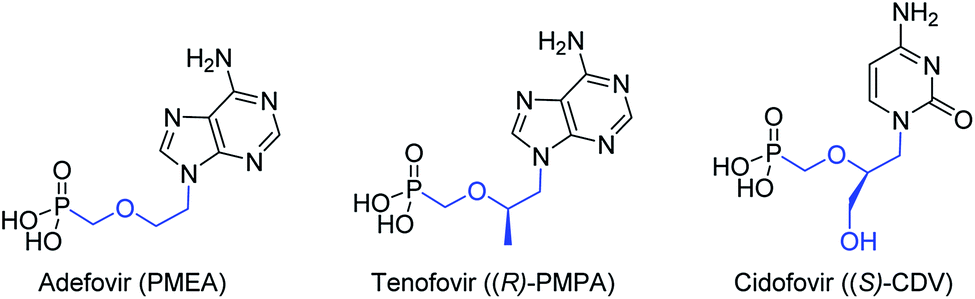 | ||
| Fig. 1 Structure of approved antiviral acyclic nucleoside phosphonates. | ||
Due to drug-resistant viruses and emerging viruses, in an effort to identify new nucleoside inhibitors of viral enzymes, new generations of ANPs, including fluorinated ANPs, were prepared and evaluated for their antiviral activity.4,5 Over the last decade, our laboratory has developed a new family of ANPs based on the (E)-but-2-enyl linker between the phosphonate moiety and the nucleobase. Several of them exhibited antiviral activity against DNA and RNA viruses in submicromolar concentrations.6 During our investigations, we have demonstrated that the N1-[(E)-4-phosphono-but-2-en-1-yl]-thymine is a substrate of human TMPK and that the (E)-but-2-enyl moiety mimics the conformation of the C1′–O4′–C4′–C5′ atoms from the natural substrate, the thymidine 5′-monophosphate, (Fig. 2).6 However, unlike natural nucleotide, our molecule missed the oxygen of the phosphate group (e.g., –O–P). Thus, we turn our attention to the introduction of a gem-difluoromethylphosphonate moiety (e.g., –CF2–P), which is isopolar and isosteric to the phosphate group.7
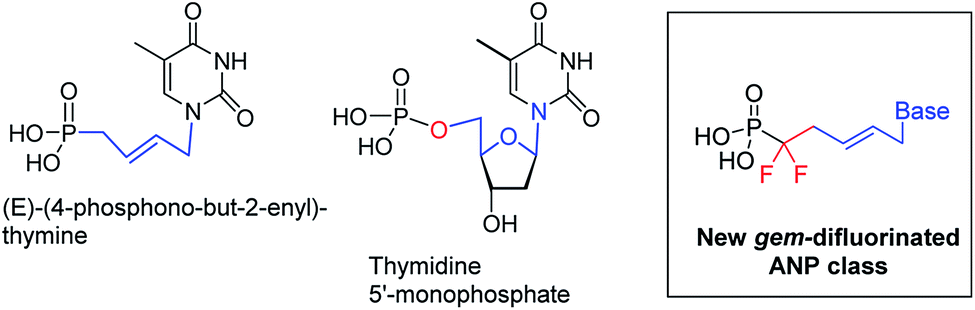 | ||
| Fig. 2 Structure of newly synthesized 5,5-difluoro-5-phosphono-pent-2-en-1-yl nucleosides as potent mimics of dNMP. | ||
In fact, due to specific properties of fluorine (high electronegativity, small steric size, hydrogen bond acceptor, …), its introduction into biologically active molecules could lead to major changes in their biological properties, such as reported by Halazy et al.8 for the 9-(5,5-difluoro-5-phosphonopentyl)-guanine, an inhibitor of purine nucleoside phosphorylase (PNP), a key enzyme in the purine metabolism.9 Therefore, based on these findings, it was interesting to design and synthesize a new type of acyclic nucleoside phosphonate, the 5,5-difluoro-5-phosphono-pent-2-en-1-yl-pyrimidines, purines and -triazole, and to evaluate their inhibitory activity against several viruses.
Results and discussion
Based on our previous work on the preparation of unsaturated acyclic nucleoside phosphonate using olefin cross-metathesis as key step,10–12 we decided to utilize this reaction between the unsaturated gem-difluorophosphonate 3 and N1-crotylated N3-protected thymine.10 The key intermediate 3 was synthesized from (diethoxyphosphinyl)difluoromethyl zinc bromide (2), allylic iodine, under CuBr catalysis, following the procedure introduced by Burton et al.13,14 Compound 3 was then engaged in the reaction of cross metathesis in presence of N3-benzoyl-N1-crotylthymine 4 with Grubbs–Nolan catalyst15 in dichloromethane. If cross-metathesis reactions were reported with fluorinated substrates,16 despite all our attempts and contrary to our results with non-fluorinated phosphonate derivatives, we never obtained the desired product (Scheme 1); this could be due to the strong electron-withdrawing effect of the 2-gem-difluoro group and to the low reactivity of both cross partners with the metal alkylidene complex.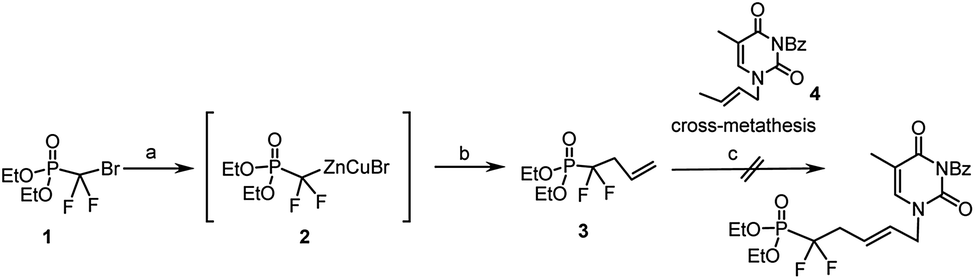 | ||
| Scheme 1 Reagents and conditions: (a) Znact, 1,2-dibromoethane, TMSCl, THFanh, 40 °C, 12 h; (b) CuBr, LiClact, allyl iodide, rt, 24 h, 35%; (c) 4 (1.5 eq.), Nolan–Grubb's II catalyst (10 mol%), CH2Cl2anh, reflux, 24 h. | ||
Alternatively we decided to introduce the nucleobase moiety through direct N-alkylation of protected and unprotected nucleobases with the corresponding (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5). Starting from bromodifluoromethylphophonate (1), the previously described organo-zinc intermediate 2 was reacted with (E)-1,4-dibromobut-2-ene14 at 0 °C, to yield the desired compound 5 in 80% with no observed isomerization of the double bond (Scheme 2).
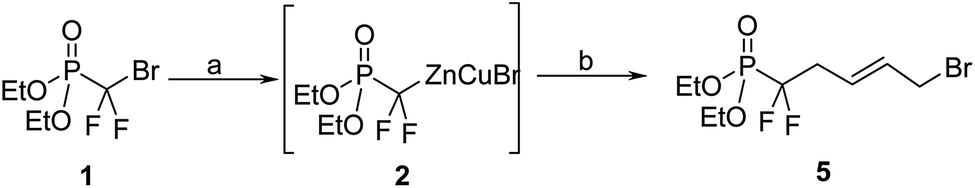 | ||
| Scheme 2 Reagents and conditions: (a) Znact, 1,2-dibromoethane, TMSCl, THFanh, 40 °C, 12 h; (b) CuBr, LiClact, trans-1,4-dibromobutene, 0 °C, 4 h, 80%. | ||
Then uracil 6a was converted to its N3-benzoyl derivative 7a through a two steps procedure involving first the formation of N1,N3-dibenzoyl derivative in presence of an excess of benzoyl chloride in CH3CN/pyridine mixture, then its selective N1-deprotection by treatment with potassium carbonate in 1,4-dioxane, (Scheme 3).17
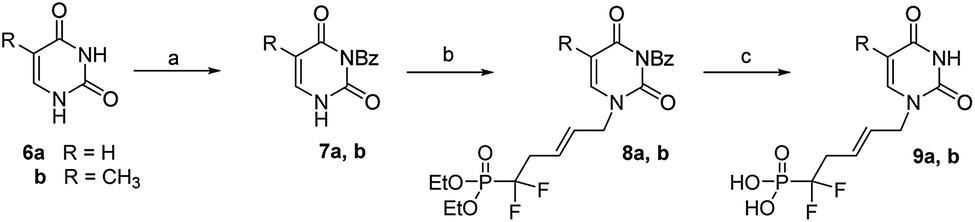 | ||
| Scheme 3 Reagents and conditions: (a) (i) BzCl, CH3CN/pyridine, rt (ii) K2CO3 (0.5 M), dioxane, 70 °C, 90% (for R = H) and 96% (for R = CH3); (b) (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5), Cs2CO3, dry DMF, 85% (for R = H) and 81% (for R = CH3); (c) TMSBr, CH2Cl2, rt, 72 h, 90% (for R = H) and 96% (for R = CH3). | ||
Similarly, thymine 6b was converted to its N3-protected derivative bromide their 7b. Finally, the successful N1-alkylation of 7a and 7b on 5 in the presence of cesium carbonate in DMF proceeded in good yields and excellent regioselectivities and afforded 8a and 8b, in 86% and 75% yields, respectively. Simultaneous deprotection of the N3-benzoyl group and phosphonic esters with TMSBr/CH2Cl2 afforded analogues 9a and 9b, in good yields, respectively. These coupling conditions were extended to other nucleic bases. Cytosine 10a and its fluorinated analogue 10b were converted to N4-bis(Boc)-cytosine derivatives 11a and 11b, respectively, in good yileds, through N1,N3,N4-tris-Boc forms followed by regioselective N1 deprotection with saturated solution of NaHCO3 in methanol.18 Alkylation at N1 position of 11a and 11b in presence of derivative 5, according to the same previous conditions using Cs2CO3, afford 12a and 12b, in 85% and 77% yield, respectively.19 Deprotection with TMSBr afforded the expected free phosphonates 13a and 13b, respectively, in quantitative yield (Scheme 4).
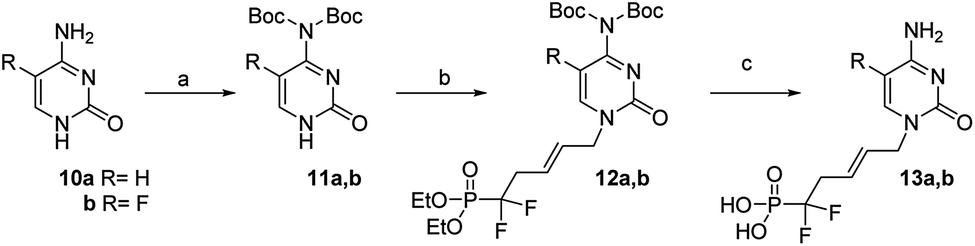 | ||
| Scheme 4 Reagents and conditions: (a) (i) Boc2O, DMAP, dry THF (ii) saturated NaHCO3, MeOH 50 °C, 62%; (b) (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5), Cs2CO3, dry DMF, 85% (for R = H) and 77% (for R = F); (c) TMSBr, CH2Cl2, rt, 72 h, quantitative for R = H and for R = F. | ||
Direct coupling of the purines 14a and 14b with difluorophosphonate derivative 5 in DMF with Cs2CO3 during 20 h at rt provided the desired N9-alkylated purine nucleotides 15a and 15b in 50 and 59% yields, respectively.20 Then, 6-chloropurine derivative 15a was converted to its hypoxanthine analogue 16a by treatment with TMSBr to release phosphonic acid followed by refluxing an aqueous solution of hydrochloric acid in 54% overall yield, (Scheme 5).21 Similarly, the 2-amino-6-chloropurine derivatives 15b gave the guanine 16b in 60% overall yields.
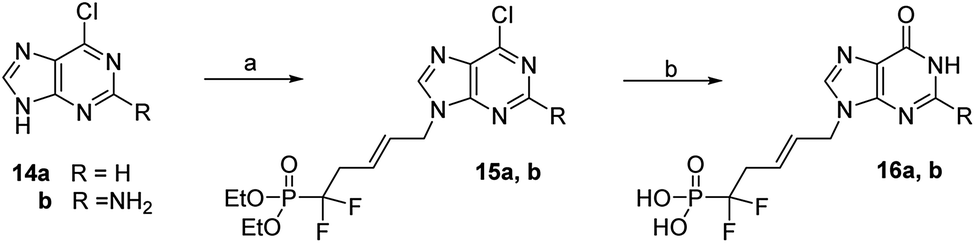 | ||
| Scheme 5 Reagents and conditions: (a) (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5), Cs2CO3, dry DMF, rt, 24 h, 50% (for R = H) and 59% (for R = NH2); (b) (i) TMSBr, CH2Cl2, rt then (ii) HCl (1 M), reflux, 54% (for R = H) and 60% (for R = NH2). | ||
Finally, with respect to the broad-spectrum antiviral drug ribavirin, which possess a 1,2,4-triazole-3-carboxamide nucleobase, its acyclic difluorinated phosphonate 19 was synthesized from 17, in a similar pathway, (Scheme 6).
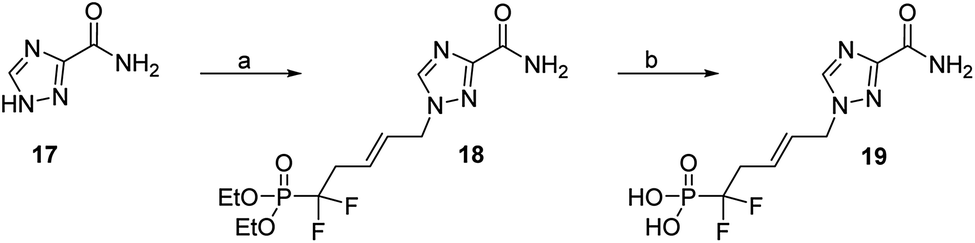 | ||
| Scheme 6 Reagents and conditions: (a) (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5), Cs2CO3, dry DMF, 42%; (b) TMSBr, CH2Cl2, rt, 72 h, 96%. | ||
All the synthesized 5,5-difluoro-5-phosphono-pent-2-en-1-yl nucleosides, 9a, b, 13a, b, 16a, b and 19, were evaluated against a wide variety of viruses, to determine their antiviral activity (EC50) in HEL, MDCK, Vero and HeLa cell lines, as the effective concentration required to reduce virus-induced cytopathicity or plaque formation by 50%. Compounds were evaluated against vaccinia virus (VV), herpes simplex virus 1 (HSV-1) (KOS strain), herpes simplex virus 2 (HSV-2) (G strain), thymidine kinase deficient (TK-) HSV-1, vesicular stomatis virus (VSV), varicella-zoster virus (VZV) (TK+ and TK− strains), human cytomegalovirus (HCMV) (AD-169 and Davis strains) in HEL, vesicular stomatitis virus (VSV), Coxsackie B4, respiratory syncytial virus in HeLa cell cultures, parainfluenza-3, reovirus-1, Sindbis virus and Coxsackie B4 in Vero cells and influenza virus in MDCK cells. All of the synthesized compounds did not exhibit promising antiviral activity.
Conclusions
In summary, a series of hitherto unknown acyclic 5,5-difluoro-5-phosphono-pent-2-en-1-yl-pyrimidines (9a, b, 13a, b), -purines (16a, b) and -(1,2,4)-triazolo-3-carboxamide (19) were successfully synthesized from (E)-1-bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5) in a convergent stereoselective manner. Surprisingly, it was discovered that cross-metathesis, in our hand, cannot afford the desired difluorinated phosphonate compounds. However, the final nucleosides were obtained, in good yields, by N-alkylation of various nucleobases with (E)-diethyl-5-bromo-1,1-difluoropent-3-enylphosphonate.However, none of the synthesized compounds showed significant antiviral activities. One plausible explanation could be due to a poor penetration to the cell and to the lack of next phosphorylation steps which could be dependent to the length of the acyclic chain.
Experimental section
General methods
Commercially available chemicals were of reagent grade and used as received. The reactions were monitored by thin layer chromatography (TLC) analysis using silica gel plates (Kieselgel 60F254, E. Merck). Column chromatography was performed on Silica Gel 60 M (0.040–0.063 mm, E. Merck). The 1H and 13C NMR spectra were recorded on a Varian InovaUnity 400 spectrometer (400 MHz) in (d4) methanol, CDCl3, shift values in parts per million relative to SiMe4 as internal reference. High resolution mass spectra were performed on a Bruker maXis mass spectrometer by the “Federation de Recherche” ICOA/CBM (FR2708) platform. The following products are known products or previously reported by our group: N3-benzoyluracil (7a), CAS registration 2775-87-3; N3-benzoylthymine (7b), CAS registration 4330-20-5; N4,N4-bis(Boc)-cytosine (11a) CAS registration 1108637-28-0; 5-fluoro-N4,N4-bis(Boc)-cytosine (11b) CAS registration: 1450880-36-0.(E)-1-Bromo-5-diethoxyphosphoryl-5,5-difluoro-pent-2-ene (5)
A suspension of zinc powder (441.3 mg, 6.75 mmol, 99.99% purity) in THF (0.4 mL) was added to a solution of 1,2-dibromoethane (0.96 mL, 1.13 mmol) in THF (5 mL) at room temperature, and was then warmed up to 65 °C. After 1 min, chlorotrimethylsilane (0.12 mL, 0.8 mmol) was added and the resulting mixture was stirred at 25 °C. After 15 min, the suspension was added dropwise to a solution of diethyl(bromodifluoro)methylphosphonate (0.8 mL, 4.5 mmol) in THF (2 mL), then the reaction mixture was stirred 12 h at 45 °C. CuBr (1.16 g, 8.1 mmol), activated LiCl (344.0 mg 8.1 mmol) and THF (4 mL) was added to the yellow solution at 0 °C under nitrogen, and then the resulting blue solution was stirred at 0 °C. After 10 min, trans-1,4-dibromo-2-butene (1.44 g, 6.75 mmol) was added and the reaction was stirred during 4 h at 0 °C. The mixture was filtered through celite and the filtrate was extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and concentrated under vacuum. The residue was purified by silica gel column chromatography with petroleum ether–EtOAc (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 2 (1.15 g, 80%) as a colourless oil. 1H NMR (400 MHz, CDCl3) δ 5.88–5.76 (m, 1H), 5.29–5.22 (m, 1H), 4.27 (q, J = 7.3 Hz, 2H), 4.25 (q, J = 7.3 Hz, 2H), 2.81 (m, 2H), 1.36 (t, J = 7.3 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 125.2, 127.1, 127.0, 126.9, 121.4, 64.6, 64.5, 39.1, 38.9, 38.7, 38.5, 16.5, 16.4; 19F NMR (376 MHz, CDCl3) δ −111.1, −111.4. 31P NMR (162 MHz, CDCl3) δ 6.9; HRMS (ESI): m/z [M + H]+ calcd for C9H17BrF2O3P: 321.006487, found: 321.006126.
:1) to give 2 (1.15 g, 80%) as a colourless oil. 1H NMR (400 MHz, CDCl3) δ 5.88–5.76 (m, 1H), 5.29–5.22 (m, 1H), 4.27 (q, J = 7.3 Hz, 2H), 4.25 (q, J = 7.3 Hz, 2H), 2.81 (m, 2H), 1.36 (t, J = 7.3 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 125.2, 127.1, 127.0, 126.9, 121.4, 64.6, 64.5, 39.1, 38.9, 38.7, 38.5, 16.5, 16.4; 19F NMR (376 MHz, CDCl3) δ −111.1, −111.4. 31P NMR (162 MHz, CDCl3) δ 6.9; HRMS (ESI): m/z [M + H]+ calcd for C9H17BrF2O3P: 321.006487, found: 321.006126.
General procedure A: alkylation with nucleobases
A solution of nucleobase (1.3 equiv.) in dry DMF (3 mL), Cs2CO3 (1.3 equiv.) and (E)-diethyl-(5-bromo-1,1-difluoro)pent-3-enylphosphonate (1 equiv.) was stirred at room temperature under argon for 16 h. After removal of DMF under vacuum, the residue was purified by silica gel column chromatography with CH2Cl2–MeOH (99:1 to 96:4) to the desired product.
General procedure B: deprotection of diethylphosphonate nucleosides
A septum-sealed microwave tube charged with diethyl phosphonate derivative and trimethylsilylbromide (10.0 equiv.) in CH3CN (0.1 M) was irradiated at 70 °C under microwave irradiation during 30 min. The progress of the reaction was monitored by TLC analysis. The reaction mixture was quenched with CH3OH and concentrated under vacuum, then deionized H2O (ELGA® water, 10 mL) was added and the aqueous layer was washed with CH2Cl2 (3 × 5 mL) and lyophilised to yield the expected phosphonic acid derivative.:2 to 1:2) to give product 12a (279 mg, 85%) as a colourless oil; 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 7.4 Hz, 1H), 6.99 (d, J = 7.4 Hz, 1H), 5.79 (m, 2H), 4.48 (d, J = 4.8 Hz, 2H), 4.27 (q, J = 7.3 Hz, 2H), 4.25 (q, J = 7.3 Hz, 2H), 2.83 (m, 2H), 1.37 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 162.3, 154.9, 149.6, 147.1, 130.2, 124.9, 96.5, 84.8, 64.5, 51.1, 37.4 (td, J = 21.5, 15.4 Hz), 27.7, 16.4; 19F NMR (376 MHz, CDCl3) δ −110.8 (t, J = 19.0 Hz), −111.1 (t, J = 19.0 Hz); 31P NMR (162 MHz, CDCl3) δ 7.09 (s), 6.44 (s), 5.78 (s); HRMS (ESI) m/z [M + H]+ calcd for C23H37F2N3O8P 552.2281, found 552.2284.:2 to 1:2) to give product 12a (149 mg, 77%) as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 4.5 Hz, 1H), 5.90–5.76 (m, 2H), 4.53 (d, J = 5.4 Hz, 2H), 4.27 (q, J = 7.1 Hz, 2H), 4.25 (q, J = 7.1 Hz, 2H), 2.87 (m, 2H), 1.47 (s, 18H), 1.37 (t, J = 7.1 Hz, 6H).;13C NMR (101 MHz, CDCl3) δ 155.95 (d, J = 14.3 Hz), 153.84, 149.02, 142.02, 139.56, 133.07 (d, J = 34.73 Hz), 129.21, 126.94 (m), 84.89, 64.80 (d, J = 8.5 Hz), 51.57, 37.60 (m), 29.82, 27.86, 16.53 (d, J = 5.5 Hz); 19F NMR (376 MHz, CDCl3) δ −110.8, −111.1, −156.23; 31P NMR (162 MHz, CDCl3) δ 6.23 (t, J = 105.8 Hz) HRMS (ESI) m/z [M + H]+ calcd for C23H35F3N3O8P 570.2189, found 570.2187.Antiviral activity assays
The antiviral assays were based on inhibition of virus-induced cytopathicity or plaque formation in HEL 299 (ATCC® CCL-137™) cell culture against herpes simplex virus 1 (HSV-1) (KOS), HSV-2 (G), vaccinia virus, vesicular stomatitis virus, cytomegalovirus (HCMV), and varicella-zoster virus (VZV). Moreover, the Vero (ATCC® CCL-81™) cell culture was utilized to test such compounds against parainfluenza-3, reovirus-1, Sindbis virus and Coxsackie B4. Furthermore, the novel compounds were evaluated in HeLa cell culture against vesicular stomatitis virus, Coxsackie virus B4, and respiratory syncytial virus or MDCK (ATCC® CCL-34™) [influenza A (H1N1; H3N2) and influenza B]. Confluent cell cultures (or nearly confluent for MDCK cells) in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or with 20 plaque forming units (PFU). After 1–2 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (200, 40, 8, 1.6, 0.32 μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or concentration required reducing virus-induced cytopathogenicity or viral plaque (VZV) plaque formation by 50%. The minimal cytotoxic concentration (MCC) of the compounds was defined as the compound concentration that caused a microscopically visible alteration of cell morphology. Alternatively, cytotoxicity of the test compounds was measured based on inhibition of cell growth. HEL cells were seeded at a rate of 5 × 103 cells per well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the cell number was determined with a Coulter counter.The cytostatic concentration was calculated as the CC50, or the compound concentration required reducing cell proliferation by 50% relative to the number of cells in the untreated controls.
Acknowledgements
This work was supported in part by the grant from LabEx SYNORG and FEDER (program COSMI). The authors's thanks are due to Mrs Leentje Persoons, Mrs Lies Van Den Heurck, Mrs Ellen De Waegenaere for the biological assays.References
- E. De Clercq, Biochem. Pharmacol., 2007, 73, 911–922 CrossRef CAS PubMed.
- E. De Clercq, A. Holý, I. Rosenberg, T. Sakuma, J. Balzarini and P. C. Maudgal, Nature, 1986, 323, 464–467 CrossRef CAS PubMed.
- S. Eriksson, B. Munch-Petersen, K. Johansson and H. Eklund, Cell. Mol. Life Sci., 2002, 59, 1327–1346 CrossRef CAS PubMed.
- F. Pertusati, M. Serpi and C. McGuigan, Antiviral Chem. Chemother., 2012, 22, 181–203 CrossRef CAS PubMed , review.
- O. Baszczynski and Z. Janeba, Med. Res. Rev., 2013, 33, 1304–1344 CAS , review.
- D. Topalis, U. Pradère, V. Roy, C. Caillat, A. Azzouzi, J. Broggi, R. Snoeck, G. Andrei, J. Lin, S. Eriksson, J. A. C. Alexandre, C. El Amri, D. Deville-Bonne, P. Meyer, J. Balzarini and L. A. Agrofoglio, J. Med. Chem., 2011, 54, 222–232 CrossRef CAS PubMed.
- V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868–3935 CrossRef CAS PubMed.
- S. Halazy, A. Ehrhard and C. Danzin, J. Am. Chem. Soc., 1991, 113, 315–317 CrossRef CAS.
- S. A. Diab, C. De Schutter, M. Muzard, R. Plantier-Royon, E. Pfund and T. Lequeux, J. Med. Chem., 2012, 55, 2758–2768 CrossRef CAS PubMed.
- H. Kumamoto, D. Topalis, J. Broggi, U. Pradère, V. Roy, S. Berteina-Raboin, S. P. Nolan, D. Deville-Bonne, R. Snoeck, D. Garin, J. M. Crance and L. A. Agrofoglio, Tetrahedron, 2008, 64, 3517–3526 CrossRef CAS.
- O. Sari, M. Hamada, V. Roy, S. P. Nolan and L. A. Agrofoglio, Org. Lett., 2013, 15, 4390–4393 CrossRef CAS PubMed.
- M. Bessières, O. Sari, V. Roy, D. Warszycki, A. J. Bojarski, S. P. Nolan, R. Snoeck, G. Andrei, R. F. Schinazi and L. A. Agrofoglio, ChemistrySelect, 2016, 1, 3108–3113 CrossRef.
- D. J. Burton and Z. Yang, Tetrahedron, 1992, 48, 189–275 CrossRef CAS.
- D. J. Burton and L. G. Sprague, J. Org. Chem., 1989, 54, 613–617 CrossRef CAS.
- J. Huang, E. D. St, S. P. Nola and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS.
- S. Fustero, A. Simon-Fuentes, P. Barrio and G. Haufe, Chem. Rev., 2015, 115, 871–930 CrossRef CAS PubMed , review.
- M. Bessières, V. Roy and L. A. Agrofoglio, RSC Adv., 2014, 4, 59747–59749 RSC.
- A. Porcheddu, G. Giacomelli, I. Piredda, M. Carta and G. Nieddu, Eur. J. Org. Chem., 2008, 5786–5797 CrossRef CAS.
- M. Hamada, V. Roy, T. R. Mcbrayer, T. Whitaker, C. Urbina-blanco, S. P. Nolan, J. Balzarini, R. Snoeck, G. Andrei, R. F. Schinazi and L. A. Agrofoglio, Eur. J. Med. Chem., 2013, 67, 398–402 CrossRef CAS PubMed.
- J. H. Hong, S.-Y. Kim, C.-H. Oh, K. H. Yoo and J.-H. Cho, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 341–350 CAS.
- S. Hikishima, M. Isobe, S. Koyanagi, S. Soeda, H. Shimeno, S. Shibuya and T. Yokomatsu, Bioorg. Med. Chem., 2006, 14, 1660–1670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 31P and 19F NMR data of selected compounds. See DOI: 10.1039/c7ra05153k |
| This journal is © The Royal Society of Chemistry 2017 |