
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnions involved in the initiation of the thermally induced SRN1 reaction for α-arylation of ketones†‡
Daniel A. Caminos *,
Marcelo Puiatti,
Javier Ivan Bardagí and
Alicia B. Peñéñory
*,
Marcelo Puiatti,
Javier Ivan Bardagí and
Alicia B. Peñéñory
INFIQC – CONICET, Instituto de Investigaciones en Fisicoquímica de Córdoba, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, X5000HUA Córdoba, Argentina. E-mail: dcaminos@fcq.unc.edu.ar; Web: www.fcq.unc.edu.ar/infiqc
First published on 19th June 2017
Abstract
The SRN1 reaction between acetophenone enolate and PhI is thermally induced and accelerated by microwave irradiation to give the corresponding substitution product 1,2-diphenylethanone in a 50% yield in DMSO at 70 °C. Regarding the mechanism of initiation, in this reaction, acetophenone enolate, tert-butoxyde anion and dimsyl anion (the ionic form of the solvent) could promote the initial electron transfer to start the radical reaction. Comparative studies on the PhI dehalogenation promoted by the different anions were conducted in DMSO under microwave irradiation and by quantum calculations. The dimsyl anion shows a higher iodide generation even at lower concentrations than acetophenone enolate and tBuO−. Likewise, DFT calculation by B3PW91, M062X and PBE0 shows the dymsyl anion to be the best electron donor. While the three anions can initiate the radical reaction, the reactivity order found locates the dimsyl anion in first place, followed by the enolate of acetophenone and then the alkoxide. The results reported herein allow a greater understanding of the initiation process with tert-butoxide solutions in DMSO.
Introduction
Microwave reactions have generated great interest in recent decades.1 While conventional cooking equipment was used in the first few years, at present there are specific microwave reactors for research, which allow adapting and broadening the detailed study of methods in organic synthesis. In the conventional heating through resistors and oil baths, the caloric energy is transferred by convection through vibrations and oscillations of matter from the heat source to reach the reagents through all the materials, containers and solvents, generating a temperature gradient from the outside to the core in the reaction.2 This means that there is a long time of gentle warming before achieving thermal equilibrium, making some reactions slow and allowing more side reactions, which reduce the reaction's yield. Furthermore, in microwave heating, the energy is transferred directly to the reagents and/or solvent through an electromagnetic pulse. Thus the heating of the reaction is intense and faster. In addition, a magnetic stirring system helps to maintain uniform heating, and with the use of sealed containers, the reactions could be performed at high temperatures and high pressures in a very short time. This microwave reactors in organic synthesis, makes it attractive to thermally induced reactions, usually slow, saving time and resources. Thus chemists have a tool to study and synthesize compounds fast, as quickly as heating up your own lunch.Radical reactions are among the wide spectrum of reactions to which microwave heating has been applied with excellent results.3 Radical reactions have been extensively developed in organic synthesis over the past twenty years since these reactions provide a powerful route for the formation of C–C bonds under mild conditions.4 Moreover, radicals are involved as intermediates in reactions such as coupling between aryl halides (ArX) and nucleophiles. These methodologies are one of the most important synthetic methods for the formation of C–N and C–S bonds and are used for the preparation of important products in the pharmaceutical and biological chemistry and materials science,5 in addition being an alternative to the Pd catalyzed reactions, which are high in cost. The reactions involving an electron-transfer step (ET) for generating radicals from radical anions, such as the unimolecular nucleophilic radical substitution or SRN1 reaction, has been successfully used to obtain new compounds.6
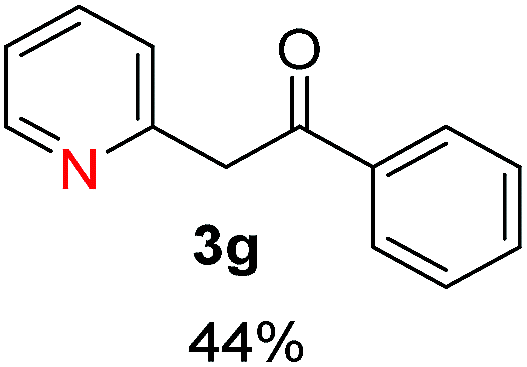
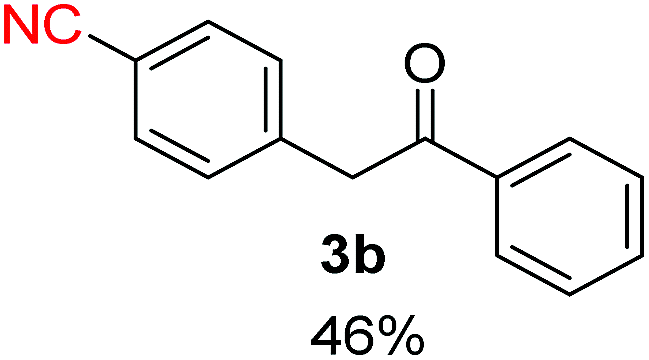
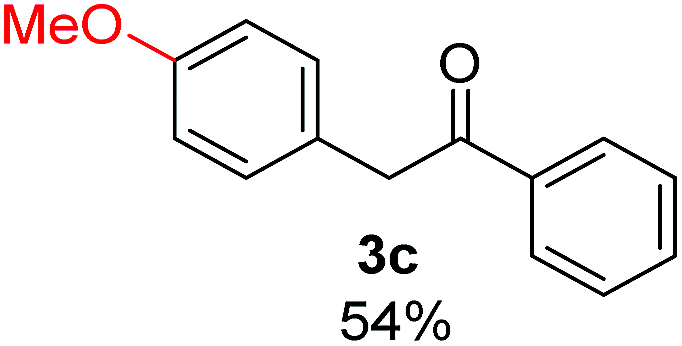
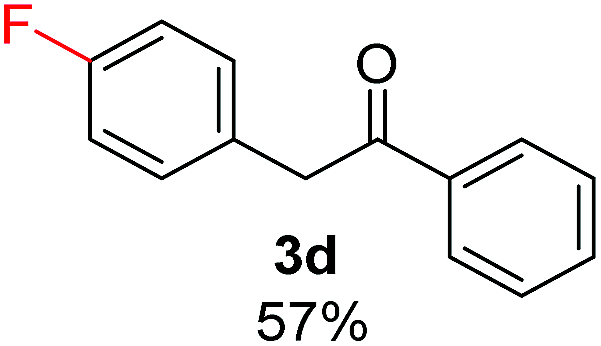
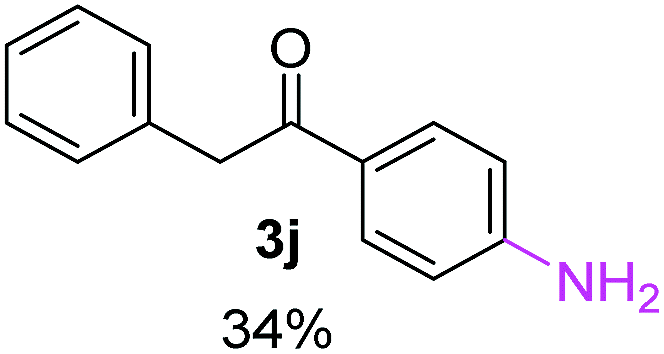
Radical reactions usually involve an initiation step to generate radicals which initiate the chain process.6 Different initiation methods include photochemical, electrochemical, thermal or chemical initiators.6 Recently we reported the α-arylation of aromatic ketones and acetamides at moderate temperatures applying microwave heating for the thermally induced SRN1 reaction, with yields ∼50% of the expected substitution product (Scheme 1A).7 On the other hand, the reaction induced by microwave offers advantages such as simplicity, shorter reaction times (1 min of microwave irradiation compared to 120 minutes under photoirradiation). It is also compatible with different substituent and shows a better performance for intramolecular cyclization reactions to get indole derivatives (Scheme 1B). In previous studies, different derivatives of the nucleophile acetophenone substituted at aryl moiety were also used (Table 1, R3).7
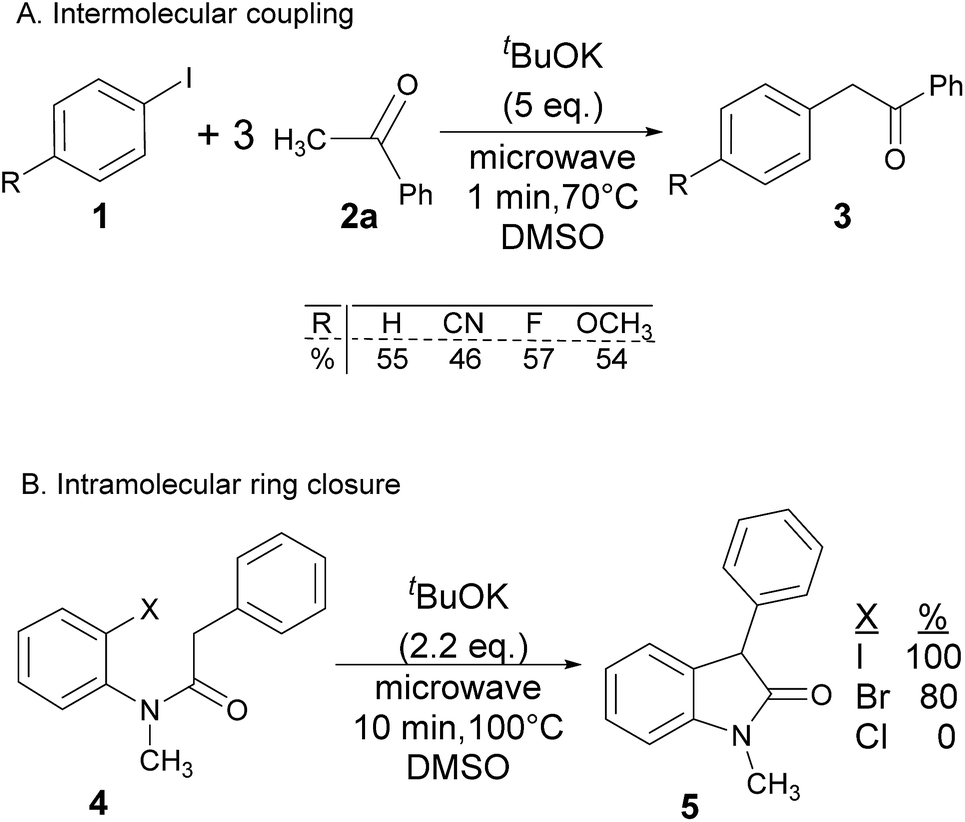 | ||
| Scheme 1 SNR1 reactions with tBuOK in DMSO under microwave irradiation. (A) intermolecular coupling of ArI (1) and acetophenone (2) to form 1,2-diphenylethanone derivatives (3), and (B) intramolecular ring closure of (2-halophenyl)phenylacetamides (4) to form indole heterocycle derivatives (5). | ||
|
||
|---|---|---|
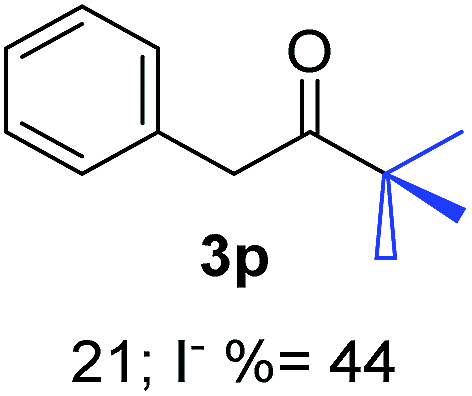
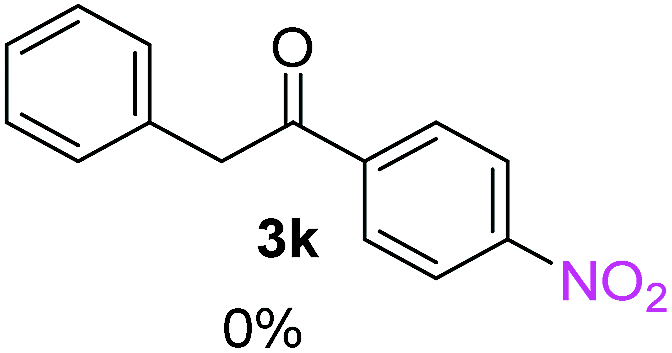
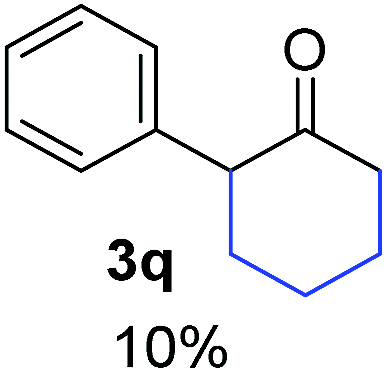
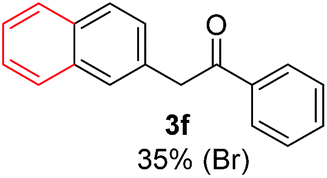
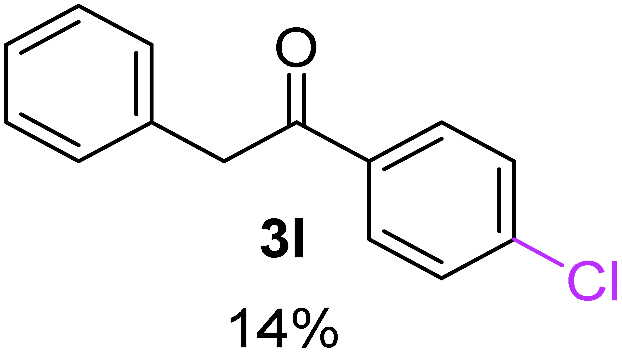
| a Coupling products between 4-substituted aryl acetophenones, and substituted haloarene, with I or Br as indicated, with tBuOK (base) in DMSO (solvent), heated by microwave. Compounds 3a–l and 3q synthesized with 3.1 eq. of tBuOK, 3 eq. of acetophenone and a reaction time of 10 min at 70 °C. The presence of electron-withdrawing groups such as Cl and NO2 in the enolate anion prevents or strongly inhibits ET pathway. A more thorough analysis can be found in from Soria-Castro et al. ref. 7. Compound 3n–p synthesized with 5 eq. of tBuOK, 3 eq. of acetophenone and a reaction time of 1 min, 100W–15s ∼70°C, compound 3p from Caminos et al., ref. 9. Product yields% quantified by 1H-NMR with internal standard. Iodide yield (% I−) determined potentiometrically using an Ag/Ag(I) electrode. | ||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
|
However, questions remain about the process of initiation of the chain reaction. There is a great interest in determining which of the species present in the reaction mixture are involved in the initiation step. In organic chemistry it is always important to determine all the steps and intermediaries involved in a reaction. This could help to a better understanding of the processes at the molecular scale, propose new experiments, allowing to improve reaction yields and extend reaction scopes.
In this paper, we extend the scope of the microwave induced α-arylation of ketones with tBuOK as base in DMSO to other α-substituted acetophenones and performed a complete mechanistic study. Particular attention is put in the initiation step performing a detailed analysis taking into account all the species present in the reaction medium that may participate in this step, with the inclusion of computational calculation in order to clarify the mechanism involved in the first step of the reaction.
Results and discussion
It is worth mention that, microwave provides a much more efficient method for heating and reducing reaction time from 1 h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 or more22 for a conventional bath to less than 1 min. In relation to the effect of the microwaves, our studies suggest that the increase in temperature, or specifically, the increase in the internal energy of an anion could provide the energy required to start the process of giving an electron that initiates the reaction.8,9 That is, the ET from the anion to ArX would be produced just by a thermal effect (either by microwave or by other heating method).
7 or more22 for a conventional bath to less than 1 min. In relation to the effect of the microwaves, our studies suggest that the increase in temperature, or specifically, the increase in the internal energy of an anion could provide the energy required to start the process of giving an electron that initiates the reaction.8,9 That is, the ET from the anion to ArX would be produced just by a thermal effect (either by microwave or by other heating method).
A comprehensive study was conducted to corroborate if other ionic species in the solution, which strongly absorb microwave radiation, help to accelerate the reaction but it was concluded they do not participate in the formation of the initial radical.9 Although the ionic or neutral species with high dipole moment in the reaction enable faster heating compared to pure DMSO solvent under microwave, none were involved in the catalysis of the reaction or acted as a radical generator to initiate the SRN1 mechanism.
The main anionic species in the reaction that could be involved in the initiation step of the radical process are the nucleophile and the tBuO− base. Furthermore, in the reaction medium another anion is present, the methyl sulfinyl carbanion or dimsyl anion formed by deprotonation reaction of DMSO with tBuO−. The dimsyl ion is an important organic agent in organic synthesis in reactions such alkylations, nucleophilic substitutions, Lewis base and condensations.10 In addition, the dimsyl anion acts as an environmentally friendly catalyzer in intermolecular cross-benzoin condensations of diaryl α-diketones (benzils) with aromatic and aliphatic aldehydes to give the corresponding aryl–aryl and aryl–alkyl benzoin.11 A recent study base-promoted arylation of 1-iodoadamantane with tBuOK in DMSO (a C–H substitution process) determine that it is the dimsyl anion responsible for initiating the reaction in a photoinduced ET process.12
Reactivity of nucleophile from ketones
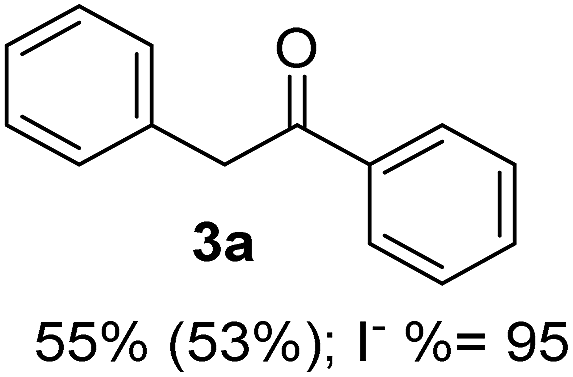
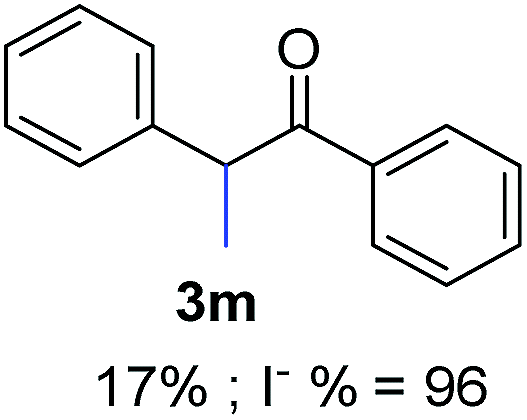
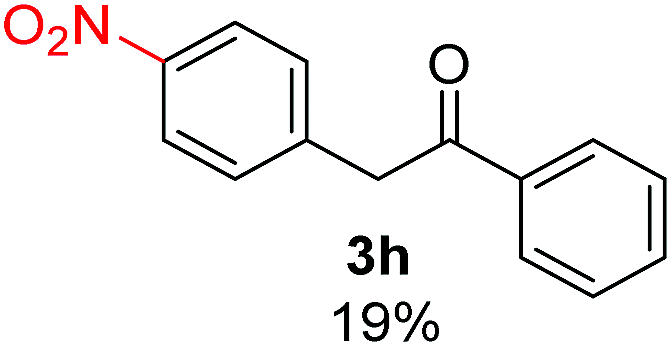
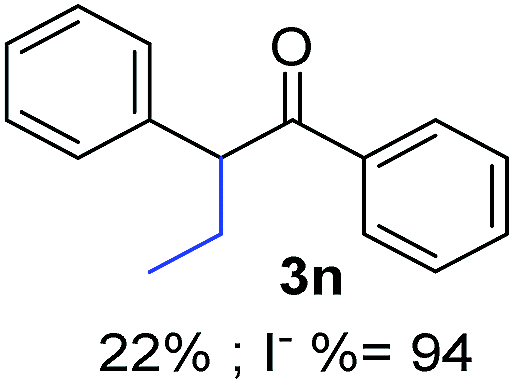
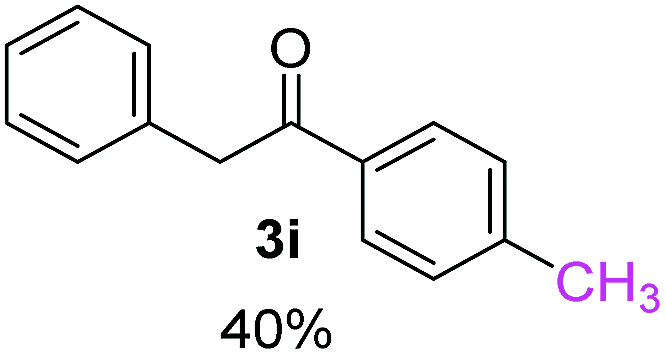
Recently, we reported reactions of PhI (or ArX) with acetophenone enolates, founding good yield of dehalogenation and moderate yields of the expected product (∼50% 3a–h)7 A minor substituent effects on the substrate was detected (Table 1, 3a vs. 3b–g), except when the p-nitro-iodobenzene was used, with 19% yield (Table 1, 3h). In addition, when any substituent was located in the aryl ring of the acetophenone, lower yields were found with a stronger effect for electro-withdrawing groups (Table 1, 3i–l).7In order to extend the scope of the reaction, we test different alkyl α-substituted acetophenones (Table 2). Here we used 5 equivalents of tBuOK, 3 equivalents of the corresponding ketone and a microwave irradiation of 100W–15s, with a temperature ranging from 70 °C to 100 °C. As we saw before, in these conditions we avoided self-condensation of the ketone and obtained full dehalogenation facilitating product purification; but did not improve product yield.9 When the acetophenone was substituted in its aliphatic extreme, yields decreased to a ∼20% (3m-17% and 3n-21%). In both cases dehalogenation was high and close to 95% (Table 2, entries 3 and 4). This indicates a lower coupling with these nucleophiles than acetophenone enolate, while the initiation step and the chain reaction continued to be favored.
| # | Nu | pKab (HA) | tBuOK eq.c | Base Nud | Prod. 3 | Yielde% | Id% |
|---|---|---|---|---|---|---|---|
| a Coupling reactions of 3 equivalents of nucleophiles and PhI with tBuOK and heated by 100 W for 15 seconds with microwave irradiation under N2 atmosphere.b From ref. 14.c Equivalents relatives to PhI, 0.5 mmol.d Relative amount of equivalents of tBuOK to nucleophile.e Quantified by NMR with internal standard. Quantified potentiometrically with Ag/Ag(I) electrode.f As evidenced by detection by GC-MS. | |||||||
| 1 | 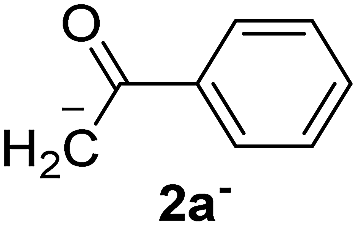 |
24.7 | 3.1 | 1.0 | 3a | 55 (ref. 7) | 78 |
| 2 | 5.0 | 1.7 | 52 (ref. 9) | 95 | |||
| 3 | 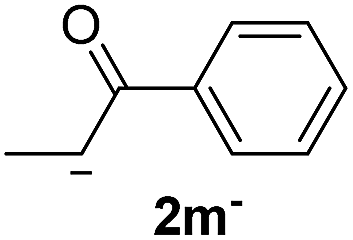 |
24.4 | 5 | 1.7 | 3m | 17 | 96 |
| 4 | 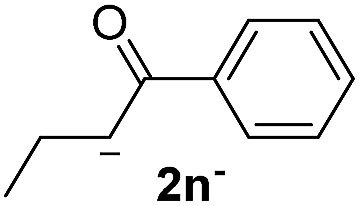 |
24 | 5 | 1.7 | 3n | 21 | 94 |
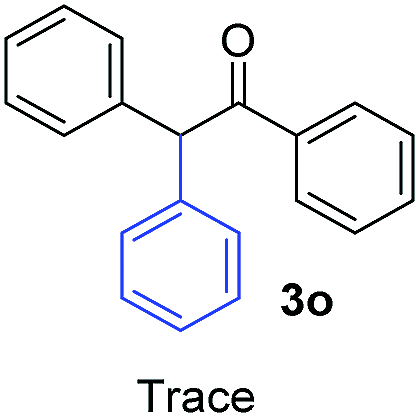
| 5 | 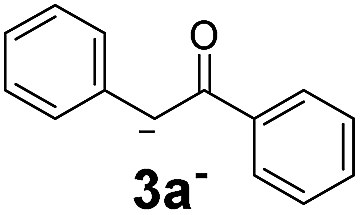 |
17.7 | 5 | 1.7 | 3o | Tracef | — |
| 6 | 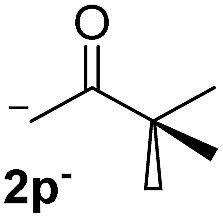 |
27.7 | 2 | 0.66 | 3p | 21 (ref. 9) | 44 |
| 7 | 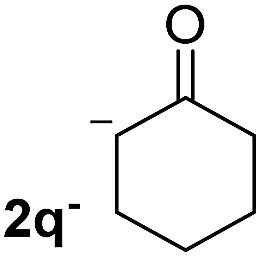 |
26.4 | 3 | 1.0 | 3q | 10 (ref. 7) | — |
The lower yields obtained for 3m–n could be explained by the presence of hydrogens in the β position (Hβ) at the carbonyl group in the nucleophile. It has been reported that Hβ abstraction by Ph˙ radical competes with the coupling reaction, decreasing the yield of the coupling product without visible effect on yield of dehalogenation13 (see ESI, Scheme S1 for details‡).
In the case of aliphatic ketones we found a low yield of arylation product. The enolate anion of 3,3-dimethylbutan-2-one (pinacolone), a good electron-donor usually used as an entrainment reagent,6a reacted poorly with PhI. That reaction achieves only 21% yield of 3p and 44% I−.9 As in the case of acetophenone, α substitution with alkyl groups produce an anion with Hβ, decreasing the yield approximately to half (Table 2, 3p vs. 3q).7
On the other hand, in the case of an aromatic substituent in the α position, only traces of product were detected (Table 2, 3o). Higher stability of nucleophile 2o− could be responsible for this behavior, which is traduced in a poor initiation since the coupling reaction of Ph radicals with nucleophiles has been reported to be not too sensitive to steric hindrance.6a
It should be noted that when we used 3.1 equiv. of tBuOK (instead of 5 equiv.) and 3 equiv. of acetophenone (in relation to PhI), the initiation was lower (compare entries 1 and 2, Table 2), a fact that indicate that the remaining tBuOK (2 equiv.) is playing a role in the initiation step. This is also supported by the reaction of PhI with 2 equiv. of tBuOK (without acetophenone) that gave benzene and I− in an 85% yield.9 At this point we found that with different conditions (with Nu or tBuOK or both present in the reaction media), the initiation step occurs when the mixture reaches 70 °C.
Mechanism reaction and radical initiation step in DMSO
In previous related works it has been determined that microwave-induced reaction of PhI and acetophenone enolate, follows an SRN1 chain mechanism. The SRN1 mechanism was well studied and involves an initiation step where radical species are produced; later it follows several propagation steps (Scheme 2). The reaction ends with termination events where radical species are consumed. In our case, in the initiation reaction, a dissociative ET occurred from some species in the reaction media to the PhI and generated Ph˙ radical and I− anion.6,15 Afterwards, the Ph˙ radical entered the propagation cycle of the reaction coupling with the enolate of acetophenone 2a. Thus a new C–C bond was formed, resulting in the radical anion of 2-phenyl-1-phenylethanone. This radical anion, by another ET to PhI, gave product 3a and regenerated radical Ph˙ which continued the chain reaction.6 Reduction of radical Ph˙ to give benzene competes with the coupling and constitute a termination step.7 Due to the low concentration of free radicals compare with the Nu and solvent concentration, the probability to find a radical–radical coupling product is really low.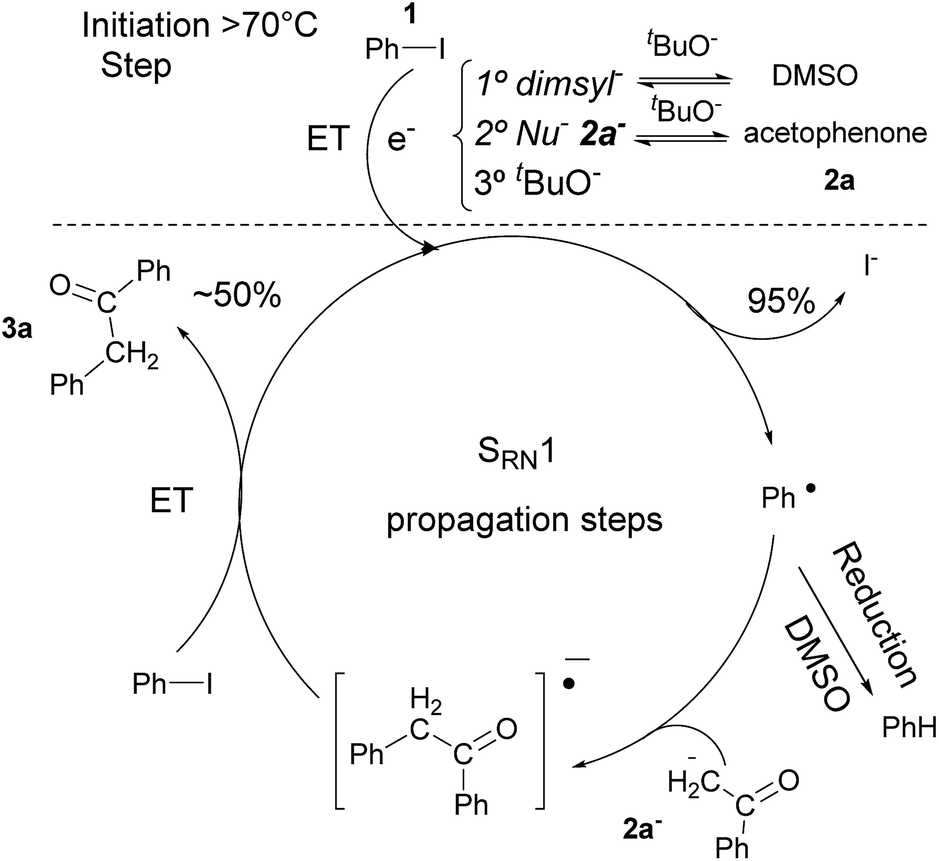 | ||
| Scheme 2 Microwave-induced coupling reaction of PhI and acetophenone with tBuOK in DMSO by SRN1 mechanism. | ||
Related to the initiation step, although it could be established that involve an ET step, a question remains about the specie/s responsible for the initiation.7,9
The initiation step in SRN1 can be photostimulated, electrochemically induced or generated by solvated electron and sodium amalgam in liquid ammonia, or inorganic salts (Fe2+; Sm2+).6a Savéants' research group has performed several studies about electrochemically induced reactions. In this case, the electron transferred to the ArX came from the electrode surface.6a Another way of producing the initiation step is “spontaneously” or thermally induced reactions. In these cases initiation depends on the relationship between the electron affinity of the substrate and the oxidation potential of the nucleophile.6a
Under microwave heating, ions (and dipolar species), are responsible for energy absorption and, therefore, heating. DMSO is a strong absorber of microwave radiation; yet, the presence of ionic and high dipole species promotes an intense and a faster heating. In Table 3 we included the different species and their concentration at the beginning of the reaction and in the equilibrium. In our previous work, we established that potassium cations, or other ions, are involved in the heating, but does not induce any kind of molecular hot spot promoting C–I bond breaking. Thus we discarded this pathway for the initiation step of the SRN1 process.9 In this highly accelerated process it is difficult to determine, with accuracy, the species that initiate the chain reaction.
| Entry# | Anion | Structure | Dipolar moment (μ) | [Ion] M |
|---|---|---|---|---|
| a The ionic and dipolar species are responsible for energy absorption and, therefore, heating under microwave irradiation. While DMSO solvent is a strong absorber of microwave radiation, the presence of this ionic species promotes an intense heating faster than the neutral counterparts. | ||||
| 1 | Dimsyl | 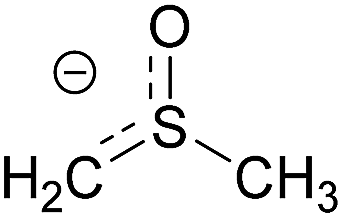 |
4.83 | 0.085 |
| 2 | tert-Butoxide | 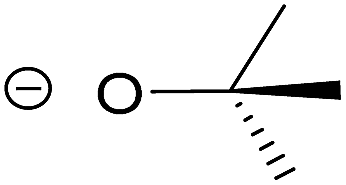 |
3.95 | 0.415 |
| 3 | Acetophenone enolate | 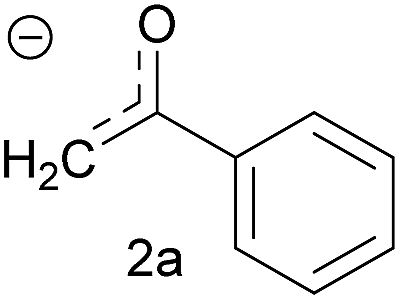 |
8.8 | 0.75 |
| 4 | Total anions | 1.25 | ||
| 5 | Cation | K+ | 1.25 | |
| 6 | Total ions | 2.5 | ||
Recently, Drapeau et al. reported results of a similar reaction in DMF with propiophenone (2m) as nucleophile obtaining a 99% yield of 3m when left for 13 h at 60 °C with conventional heating,16 in clear contrast to the reaction in DMSO as solvent at 60 °C, where yield was less than 1%. Dapreau et al., propose a radical mechanism with the initiation process depended on the exclusive participation of DMF as solvent in the catalytic cycle, supported only by density functional theory (DFT) calculations.
However, these results come into controversy with our previous results where the coupling reaction between PhI and acetophenone under microwave with DMF as solvent at 70 °C gave a 7% yield and a lower initiation 10%.7 Yet, Dapreau's work does not show optimization of time or temperature. Considering the similarity between both reactions, it would be elegant to consider that, since DMF and DMSO are polar aprotic solvents, the initiator species could be the same. However, the initiation process in their mechanism does not match with that in our proposal where initiation is attributed to the ET from an anion. In order to compare both methods and to study possible mechanisms, several reactions were performed. Nevertheless, all attempts to reproduce Drapeau's reaction yields have so far been unsuccessful (see ESI, Table S2, entries 2, 5, 10 and 11‡).17
Despite the previous inconsistencies, we assume that tBuO− could act alone as an electron donor to initiate the reaction or in complex with the solvent like that proposed by Drapeau16 and other authors.18 Recently Murphy et al. reported a study of tBuOK and its role in different reactions involving ET.18a It was proposed that ET came from “tBuO− alone or as part of a complex”. In some cases tBuOK forms a complex with an additive in the reaction media, which acts as an organic electron donor, much better than the alkoxide anion itself. However in our case, there are no additives present in the reaction medium. In this scenario, Murphy points to an optional mechanism via benzine, which acts as a diradical that initiates the process. In our previous report, we conducted a test for the benzine mechanism using p-iodotoluene, and only found 0.4% of the meta product. With this result we discarded the benzine mechanism as the main contributor to the generation of product 3a (see ESI Scheme S3‡).
The initiation step by anions
To determine the full initiation mechanism, it was necessary to determine if the anionic species: tBuO− anion, enolate of acetophenone, and dimsyl anion, or their combined effects, are responsible for the initial ET to PhI.With this in mind, we prepared solutions of each anion (acetophenone enolate, tBuO−, or dimsyl) of different concentration in DMSO and evaluated the iodide liberation from PhI, after microwave irradiation (100W–15s). Fig. 1 shows the results. In general, all reactions reached a temperature of 70 °C (see ESI Fig. S1 for details‡).19
 | ||
| Fig. 1 Reactions of PhI 0.25 M with different concentrations of dimsyl (black squares), tBuOK (red circles) and acetophenone enolate (blue triangles). Heated by microwave irradiation (100W–15s) under N2(g) atmosphere. The % I− was determined potentiometrically using an Ag/Ag(I) electrode. | ||
For the case of nucleophile 2a− (blue triangle), the enolate acetophenone was formed with tBuOK in default (0.9 equiv.).
The formation of acetophenone enolate by tBuOK is highly favored (Scheme 3, eqn (1)) and the formation of dimsyl by the presence of acetophenone enolate in DMSO is prevented since this equilibrium has an unfavorable Keq = 4 × 10−11. As a consequence, it could be consider that 2a− is the only anion present even by raising the temperature to >70 °C. It is important to note that the yield of iodide will be the sum of initiation and propagation since an SRN1 reaction will process, and the real initiation will be equal (in case of no chain) or lower than this value. At low concentrations of enolate anion (from 0.01 to 0.04 M), release of iodide was not detected. Only 1.3% I− was detected when we used a concentration of 0.085 M. At higher concentrations, the amount of released iodide increased describing a curve with an exponential grow, due to the chain reaction that is taking place.
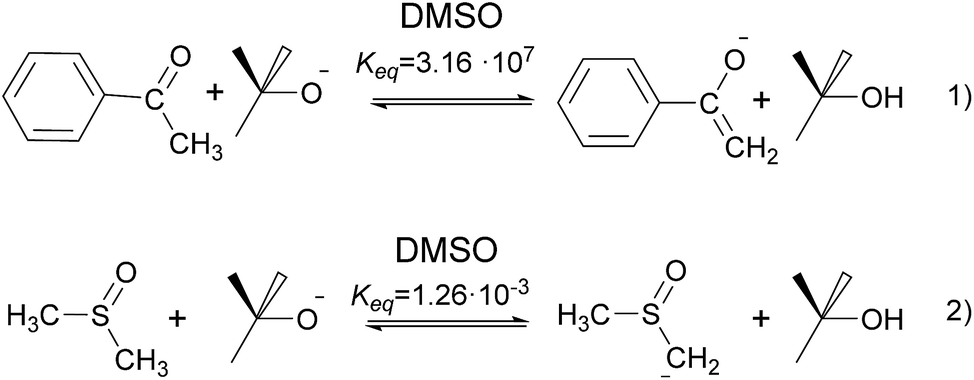 | ||
| Scheme 3 Anions formation by tBuOK. Due to the Keq constant of 3.16 × 107, we consider eqn (1) an irreversible reaction. While the value of 1.26 × 10−3 for the dimsyl formation, we assume eqn (2) as equilibrium. | ||
In the case of the dimsyl anion (black squares), substrate dehalogenation was detected, even with small dimsyl concentrations (0.01–0.085 M). These dimsyl solutions are difficult to work with, especially with high dimsyl anion concentrations (0.085 M), because the mixture is highly viscous, preventing a uniform heating and producing an average temperature in the reaction vessel of only 40 °C (the microwave vessel presents clearly DMSO around a black-brown core of superheated solution. See ESI, Fig. S5‡).20 In despite of this complication, it is clear that dimsyl anion is capable of a thermal ET to PhI.
When we used tBuOK in DMSO (Fig. 1, red circles), we observed that, at the same low concentration (0.01–0.085 M) conversion is in between the two previous cases; iodide anion reached 10% at 0.085 M tBuOK. At higher concentrations (0.5–1.25 M), the amount of iodine anion increased with an asymptotic growth until 80%. It is important to note that there is always dimsyl anion in equilibrium with tBuO−. For instance with an initial concentration of tBuOK of 0.5 M, and according to pKa values in DMSO,14 concentrations at the equilibrium would be [dimsyl]eq = ∼0.085 M and [tBuOK]eq = ∼0.415 M. However, we must consider that at higher temperatures (70–100 °C), the dimsyl–alkoxide balance is displaced forming a greater amount of dimsyl anion. Similar concentration of “pure” dimsyl anion and dimsyl in equilibrium with tBuOK gave higher dehalogenation yields, for example a solution of 0.012 M of dimsyl anion gave 17% I− and the same amount of iodide is obtained with a mixture of [dimsyl]eq = ∼0.050 M and [tBuO−]eq = ∼0.14 M (see ESI, Fig. S6‡). These results seem to indicate that tBuO− do not participate in the initiation but inhibit the action of dimsyl anion. Nevertheless due to the complexity of the experimental conditions its participation could not be totally discarded.
Another phenomenon that could be considered is the thermal decomposition of dimsyl anion. This phenomenon generates other new species in the solution. Previous reports on dimsyl solutions in DMSO indicate that the anion was stable at 25 °C,21 but at temperatures higher than 70 °C, sodium dimsyl solution was mainly decomposed in a mixture of methanesulfonate and sulfur (Scheme 4).21 These studies have been conducted by conventional heating; and under microwave irradiation dimsyl decomposition could be accelerated forming other species that help initiation. In some cases of overheating of the model reaction (Scheme 1), traces of Ph–S–Ph were detected by CG-MS.7 This leads us to think that these species of sulfur might be present, but studies carried out in parallel on a photochemical reaction allow discarding these species as those responsible for initiation.12
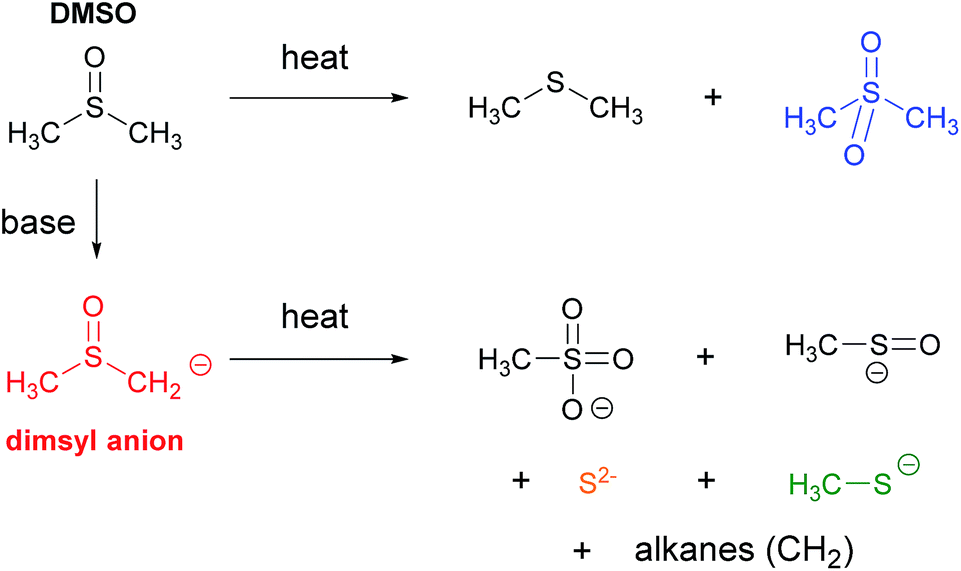 | ||
| Scheme 4 DMSO (up) and dimsyl (down) thermo decomposition at temperatures higher than 70 °C. DMSO produces dimethylsulfane and (methylsulfonyl)methane, while the dimsyl anion produce methanesulphonate, methylsulphanolate, sulfur, methanethiolate and a mixture of alkanes. | ||
Computational approach
Due to the complexity of experimental results, we were left with some unresolved questions about the initiating species. The rapid absorption of energy, the change in temperature and a possible change in the concentration of the species involved drive our attention to computational studies. In this case, our interest lies in determining, through this tool, which anionic species could participate as an electron donor in the process of radical initiation. Unlike experiments in solution, this methodology allows us to analyze each anion separately.In order to estimate the differences in the reductive power of the different electron donors (tBuOK, acetophenone and acetone enolates, and dimsyl anion, all in DMSO) DFT calculations were carried out.22 We computed different parameters such as difference in the oxidation potentials of the donors, ΔG of the ET reaction with PhI as an acceptor,23 and the activation energy for ET process ΔG‡ using Saveant's approximation for a dissociative ET (Scheme 5a).24,25 For this, we use the Tomasi's polarized continuum model (IEFPCM) and B3PW91, M062X and PBE0 DFT functionals. Results are presented in Table 4, and extra details are provided in Table S7 of the ESI.‡ The values obtained with the three evaluated DFT functionals follow a similar trend.
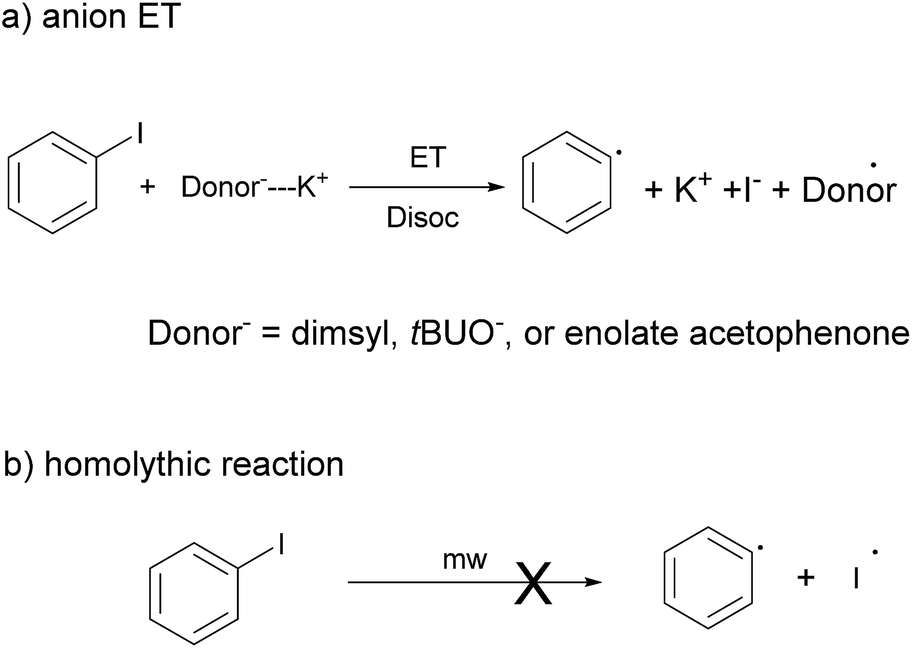 | ||
| Scheme 5 Processes considered for initiation of the radical reaction. (a) An ET from an anion acting as donor specie to PhI that generates the Ph˙, the iodide and the radical donor˙; and (b) a homolythic CAr–I bond breaking in PhI. | ||
| Anion/donor | B3PW91 | M06-2X | PBE | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔΔEred | ΔGET | EETact | ΔΔEred | ΔGET | EETact | ΔΔEred | ΔGET | EETact | |
| a Computed properties of the anions by DFT with DMSO as an implicit solvent using 6-311+G(d,p) as basis set. ΔΔEred difference in the oxidation potentials of the donors, ΔGET difference in Gibs free energy for the ET reaction with PhI as an acceptor and EETact the activation energy for ET process ΔG‡ using Saveant's approximation. | |||||||||
| tBuOK | 0 | 25.5 | 33.8 | 0 | 32.8 | 40 | 0 | 26.8 | 35.1 |
| Potassium acetophenone enolate (2a) | 0.35 | 18.9 | 29.4 | 0.31 | 27 | 35.8 | 0.34 | 20.4 | 30.5 |
| Potassium acetone enolate | 0.50 | 17.9 | 28.9 | 0.4 | 25.5 | 34.9 | 0.49 | 19.4 | 30.2 |
| Potassium dimsylate | 0.76 | 11.3 | 25.5 | 0.65 | 20.7 | 32.4 | 0.76 | 13.5 | 27.2 |
| RA intermediate of the reaction (3a−) | 2.18 | −22.2 | 8.85 | 2.36 | −18.1 | 11.6 | 2.15 | −20.5 | 9.9 |
According to the relative values of the oxidation potentials in the compounds analyzed, computed following a method reported lately,23 dimsyl anion should be the best electron donor in DMSO, followed by enolates, and then tert-butoxide anion. This is agreement with the experimental results depicted in Fig. 1, for the dimsyl and enolate anion. Computations of the radical anion of the product support a chain mechanism being the ET to the substrate from this intermediate faster than any initiation ET event. The presence of a dimmer coming from the radicals formed after ET would be a proof that these processes are occurring. Unfortunately none of these compounds could be detected in the experiments under microwave irradiation (see ESI pag. 26‡).
In our previous work we discarded the homolytic bond rupture between CAr–I in PhI by effect of microwave heating as a radical source for initial step (Scheme 5b).9 Here we also found that energy barrier is ∼60 kcal mol−1,26 too high in comparison to that of an ET from an anion present in solution.
According to our analysis, the dimsyl anion is the most effective species for initiation, followed the enolate nucleophile. In our model reaction conditions, tBuOK excess (1 mmol remain, 0.5 M) reacts with the solvent to form the dimsyl anion (Scheme 3, eqn (2)). In the equilibrium at 25 °C, the dimsyl concentration will be ∼0.085 M and tBuO− ∼ 0.415 M (Table 3). Although we have found that the order of reactivity of the anions that act as donors in the ET resulting in the initiation stage is: dimsyl > acetophenone enolate > tBuO−; it should also be considered that not all the anionic species have the same concentration; thus a shared contribution from the three anions should be considered.
Experimental
Chemicals and general methods
Potassium tert-butoxide (tBuOK), iodobenzene, acetophenone, nBuLi (in hexane), 4-iodo-toluene, bromobenzyl, potassium chloride were all high-purity commercial samples used without further purification. DMSO absolute grade was used without further purification and stored over molecular sieves (4 Å).The 1H and 13C NMR spectra were recorded at 400.16 and 100.62 MHz, respectively, on a Bruker 400 spectrometer, and all spectra were reported in δ (ppm) relative to Me4Si, with CDCl3 as a solvent. Gas chromatographic analyses were performed with a flame-ionization detector, on 30 m capillary column of a 0.32 mm × 0.25 μm film thickness, with a 5% phenylpolysiloxane phase. GC-MS analyses were performed employing a 25 m × 0.2 mm × 0.33 μm with a 5% phenylpolysiloxane phase column.
Representative experimental procedure for PhI and acetophenone enolate coupling
The reactions were carried out in a 10 mL CEM Discover microwave glass vessel, filled with nitrogen and a magnetic stirrer and 2.5 mmol of tBuOK. The tube was dried under vacuum, filled with nitrogen, and then charged with dried DMSO (2 mL) and degassed. Then 1.5 mmol of the α-acetophenone (2a–m, n and 3a) and iodobenzene 1 (0.5 mmol) were added to the degassed solvent under nitrogen atmosphere. Microwave-induced reactions were performed in a single-mode instrument equipped with a noncontact infrared temperature sensor, direct pressure control system for measuring the pressure of the reaction vessel contents and a cooling system by compressed air. The sample vessels were irradiated by microwave at 100 W for 15 seconds. Temperature was recorded by the internal IR sensor in the bottom of the reactor chamber. After irradiation, the device cooled the tube to 50 °C with compressed air above 1 min (−0.5 °C s−1). The average pressure was 1.7 atm in the vessel during the reaction time. After completion of the reaction, the vessel was removed from the microwave cavity and opened to the atmosphere. The reaction was subsequently quenched by addition of water (10 mL) and NH4NO3 in excess, and the mixture was extracted with 10 mL ethyl acetate and water (2 × 10 mL). The combined organic extract was dried over anhydrous CaCl2, and completed with ethyl acetate to get 10 mL; the water extract was filled to 100 mL for further quantification. The products were quantified by GC or NMR by the internal standard method or isolated by silica gel chromatography from the crude product reaction mixture. Water layer was recovered to quantify halide ions by potentiometric titration with an AgNO3 standard solution (0.01 M).Dimsyl solutions in DMSO
The dimsyl anion was generated by the reaction of DMSO with butyl lithium (nBuLi), (Scheme S2, ESI‡).10 The solutions were prepared in a 25 mL Schlenk, filled with nitrogen and a magnetic stirrer. The Schlenk was dried under vacuum, filled with nitrogen, and then charged with dried DMSO (10 mL) and degassed. Then, nBuLi solution in hexane was added slowly in portions (<0.5 mL each) without stirring to avoid mixing with DMSO. After addition of each portion, hexane was evaporated with vacuum (without stirring) until the upper phase disappear and then the mixture was stirred by 1 min. An aliquot was taken with a syringe and carried to titration vessel or to a 10 mL CEM Discover microwave glass vessel. The concentration of dimsyl solutions was determined by titration with carbazole [0.4008 M] (pKa = 19.9), using Ph3CH (pKa = 30). In this compound, the proton loss, form the intense red anion Ph3C−. The dimsyl solutions obtained were 30 and 85 mM.Initiation experiments
The reactions were carried out in a microwave reactor as described in the previous section. Likewise, after completion of the reaction, the vessel was removed from the microwave cavity and opened to the atmosphere, quenched by addition of water (10 mL) and NH4NO3 in excess, and the mixture was extracted with 10 mL ethyl acetate and water (2 × 10 mL). The combined organic extract was dried over anhydrous CaCl2, and completed with ethyl acetate to get 10 mL; the water extract was filled to 100 mL for further quantification. The products were quantified by GC or NMR by the internal standard method. Water layer was recovered to quantify halide ions by potentiometric titration with an AgNO3 standard solution. Fig. 1 shows the results of % I− vs. anion concentration.Products identification
All the products in Table 2 were obtained following the general procedure, quantified by NMR or GC. Products 3a, m–o, are known compounds and present spectral data as shown in the literature, in agreement with the structures proposed.Computational calculations
Computational studies were carried out using Gaussian 09 package.29 Calculations were performed with full geometry optimization including in all cases the effect of the solvent (DMSO as polar solvent) through the Tomasi's polarized continuum model (IEFPCM),30 B3PW9131 M062X32 and PBE033 DFT functionals and 6-311+G(d,p) as basis set.34 We checked that the conformations obtained were minima by running frequency calculations. No imaginary vibrational frequencies were found. All energy values include zero point correction.
Conclusions
In this work, we determinate that dimsyl, tBuO− and enolate anion presents in the mixture are able to start the reaction. Experimental results and computational calculations indicate that priority to ET comprises: 1° dimsyl anion, 2° enolate ketone anion and then tBuO−. We might consider an order of reactivity, but cannot refer to an exclusive role of one anion in the initiation step. While in photochemical processes, we could attribute the role of electron donor to a particular species, by a direct excitation of the same; in these thermal reactions under microwave irradiation, a multifactorial initiation process should be considered.Thus, tBuO− solutions in DMSO are a powerful reactive,35 it is highly basic and for radical reactions it is capable to initiate an ET at 70 °C mainly by the generation of the dimsyl anion in situ, which is a more powerful electron donor.
Finally, the methodology of microwave-induced ET may be applicable to other examples of SRN1, mainly in the intramolecular ring closure and other reactions involving molecular rearrangements and addition of radicals to neutral molecules and homolytic aromatic substitution reactions (HAS).36 It is highly probable that in intramolecular ring closures, the use of two equivalents of tBuOK leads to the formation of the anion and the remaining excess reacts with the solvent and allows the formation of dimsyl which acts as a radical initiator. A comparative study between photo- and thermal-induced radical cyclizations promoted with tBuOK taking parameters like yield, energy consumption, atom economy, costs and waste generation is being undertaken in our lab.
Acknowledgements
This work was supported by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Agencia Nacional de Promoción Científica y Técnica (ANPCyT), Ministerio de Ciencia y Tecnología de la Provincia de Córdoba, Argentina and SeCyT-UNC. All authors are researchers from CONICET. Authors thank PhD Maria Eugenia Budén for assistance and discussion.Notes and references
- (a) C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed; (b) C. R. Strauss and R. S. Varma, Top. Curr. Chem., 2006, 266, 199–231 CrossRef CAS; (c) C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
- (a) C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127–1139 RSC; (b) B. L. Hayes, Microwave synthesis, chemistry at speed of light, CEM Publishing, USA, 2002 Search PubMed.
- (a) K. Olofsson, S.-Y. Kim, M. Larhed, D. P. Curran and A. Halberg, J. Org. Chem., 1999, 64, 4539 CrossRef CAS; (b) H. Akamatsu, K. Fukase and S. Kusumuto, Synlett, 2004, 1049–1053 CAS.
- Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- (a) A. B. Peñeñory and J. E. Argüello, Aromatic and Heteroaromatic Substitution by SRN1 and SN1 Reactions, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2010, ch. 10, pp. 319–346 Search PubMed; (b) R. A. Rossi and A. B. Peñeñory, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, CRC Press Inc., Boca Raton, 2nd edn, 2003, ch. 47, p. 47 Search PubMed; (c) R. A. Rossi, A. B. Pierini and A. N. Santiago, in Organic Reactions, ed. L. A. Paquette and R. Bittman, Wiley & Sons, 1999, pp. 1–271 Search PubMed.
- (a) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71–167 CrossRef CAS PubMed; (b) R. A. Rossi and A. B. Peñéñory, Curr. Org. Synth., 2006, 3, 347–351 Search PubMed.
- S. M. Soria-Castro, D. A. Caminos and A. B. Peñéñory, RSC Adv., 2014, 4, 17490–17497 RSC.
- L. C. Schmidt, J. E. Argüello and A. B. Peñéñory, J. Org. Chem., 2007, 72, 2936–2944 CrossRef CAS PubMed.
- D. A. Caminos, A. D. Garro, S. M. Soria-Castro and A. B. Peñéñory, RSC Adv., 2015, 5, 20058 RSC.
- (a) C. Corey, J. Am. Chem. Soc., 1965, 87, 6 Search PubMed; (b) D. Ragno, O. Bortolini, G. Fantin, M. Fogagnolo, P. P. O. Giovannini and A. Massi, J. Org. Chem., 2015, 80, 1937–1945 CrossRef CAS PubMed.
- (a) O. Bortolini, G. Fantin, V. Ferretti, M. Fogagnolo, P. P. Giovannini, A. Massi, S. Pacifico and D. Ragno, Adv. Synth. Catal., 2013, 355, 3244–3252 CrossRef CAS; (b) D. Ragno, O. Bortolini, P. P. Giovannini, A. Massi, S. Pacifico and A. Zaghi, Org. Biomol. Chem., 2014, 12, 5733 RSC.
- M. E. Budén, J. I. Bardagí, M. Puiati and R. A. Rossi, J. Org. Chem., 2017 DOI:10.1021/acs.joc.7b00822.
- (a) M. F. Semmelhack and T. M. Bargar, J. Org. Chem., 1977, 42,(8), 1481–1482 CrossRef; (b) A. E. Lukach, A. N. Santiago and R. A. Rossi, J. Org. Chem., 1997, 62, 4260–4265 CrossRef CAS PubMed; (c) C. Dell'Erba, M. Novi, G. Petrillo and C. Tavani, Tetrahedron, 1993, 49(1), 235–242 CrossRef.
- All pKa values in DMSO from Bordwell pKa table (acidity in DMSO) by Hans J. Reich and references therein, http://www.chem.wisc.edu/areas/reich/pkatable, accessed 04-2017.
- In PhI it has been proposed that the ET is concerted with the fragmentation of the C-I generating the radical Ph and iodide anions. See ref. 6 and A. B. Pierini and D. M. A. Vera, J. Org. Chem., 2003, 68(24), 9191–9199, DOI:10.1021/jo035087w.
- M. P. Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Angew. Chem., Int. Ed., 2015, 54, 10587–10591 CrossRef PubMed.
- All reagents and solvents were analyzed by 1H and 13C-NMR, and contaminants were not detected. The reagents were newly opened and further analyzed by GC-FID and GC-MS, obtaining similar information. Also, this reagents were successfully employed for reproduce reactions with both, microwave irradiation and conventional oil bath heating, Table 1, 3a, 3m and 3n.
- (a) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410, DOI:10.1021/jacs.6b03282; (b) D. E. Stephens, J. Lakey-Beitia, J. E. Burch, H. D. Arman and O. V. Larionov, Chem. Commun., 2016, 52, 9945–9948, 10.1039/c6cc04816a; (c) L. Zhang, H. Yang and L. Jiao, J. Am. Chem. Soc., 2016, 138, 7151–7160, DOI:10.1021/jacs.6b03442.
- Alternatively, we developed a chromatographic method for the quantification of PhI using PhBr and I-toluene. Although very reproducible calibration lines were obtained, the quantification of PhI was deficient since during the work-up, this volatile species was lost, despite the fact that all possible precautions were taken. See ESI‡ for more details.
- It is possible that this effect is attributed to lithium cation. Taking this with some concern, we performed tests. We believe that a close ion pair Li+–tBuO–(DMSO) was formed in this condition precluding the formation of dimsyl anion, see ESI,‡ Table S4.
- K. Sjoberg, Tetrahedron Lett., 1966, 51, 6383–6384 CrossRef CAS; G. G. Price and M. C. Whiting, Chem. Ind., 1963, 775–776 Search PubMed.
- In addition we calculated the properties of the electron donors proposed for ET reaction in DMF, namely, tBuOK anion, the anion of DMF, the complex DMF–tBuOK recently proposed in ref. 16 and the super-electron-donors (SED) proposed in ref. 18a, see ESI Table S8‡.
- J. L. Borioni, M. Puiatti, D. M. A. Vera and A.-B. Pierini, Phys. Chem. Chem. Phys., 2017, 19, 9189–9198, 10.1039/c6cp06163j.
- J.-M. Savéant, in Advances in Physical Organic Chemistry, ed. T. T. Tidwell, Academic Press, New York, 2000, p. 35 Search PubMed.
- For the complexes DMF–tBuOK and for the SEDs the calculations used the published structures in ref. 16 and 18a as starting points.
- This value considers the formation of radicals in addition to their solvation energy and this value is according to the 67 kcal per mole for the Ph˙ radical in the gas phase, found in S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36(4), 255–263, DOI:10.1021/ar020230.
- J.-C. Hsieh, Y.-C. Chen, A.-Y. Cheng and H.-C. Tseng, Org. Lett., 2012, 14, 1282–1285 CrossRef CAS PubMed.
- DBS-mass, MS-IW-5435 SDBS no. 32458, 2,2-diphenylacetophenone, National Metrology Institute of Japan (NMIJ); © National Institute of Advanced Industrial Science and Technology (AIST) 04-2016 last accessed URL: http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_disp.cgi?sdbsno=32458.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105(8), 2999–3093 CrossRef CAS PubMed; (b) G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132(11), 114110 CrossRef PubMed.
- (a) J. P. Perdew, in Electronic Structure of Solids '91, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991 Search PubMed; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef; (c) J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4978 CrossRef CAS; (d) J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS; (e) K. Burke, J. P. Perdew and Y. Wang, in Electronic Density Functional Theory: Recent Progress and New Directions, ed. J. F. Dobson, G. Vignale and M. P. Das, Plenum, 1998 Search PubMed; (f) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, J. Phys. Chem., 2006, 110, 5121–5129 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef CAS PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS; (c) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (b) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- D. Caine, Potassium tert-Butoxide, e-EROS Encycl. Reagents Org. Synth., 2006 DOI:10.1002/047084289X.rp198.pub2.
- (a) M. E. Budén, V. A. Vaillard, S. E. Martin and R. A. Rossi, J. Org. Chem., 2009, 74, 4490–4498 CrossRef PubMed; (b) M. E. Budén, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206–2218 CrossRef PubMed; (c) J. I. Bardagí, S. E. Vaillard and R. Rossi, Tetrahedron Lett., 2006, 47, 3149–3152 CrossRef; (d) Z.-L. Xue, Y. Y. Qian and K. S. Chan, Tetrahedron Lett., 2014, 55, 6180–6183 CrossRef CAS.
Footnotes |
| † This paper is dedicated to the memory of Adriana Beatriz Pierini 1953–2016, who passed away during the preparation of the manuscript. Those who have worked with her and shared her wisdom will remember her forever. |
| ‡ Electronic supplementary information (ESI) available: Experimental details including microwave reaction profiles, iodine determination, NMR spectra, computational calculations and tables, figures and graphics with additional information are available. See DOI: 10.1039/c7ra05156e |
| This journal is © The Royal Society of Chemistry 2017 |