
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of carbonization temperature and press processing on the electrochemical characteristics of self-standing iron oxide/carbon composite electrospun nanofibers†
J. Abe,
K. Kawase ,
N. Tachikawa,
Y. Katayama and
S. Shiratori*
,
N. Tachikawa,
Y. Katayama and
S. Shiratori*
Department of Integrated Design Engineering, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, Kanagawa 223-8522, Japan. E-mail: shiratori@appi.keio.ac.jp
First published on 27th June 2017
Abstract
Lithium ion batteries (LIBs) are popular energy storage devices used in various fields such as electronics, mobilities, and power devices. In recent years, LIBs have been used in applications that require high energy densities to improve device performance metrics such as weight saving and miniaturization, as well as for reducing the cost. In this study, we propose two solutions for meeting the high energy density demands of these applications, namely (i) the use of active materials with high energy density and (ii) reduction of non-active materials such as the conductive agent, binder, separator, and electrolyte in the battery systems. In this study, we investigate the role of carbonization temperature and press processing of iron oxide/carbon composite nanofibers in improving the electrochemical characteristics of these electrode materials. The results of the study indicate that increasing the carbonization temperature improves the energy density per unit weight and unit volume as well as the rate capabilities, whereas press processing improves the energy density per unit volume, but reduces the rate capabilities. The investigation is useful for improving the performance of iron oxide/carbon composite nanofibers as the anode for LIBs.
Introduction
Development of energy storage technologies is one of the major topics of interest in the 21st century. Among the various battery systems developed so far, lithium ion batteries (LIBs) exhibit superior performance compared to other rechargeable battery systems such as nickel metal hydride (NiMH), nickel–cadmium (NiCd), and lead acid batteries from the points of view of voltage, energy density, and cycle capability.1,2 Therefore, LIBs have been very popular in various fields such as electronics, mobilities, and power devices for several decades. In recent years, LIB use has expanded to the areas of drones, wearable devices, electrical vehicles, and so on. In these applications, high energy densities are required for achieving improved performance metrics such as weight saving, miniaturization, and low cost. In this study, we propose two battery design solutions for meeting these demands.The first solution involves the use of active materials with high energy density. Currently, lithium metal oxide and carbon are used as the typical cathode and anode materials, respectively, in LIBs. This combination has remained unchanged from that used in the first commercial LIB manufactured by Sony Corporation in the early 1990s,3 owing to problems such as cycle capability and cost when used with other material combinations. If conventional active materials are replaced with new materials with higher energy densities, it is possible to significantly improve the energy density of the LIBs. Therefore, in this study, we have examined the use of iron oxide as a high-energy density anode material. While iron oxides are abundant, inexpensive, and environmentally friendly, they are fraught with several problems, the most important of which is cycle deterioration. Cycle deterioration is caused by several factors such as the decomposition of the electrolyte solution and loss of the conductive path owing to electrode collapse.4 In particular, large volume change of the anode material during cycling causes the destruction of the solid electrolyte interface (SEI)5 and loss of the conductive path.6 Reducing the extent of volume change would suppress these problems; nano-scale processing of active material particles has been studied for this purpose.7–9 Although the volume change ratio depends on material-specific quantities, it is possible to control the extent of volume change by controlling the particle size of the active material. Further, nano-scale processing decreases the chemical reaction resistance owing to the short ion diffusion distance. However, there are various other problems with using nano-sized active material particles such as poor dispersibility owing to an increase in the van der Waals force, low initial coulombic efficiency owing to high specific surface area, and low safety owing to high chemical activity. In order to solve these problems, electrodes prepared using composites of nano-scale active materials and carbon have been proposed in a previous study,9 since the interfacial area between the electrolyte and active material is reduced by the composite effect. This suppresses the SEI growth, which improves the coulombic efficiency.
The second solution for achieving superior performance metrics in batteries is reduction in the amount of non-active materials such as conductive agents, binders, separators, and electrolyte. In general, reducing the amounts of these materials causes some performance degradation owing to poor strength and poor adhesion to the collector. Hence, non-woven carbon electrodes have been proposed as self-standing electrodes in previous studies.10,11 These electrodes do not need a current collector and binder because they are free-standing and exhibit good conductivity. Non-woven carbon anodes also act as active materials.
Electrospinning has been proposed as a method for fabricating non-woven carbon electrodes with high specific surface area. It is a facile method for fabricating nanofibrous non-woven polymer membranes, which are used in various applications such as anti-fouling12,13 as well as in the electronics,14,15 tissue engineering,16,17 and filtration field.18,19 The electrospun nanofiber membranes possess high specific surface area, high porosity, and low weight, and are self-standing and are prepared by the carbonization of electrospun PAN nanofibers. They exhibit high specific surface area and high conductivity. Self-standing metal oxide/carbon composite nanofibers prepared by electrospinning using a polymer containing a metal precursor followed by carbonization have also been proposed,20–22 as shown in Fig. 1.
 | ||
| Fig. 1 Schematic illustration of self-standing active material composite carbon nanofibrous electrodes for lithium ion battery applications. | ||
In this study, we investigate the influence of carbonization temperature and press processing on the electrical characteristics of iron oxide/carbon composite nanofibers.
Experimental
Materials
Polyacrylonitrile (PAN, average Mw ∼150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Sigma-Aldrich (USA). N,N-dimethylformamide (DMF; 99.5%) was purchased from Tokyo Chemical Industry Co. Inc. (Tokyo, Japan). Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O) was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). Plastic syringes and needles (21G 1/2) were purchased from Terumo (Tokyo, Japan). LiPF6 (1 mol L−1) in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v%) solution was purchased from Kishida Chemical (Osaka, Japan).
000) was purchased from Sigma-Aldrich (USA). N,N-dimethylformamide (DMF; 99.5%) was purchased from Tokyo Chemical Industry Co. Inc. (Tokyo, Japan). Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O) was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). Plastic syringes and needles (21G 1/2) were purchased from Terumo (Tokyo, Japan). LiPF6 (1 mol L−1) in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v%) solution was purchased from Kishida Chemical (Osaka, Japan).
Electrospinning of PAN/Fe(NO3)3·9H2O composite nanofibers
PAN (10 wt%) was dissolved in DMF containing 15 wt% Fe(NO3)3·9H2O with stirring for at least 48 h at 60 °C. The solution was loaded into a plastic syringe and Al substrates were mounted on a metal collector. The applied voltage was set to 10 kV, the distance between the needle tip and collector was set to 20 cm, the solution flow rate was 1.0 mL h−1, and humidity was maintained at 30–40% by silica gel. After electrospinning, the as-spun membranes were removed from the Al collector and cut into 3 cm × 3 cm pieces. The membranes were then sandwiched between flat metal plates covered by a PTFE film and placed in a hot press set at zero pressure until the temperature of the plates reached 110 °C. Then, 2 MPa pressure was applied between the plates for 3 min. Next, the pressure was released completely, and the sample was allowed to cool to room temperature.Carbonization of electrospun nanofibers
The PAN/Fe(NO3)3·9H2O component nanofibers were first stabilized in air at 280 °C for 2 h (heating rate: 1 °C min−1) and then carbonized in N2 atmosphere at 600, 700, and 800 °C for 1 h (heating rate: 5 °C min−1), as shown in Fig. 2.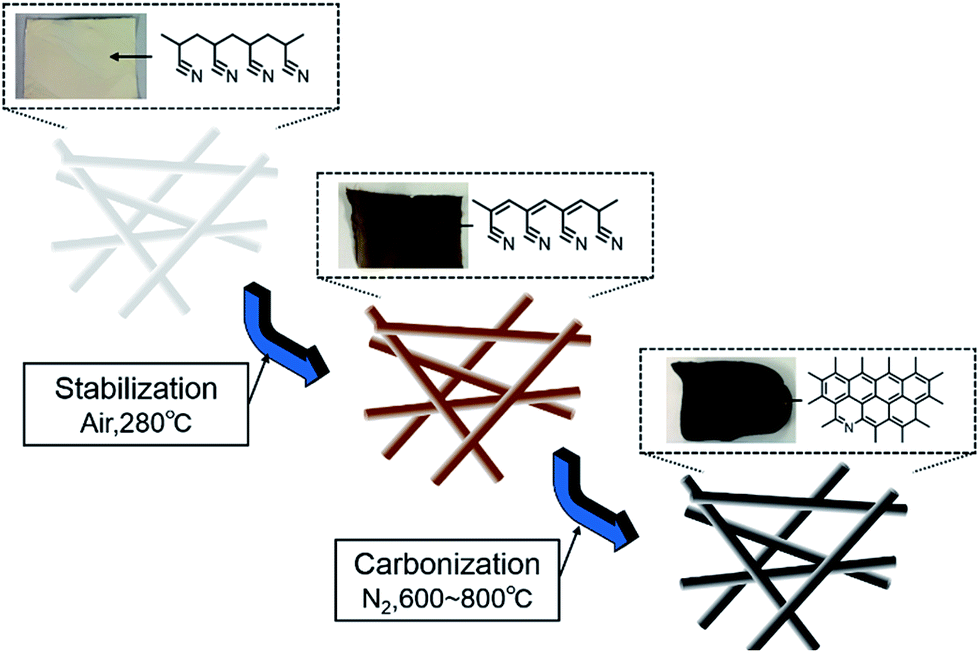 | ||
| Fig. 2 Schematic illustration of the electrospun carbon nanofiber fabrication process. | ||
Characterization of composite nanofibers
The surface morphology of the self-standing iron oxide carbon composite nanofibers was determined with a field emission scanning electron microscope (S-4700, Hitachi, Tokyo, Japan) and accelerated surface area and porosimetry system (ASAP, shimadzu, Kyoto, Japan). The carbon microstructure of these nanofibers was measured using Raman spectroscopy (inVia confocal Raman microscope, Renishaw, Gloucestershire, United Kingdom), whereas the thickness of the nanofibers was measured using a Digimatic Outside Micrometer (MDE-25MJ, Mitutoyo, Kanagawa, Japan).The electrochemical properties of the self-standing iron oxide/carbon composite nanofibers were characterized using 2032 coin cells with a lithium metal foil as the anode. Copper foil collector and poly (vinylidene fluoride) binder were not used in the preparation of the working electrodes for these 2032 coin cells. The separator consisted of a 25 μm microporous monolayer membrane (Celgard 2500, Celgard, Charlotte, North Carolina, USA), whereas the electrolyte was 1 mol L−1 LiPF6 in EC/DMC (1:1 v/v%). The cells were assembled in a dry air-filled glove box (Galaxy, Matsuura Manufactory Corporation, Tokyo, Japan) and cycled in the voltage range of 3.0–0.05 V with a multi-channel charge–discharge device (HJ-1001SMB, Hokuto Denko Corporation, Tokyo, Japan). The current value for the 1st to 5th charge–discharge cycles was 100 mA g−1. The current was then increased to 250, 500, 1000, and 2500 mA g−1 every three cycles for examining the current load characteristics. Finally, the current value reverted to 100 mA g−1. We calculated the direct current resistance from the IR drop after 1 s at 1000 mA g−1.
Results and discussion
FE-SEM images of the surface morphology of the iron oxide/carbon composite nanofibers are shown in Fig. 3. Under identical carbonization conditions, a larger number of pores form on the surface of the iron oxide/carbon composite nanofibers subjected to press processing compared to those not subjected to press processing. These differences are attributed to the reactivity differences between iron oxide and the carbon nanofibers. We believe that two factors cause reactivity differences in this reaction. The first is the influx of atmospheric oxygen into the experimental system, whereas the second factor is the presence of oxygen-containing materials in the reaction system. In this study, we think that the second factor is crucial. Oxygen present in iron oxide likely reacts with the carbon nanofibers and nano-scale processing promotes the reaction owing to increased specific surface area. Thus, nano-sized iron oxide decomposes the carbon nanofibers to carbon dioxide during carbonization. This decomposition reaction was confirmed by the weight retention measurements obtained at various carbonization temperatures as shown in Fig. S1.† These results show that while the presence of iron oxide and increase in carbonization temperature promote carbon decomposition, press processing does not promote carbon decomposition. On the other hand, under identical press processing conditions, the number of pores increases with increase in the carbonization temperature. While the iron oxide/carbon composite nanofibers subjected to press processing exhibit pores, regardless of the carbonization temperature, the self-standing iron oxide/carbon composite electrospun nanofibers not subjected to press processing exhibit pores at a carbonization temperature of 800 °C. Press processing is found to promote oxidation degradation by iron oxide. Fig. S2† shows that the density of the nanofibers increases with decreasing thickness at all carbonization temperatures. The increasing density enhances the interactions between the iron oxide and electrospun carbon nanofibers. | ||
| Fig. 3 FE-SEM images of self-standing iron oxide/carbon composite electrospun nanofibers: (a) before carbonization, without press processing; (b) carbonization at 600 °C, without press processing; (c) carbonization at 700 °C, without press processing; (e) carbonization at 800 °C, without press processing; (e) before carbonization, with press processing; (f) carbonization at 600 °C, with press processing; (g) carbonization at 700 °C, with press processing; (h) carbonization at 800 °C, with press processing. | ||
Raman spectra of the self-standing iron oxide/carbon composite electrospun nanofibers under various preparation conditions are shown in Fig. S3.† The extent of graphitization of the samples was estimated by considering the ratio between the D-band (disorder-induced phonon mode; 1250 and 1450 cm−1) and G-band (graphite band; 1550 and 1660 cm−1) intensities. The former can be attributed to defects and disordered portions of carbon (sp3-coordinated), whereas the latter can be attributed to ordered graphitic crystallites of carbon (sp2-coordinated). The ratio between the D-band and G-band intensities (ID/IG) can be used to analyze the number of carbon defects in the self-standing iron oxide/carbon composite electrospun nanofibers. A low ID/IG ratio indicates a large amount of sp2-coordinated carbon. The ID/IG ratios of self-standing iron oxide/carbon composite electrospun nanofibers under various preparation conditions are shown in Fig. 4.
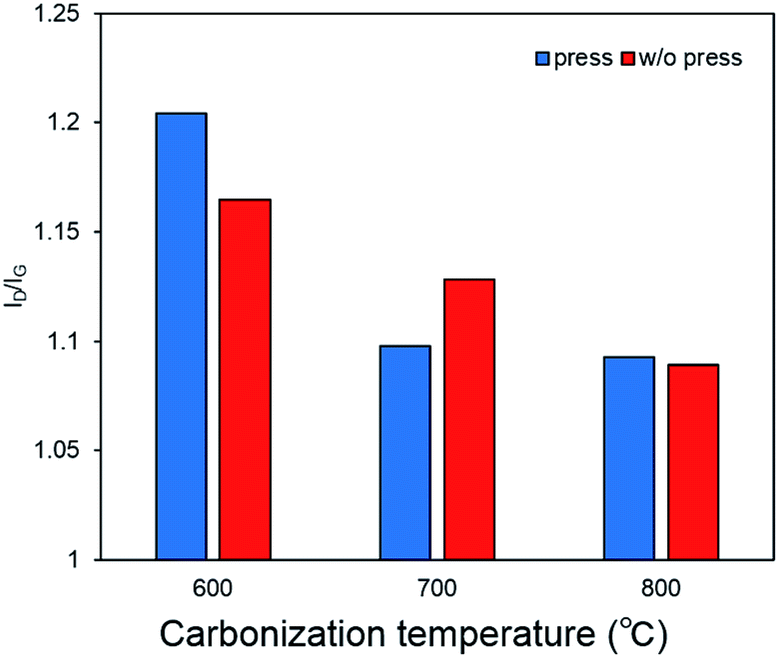 | ||
| Fig. 4 Raman spectra of self-standing iron oxide/carbon composite electrospun nanofibers; (red) carbonization at 600, 700, and 800 °C without press treatment; (blue) carbonization at 600, 700, and 800 °C with press treatment. | ||
The ID/IG ratio decreases as the carbonization temperature increases from 600 to 800 °C. From these results, it may be concluded that increasing the carbonization temperature promotes graphitization, with the ID/IG ratio decreasing as the carbonization temperature increases from 600 to 800 °C.
Fig. 5 shows the 1st to 3rd charge/discharge curves of the self-standing iron oxide/carbon composite nanofibers prepared under various conditions. The initial discharge capacity is observed during the 1st discharge for all the samples owing to the initial SEI generation at the surface and the presence of mesopores in the carbon nanofibers.23 On the other hand, the 2nd and 3rd charge/discharge curves for the nanofibers overlap, suggesting that the SEI is formed during initial discharge and becomes stable after the 1st cycle. The capacity after stabilization is the sum of the electrode capacities of the carbon nanofibers and iron oxide. The iron oxide reaction is represented by the following equation:
| Fe2O3 + 6Li+ + 6e− → 2Fe + 3Li2O | (1) |
| Fe3O4 + 8Li+ + 8e− → 3Fe + 4Li2O | (2) |
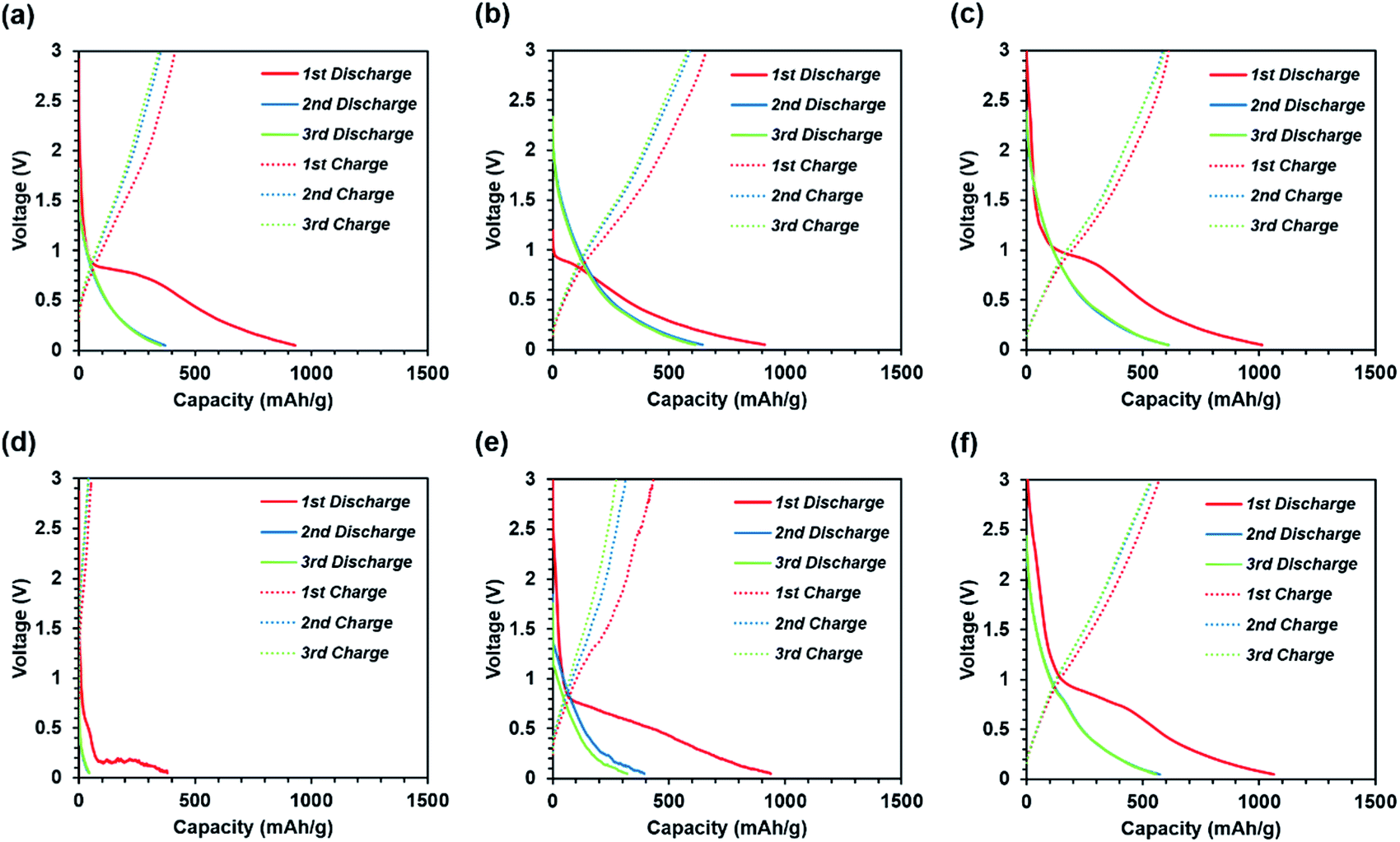 | ||
| Fig. 5 1st to 3rd charge–discharge curves of self-standing iron oxide/carbon composite electrospun nanofibers at 100 mA g−1; (a) carbonization at 600 °C, without press treatment; (b) carbonization at 700 °C, without press treatment; (c) carbonization at 800 °C, without press treatment; (d) carbonization at 600 °C, with press treatment; (e) carbonization at 700 °C, with press treatment; and (f) carbonization at 800 °C, with press treatment. | ||
The theoretical reversible capacity of Fe2O3 and Fe3O4 are 1007 and 926 mA h g−1, respectively. On the other hand, the theoretical reversible capacity of carbon nanofiber is about 400 mA h g−1, as shown in Fig. S4.† In the past paper, type of iron oxide was referred to as Fe3O4.24,25 The self-standing iron oxide/carbon composite nanofibers subjected to carbonization at 600 °C and press processing exhibit the lowest capacity during the 1st discharge cycle owing to the poor conductivity of the fibers, as shown in Fig. S5.† These conductivity differences can be observed from the change in the position of the initial discharge plateau due to ohmic drop. The pores caused by the presence of iron oxide and the disordered portions of carbon caused by low-temperature carbonization reduce the conductivity of the nanofibers. Thus, the total capacity of carbon nanofibers and iron oxide decreases at low carbonization temperatures and press treatment. The results indicate that a certain level of graphitization is necessary to achieve a certain capacity.
Fig. 6 shows the capacity per unit weight and volume of the self-standing iron oxide/carbon composite electrospun nanofibers under all the preparation conditions. The results indicate that carbonization and press processing increase the capacity per unit volume of the nanofibers. Carbonization shrinks the nanofibers, whereas press processing increases the nanofiber density, owing to improved electrode density as shown in Fig. S2.† In addition, Fig. S2† also indicates that the nanofiber electrodes exhibit low electrode density and consequently, low energy density, in general. This fact has not been discussed in most of the previous reports. Therefore, research into nanofiber electrodes requires investigation into techniques for electrode density enhancement, such as press processing.
 | ||
| Fig. 6 Capacity of self-standing iron oxide/carbon composite electrospun nanofibers carbonized at 600, 700, and 800 °C, with and without press treatment expressed in (a) per unit weight and (b) per unit volume. | ||
Fig. 7 shows that self-standing iron oxide/carbon composite electrospun nanofibers prepared by carbonization at 800 °C without press processing exhibit the highest capacity retention of 61% at 2500 mA g−1. Increasing the carbonization temperature increases the capacity retention, whereas press processing decreases the retention of rate capability. These differences in rate capability retention are caused by differences in electrode resistance, as can be observed from direct current resistance in Fig. 8. These can be attributed to change of the pore structure in Fig. S7.† The pore structure of self-standing iron oxide/carbon composite nanofiber carbonized at 800 °C, with press treatment changes from type II to type IV.
 | ||
| Fig. 7 Rate capabilities of self-standing iron oxide/carbon composite nanofibers: (a) carbonization at 600 °C, without press treatment; (b) carbonization at 700 °C, without press treatment; (c) carbonization at 800 °C, without press treatment; (d) carbonization at 600 °C, with press treatment; (e) carbonization at 700 °C, with press treatment; (f) carbonization at 800 °C, with press treatment. | ||
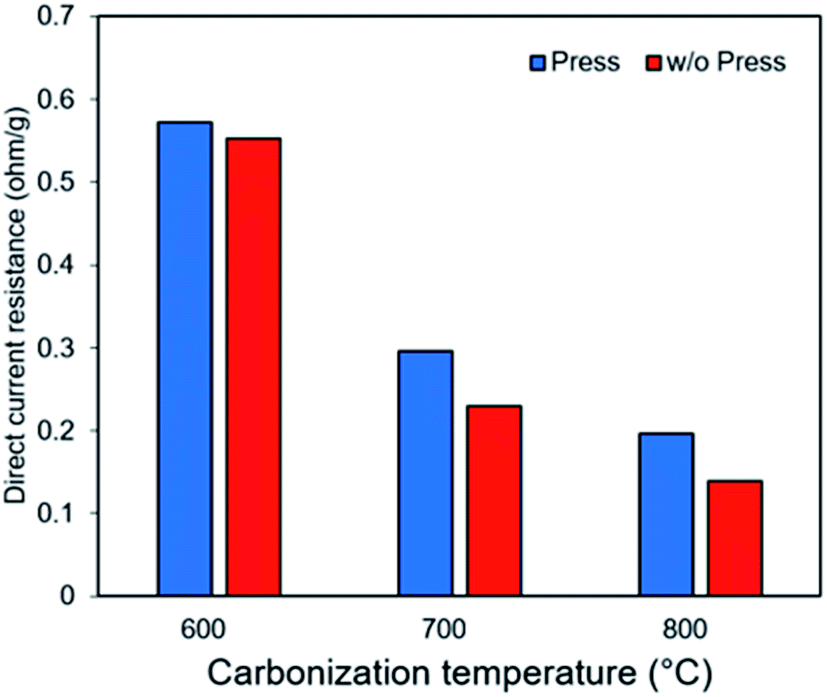 | ||
| Fig. 8 Direct current resistance of self-standing iron oxide/carbon composite electrospun nanofibers: carbonization at 600, 700, and 800 °C with and without press treatment. | ||
Fig. 9 shows the discharge curves measured during the cycle tests. In general, a high-capacity electrode has lower cycle durability owing to the higher volume change as compared to that of graphite.26,27
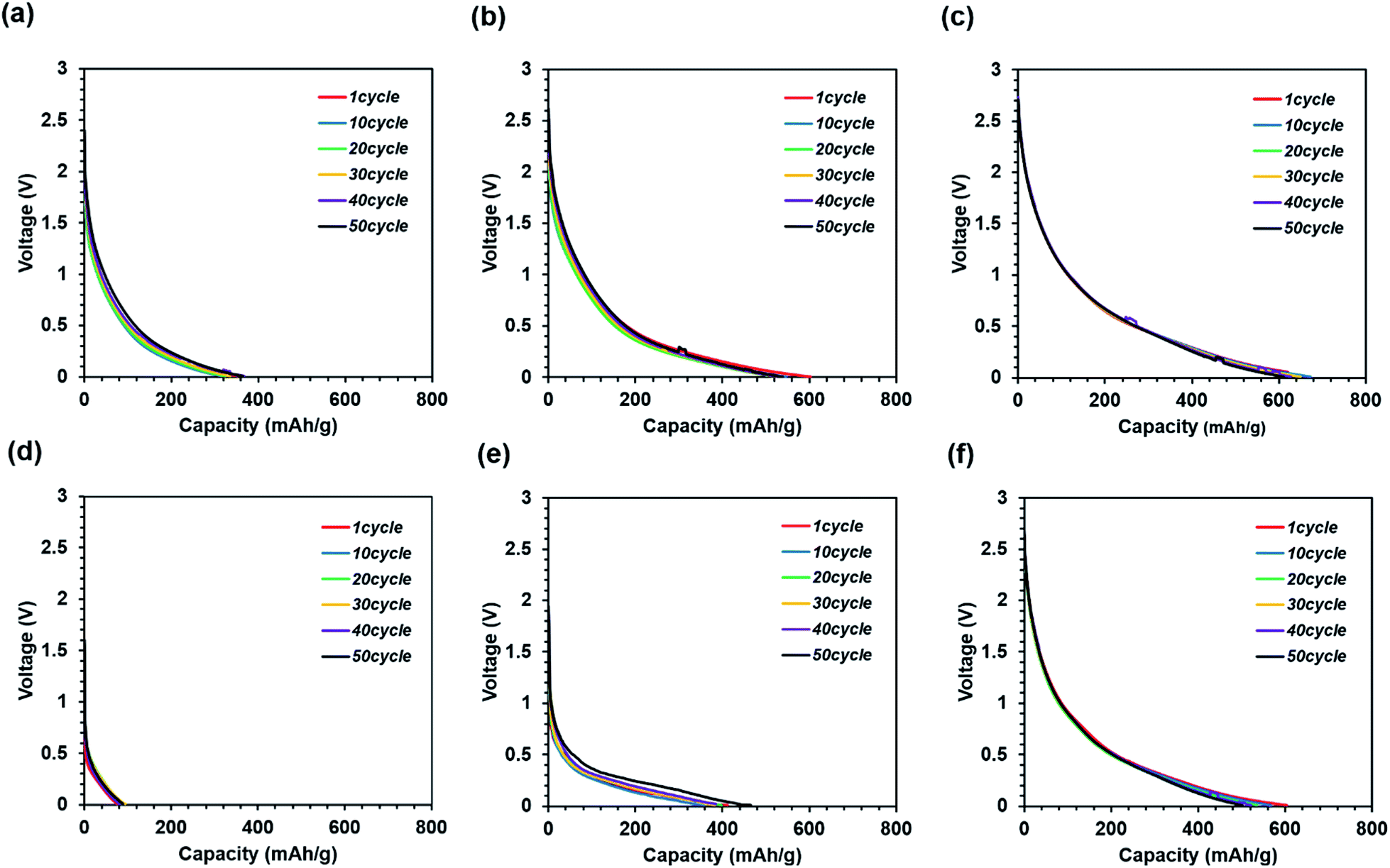 | ||
| Fig. 9 Cycle test discharge curves of self-standing iron oxide/carbon composite nanofibers; (a) carbonization at 600 °C, without press treatment; (b) carbonization at 700 °C, without press treatment; (c) carbonization at 800 °C, without press treatment; (d) carbonization at 600 °C, with press treatment; (e) carbonization at 700 °C, with press treatment; (f) carbonization at 800 °C, with press treatment at 100 mA g−1. | ||
These results indicate that all the samples exhibit high cycle durability, regardless of calcination and press conditions. Such high cycle durability is thought to suppress electrode destruction by the nano-iron oxide and carbon nanofiber composites. It is difficult to achieve cycle durability in conventional electrode structures in the absence of a binder and current collector. In addition, it can be seen that the resistance increases only slightly since the discharge cycle curves overlap. If there is a large increase in resistance, a voltage peak is expected to appear on the discharge curve. This would suppress the destruction of the conductivity path between the nano-iron oxide and carbon nanofiber composite. Thus, the nano-iron oxide and carbon nanofiber composite suppresses the increase in resistance due to cycling.
Increase in the carbonization temperature increases the rate capabilities. This causes more ordered graphitic crystallite growth when the carbonization temperature is increased.28 On the other hand, press treatment significantly improves the volumetric energy density. Further, nanofibers exhibit high cycle durability under all the preparation conditions. Knowledge of such processing parameters is important for the development of electrode materials in the future.
Conclusions
We fabricated the iron oxide/carbon composite nanofibrous electrode with excellent cycle characteristics. Some electrodes showed the capacity of about 600 mA h g−1 without using non-active materials such as conductive agents, binders, and separators. The results of the rate capabilities indicate that increasing the carbonization temperature improves the electrical characteristics of the electrode such as capacity and rate capabilities. On the other hand, press processing improves the capacity density per unit volume, whereas it reduces the rate capabilities of the electrode. It is found that the nano-iron oxide in the electrode oxidizes carbon nanofibers with press processing. In the future, we plan to propose a fabrication method in which such oxidation does not occur. Such investigations are useful for improving the performance of iron oxide/carbon composite nanofibers for LIB anodes.Acknowledgements
This work is supported by the Graduate School Doctoral Student Grant-in-Aid Program 2016 (Keio University). We are grateful to Shiratori Laboratory Members, Yuka Abe and Fumika Abe.Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- B. Scrosati, J. Solid State Electrochem., 2011, 15, 1623–1630 CrossRef CAS.
- J. Vetter, P. Novák, M. Wagner, C. Veit, K. Möller, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, et al., J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- J. Cho and S. T. Picraux, Nano Lett., 2014, 14(6), 3088–3095 CrossRef CAS PubMed.
- O. O. Taiwo, J. M. Paz-garcía, S. A. Hall, T. M. M. Heenan, A. Patera, D. P. Finegan, R. Mokso, P. Villanueva-p, D. J. L. Brett and P. R. Shearing, J. Power Sources, 2017, 342, 904–912 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- L. Xia, S. Wang, G. Liu, L. Ding, D. Li, H. Wang and S. Qiao, Small, 2016, 12, 853–859 CrossRef CAS PubMed.
- C. Kim, K. S. Yang, M. Kojima, K. Yoshida, Y. J. Kim, Y. A. Kim and M. Endo, Adv. Funct. Mater., 2006, 16, 2393–2397 CrossRef CAS.
- J. Abe, M. Tenjimbayashi and S. Shiratori, RSC Adv., 2016, 6, 38018–38023 RSC.
- V. A. Ganesh, A. S. Nair, H. K. Raut, T. T. Yuan Tan, C. He, S. Ramakrishna and J. Xu, J. Mater. Chem., 2012, 22, 18479 RSC.
- H. Wu, L. Hu, M. W. Rowell, D. Kong, J. J. Cha, J. R. McDonough, J. Zhu, Y. Yang, M. D. McGehee and Y. Cui, Nano Lett., 2010, 10, 4242–4248 CrossRef CAS PubMed.
- K. Onozuka, B. Ding, Y. Tsuge, T. Naka, M. Yamazaki, S. Sugi, S. Ohno, M. Yoshikawa and S. Shiratori, Nanotechnology, 2006, 17, 1026–1031 CrossRef CAS PubMed.
- W. J. Li, C. T. Laurencin, E. J. Caterson, R. S. Tuan and F. K. Ko, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef CAS PubMed.
- F. Yang, R. Murugan, S. Wang and S. Ramakrishna, Biomaterials, 2005, 26, 2603–2610 CrossRef CAS PubMed.
- R. Gopal, S. Kaur, C. Y. Feng, C. Chan, S. Ramakrishna, S. Tabe and T. Matsuura, J. Membr. Sci., 2007, 289, 210–219 CrossRef CAS.
- M. Tenjimbayashi, K. Sasaki, T. Matsubayashi, J. Abe, K. Manabe, S. Nishioka and S. Shiratori, Nanoscale, 2016, 8, 10922–10927 RSC.
- C. T. Cherian, J. Sundaramurthy, M. Kalaivani, P. Ragupathy, P. S. Kumar, V. Thavasi, M. V. Reddy, C. H. Sow, S. G. Mhaisalkar, S. Ramakrishna and B. V. R. Chowdari, J. Mater. Chem., 2012, 22, 12198 RSC.
- X. Zhang, P. Suresh Kumar, V. Aravindan, H. H. Liu, J. Sundaramurthy, S. G. Mhaisalkar, H. M. Duong, S. Ramakrishna and S. Madhavi, J. Phys. Chem. C, 2012, 116, 14780–14788 CAS.
- X. Li, Y. Chen, H. Huang, Y.-W. Mai and L. Zhou, Energy Storage Mater., 2016, 5, 58–92 CrossRef.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- L. Ji, O. Toprakci, M. Alcoutlabi, Y. Yao, Y. Li, S. Zhang, B. Guo, Z. Lin and X. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 2672–2679 CAS.
- S. Gu, Y. Liu, G. Zhang, W. Shi, Y. Liu and J. Zhu, RSC Adv., 2014, 4, 41179–41184 RSC.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- M. T. McDowell, S. Xia and T. Zhu, Extreme Mech. Lett., 2016, 9, 480–494 CrossRef.
- J. K. Lee, K. W. An, J. B. Ju, B. W. Cho, W. Il Cho, D. Park and K. S. Yun, Carbon, 2001, 39, 1299–1305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05301k |
| This journal is © The Royal Society of Chemistry 2017 |