
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sub-5 μm balls possessing forest-like poly(methyloxazoline)/polyethyleneimine side chains and templated silica microballs with unusual internal structures†
Daiki Soma and
Ren-Hua Jin *
*
Department of Material and Life Chemistry, Faculty of Engineering, Kanagawa University, 3-2-7, Rokkakubashi, Yokohama 221-8686, Japan. E-mail: rhjin@kanagawa-u.ac.jp
First published on 20th July 2017
Abstract
Herein, sub-5 μm microballs, with unusual forest-like structures consisting of a polystyrene network and forest-like poly(2-methyl-2-oxazoline) (PMOZ) and/or linear polyethyleneimine (LPEI) side chains, were synthesized by combining two isolated processes. The first process is the dispersion radical polymerization of 4-chloromethylstyrene (CMS) and divinylbenzene (DVB) in the presence of a stabilizer, polyvinylpyrrolidone (PVP), using 2,2′-azobis(isobutyronitrile) (AIBN) as an initiator, which produces a poly(4-chloromethylstyrene) type microgel (μ-PStCl). The second process is the cationic ring-opening polymerization of 2-methyl-2-oxazoline (MOZ) using μ-PStCl as a solid initiator, which produces poly(2-methyl-2-oxazoline)-grafted microballs (μ-PSt-g-PMOZ). The latter, possessing PMOZ side chains, was transformed into poly(ethyleneimine)-grafted microballs (μ-PSt-g-PEI) via treatment with HCl (aq). Moreover, the three types of microballs μ-PStCl, μ-PSt-g-PMOZ, and μ-PSt-g-PEI of 1–3 micrometer in diameter were characterized by FT-IR, 13C CP/MAS NMR, elemental analysis, XRD, and SEM. Both the microballs μ-PSt-g-PMOZ and μ-PSt-g-PEI, which resembled the assemblies of hydrophilic comb polymers in their chemical structure, exhibited good wettability in an aqueous phase. In particular, the μ-PSt-g-PEI microballs, which have forest-like basic polyamine chains throughout the microballs, act as catalytic templates in the hydrolytic polycondensation of tetramethoxysilane (TMOS) at room temperature to produce polymer/silica hybrid microballs of μ-PSt-g-PEI@SiO2 with 2.5–3.5 μm diameter and 35–70 wt% silica content depending upon the mediation conditions. Calcination of the μ-PSt-g-PEI@SiO2 hybrid microballs at 700, 800, and 900 °C resulted in silica microballs possessing 2–4 nm mesopores with reduced diameter from 3.5 to 1.2 μm and reduced BET surface area from 582 to 189 m2 g−1. It was confirmed that the sub-5 μm microballs of μ-PSt-g-PEI were very effective catalytic templates for the construction of silica microballs of different sizes (1–4 μm), different surface areas, and different (large hollow or co-continuous) internal structures.
Introduction
Spherical microgels, including aqueous microgels and non-aqueous microgel particles, have been widely used for various applications such as in coating films, catalyst supports, and biomedical materials.1–9 Various polymerization methods such as emulsion polymerization,10,11 dispersion polymerization,12–14 precipitation polymerization,15,16 suspension polymerization,17,18 and seed dispersion polymerization19 have been reported for the preparation of well-defined microgels. In the design of functional non-aqueous microgels via the abovementioned polymerization systems, styrene (St) and 4-chloromethylstyrene (CMS) are often used as monomers for constructing the network backbone frame of the gels.20,21 Although they are suitable for the preparation of nano-scaled spheres, for the abovementioned monomers, emulsion polymerization and precipitation polymerization are not utilized in the synthesis of well-controlled microgels; this is because of the fact that a number of synthesis factors need to be controlled in this case.11,16 In comparison, dispersion polymerization of 4-chloromethylstyrene (CMS) is a facile preparation approach to obtain microgels with the functional groups –CH2Cl and well-controlled particle size of 1–3 μm.21,22 We were particularly interested in the preparation of non-aqueous microgels possessing –CH2Cl groups and their transformation into aqueous microgels via cationic ring-opening polymerization (CROP) of cyclic 2-methyl-2-oxazoline using the non-aqueous microgels as polymerization initiators.Poly(2-oxazoline)s (POZs) are well known as smart polymers. Depending on their molecular architectures, POZs behave as water-soluble or amphiphilic polymers that can respond to external stimuli such as heat, pH, and solvents.23 POZs can be easily obtained via cationic ring-opening polymerization (CROP) of cyclic 2-oxazolines (nucleophiles) in the presence of usual electrophiles or Lewis acids.24–26 This is a very simple and effective living polymerization system and thus has been divergently used in the design of POZs with different molecular structures and dimensions, such as di- and tri-block copolymers,26–29 star polymers,30–33 comb polymers,34–37 and their applications as soft matter.38,39 In terms of the molecular design of POZs, in the early 1980's, Kobayashi et al. established a process for the preparation of amphiphilic block copolymers by combining hydrophilic monomers, such as 2-methyl-2-oxazoline and 2-ethyl-2-oxazoline, and hydrophobic monomers such as 2-phenyl-2-oxazoline, 2-n-butyl-2-oxazoline, as well as 2-n-octyl-2-oxazoline.40 Nuyken et al. reported the preparation of amphiphilic graft copolymers via CROP of 2-methyl-2-oxazoline using poly(isobutene-co-(p,m-chloromethylstyrene)) as a macroinitiator.41 We also reported the synthesis and functionalities of porphyrin core star poly(oxazoline)s with homopolymeric31,42 and block-copolymeric arms.42–44 Since the 2000's, Hoogenboom et al. developed a new method of polymerization of 2-oxazolines using microwave45,46 and robot synthesizer techniques47 and prepared a series of new poly(2-oxazoline)s. Their study exhibited strong impact on the chemistry of poly(2-oxazoline)s and shed light on poly(2-oxazoline)s as new biomimetic polymers. In the polymerization of 2-oxzolines, benzyl halide-type electrophiles are often used as initiators to obtain the target poly(2-oxazoline)s. Therefore, polymers consisting of chloromethylstyrene residues are desirable candidates as macroinitiators for the polymerization of 2-oxazolines.34,36 We reported that homopolymer and/or block copolymers having a poly(chloromethylstyrene) block could be used as macroinitiators to produce comb-like poly(2-oxazoline)s in a controllable fashion.48 In this sense, microgels with poly(chloromethylstyrene) chains are reasonably capable of acting as solid polymeric initiators for the preparation of microballs bearing poly(2-oxazoline)s brushes.
In the structure, the backbone of poly(2-oxazoline)s is characteristically polyethyleleimine with side chains consisting of acyl groups. Therefore, poly(2-oxazoline)s can be easily hydrolyzed to the corresponding polyamine, having only secondary-amine in the polymer backbone, that is named linear poly(ethyleneimine) [–CH2CH2–NH–] (PEI).49 LPEI is a derivative of poly(2-oxazoline) showing a very unique feature in aqueous media; that is, it can self-assemble into crystalline-nanostructure-based aggregates with various morphologies because crystallization acts as a driving force for self-organization. We focused our attention on this feature and systematically developed LPEI-based chemistry via a biomimetic mineralization process and established a new process to design morphology-controlled nanostructured silica and titania materials using nanocrystalline aggregates of LPEI as catalytic templates in silica and titania deposition. In this process, LPEIs with different structural dimensions, such as straight, star, and comb, could result in morphologically different silica materials.50–52
There is no doubt that the design of LPEIs is same as the design of poly(2-oxazoline)s. In other words, a topological architecture of poly(2-oxazoline)s must result in the corresponding LPEI with the same topological architecture. A further benefit of LPEI is its great potential in the transformation of various side structures; this is because it is very easy to introduce side-group bonding into the nitrogen atoms via the reaction of the secondary amine in LPEI with many functional groups. Unambiguously, both the polymers poly(2-oxazoline) and LPEI will become significantly important in soft matter science, as recently indicated by Hoogenboom in his editorial comments, “50 years of poly(oxazoline)s”.39
As a strategy of our LPEI chemistry, herein, the synthesis of microballs of poly(2-methyloxazoline) using a sub-5 μm mirogel (i.e., cross-linked microball) as an initiator was particularly utilized. As reported in our recent study, comb-like LPEI can be easily obtained via polymerization of 2-methyl-2-oxazoline using poly(4-chloromethylstyrene) as a macroinitiator followed by hydrolyzation of the acetyl side chains of poly(2-methyloxazoline). Therefore, the microballs containing 4-chloromethylstyrene and a cross-linker would be reasonably utilizable as a solid-type initiator to synthesize poly(2-oxazoline)-grafted microballs that are forest-like in structure possessing non-cross-linked poly(2-oxazoline) side chains through the microballs from exterior to interior. From this idea, we synthesized sub-5 μm microballs (e.g., 1–2 μm in diameter) consisting of 4-chloromethylstyrene and 2 mol% of divinylbenzene (cross-linker) and used them in the polymerization of 2-methyl-2-oxazoline (MOZ) with different polymerization degrees and finally changed the PMOZ type microballs into LPEI type microballs. All the microballs with different chemical components were characterized by FT-IR, solid state 13C NMR, elemental analysis, XRD, and wettability studies. Furthermore, we performed silica deposition in the presence of microballs with a forest-like structure and having LPEI and found that polymer/silica hybrid microballs with the silica contents within 35–65 wt% were effectively produced. Calcination of these hybrid balls at different temperatures resulted in silica microballs whose spherical shape was retained, but they possessed remarkably different diameters, different BET surface areas, and different internal structures; this indicated the effectiveness of the microballs as catalytic templates in the deposition of silica microballs in a controlled fashion.
Experimental
Materials
4-Chloromethylstyrene (CMS, > 90%, TCI) was purified by column chromatography with basic aluminum oxide (Aldrich) before use. 2-Methyl-2-oxazoline (MOZ, 98%, Aldrich) was distilled from sodium hydroxide under atmospheric pressure and then stored under a nitrogen atmosphere prior to use. Divinylbenzene (DVB, m- and p- mixture, >50%, TCI) was extracted from a sodium hydroxide aqueous solution (1 M) to remove the polymerization inhibitor. Azobis(isobutyronitrile) (AIBN, >98%, TCI) was crystallized from ethanol. Potassium iodide (KI, >99.5%, Wako), tetramethoxysilane (TMOS, >98%, TCI), dimethylacetamide (DMAc, Super dehydrated, >99.5%, Wako), polyvinylpyrrolidone K25 (PVP, Wako), hydrochloric acid (5 M, Wako), ammonium solution (28 vol%, Wako), and other reagents were used as received without further purification.Synthesis of the poly(p-chloromethylstyrene) microgel (μ-PStCl)
By modifying the method reported in ref. 21, the dispersion polymerization was performed as follows. To a 100 mL three-necked round-bottom flask equipped with a condenser, ethanol (solvent, 50 mL), DMAc (co-solvent, 3 mL), and polyvinylpyrrolidone (PVP K25, surfactant, 0.6 g) were added, and the mixture was stirred at room temperature until PVP was completely dissolved. Then, DVB (cross-linker, 25 μL) and CMS (monomer, 1.4 g) were added, followed by bubbling nitrogen through the solution for 30 min to exclude the dissolved oxygen. After heating the solution to 60 °C, AIBN (initiator, 0.03 g) dissolved in 2 mL of ethanol was added, and then, the mixture was stirred at the same temperature for 24 h. After polymerization, the obtained suspension products were corrected by centrifugation (4000 rpm, 5 min), washed with methanol to remove excess amounts of PVP and CMS, and then dried under reduced pressure at 50 °C for 6 h. White powders of μ-PStCl were obtained in 0.8 g (57%) yield.Synthesis of the polystyrene microgel grafting poly(2-methyl-2-oxazoline) (μ-PSt-g-PMOZ)
Polymerization of MOZ using μ-PStCl as a solid initiator was performed as follows. μ-PStCl 0.2 g (–CH2Cl unit: 1.2 mmol), KI (0.6 mg), and DMAc (15 mL) were added to a two-necked round-bottom flask and stirred at 100 °C for 20 min under a nitrogen atmosphere. Then, 5.5 mL of MOZ (65 mmol) was added, and the mixture was stirred at 100 °C for 24 h. After polymerization, the solid fractions were obtained via centrifugation (4000 rpm, 5 min), washed with water and methanol, and then dried under reduced pressure. White powders of μ-PSt-g-PMOZ were obtained in a 2.0 g yield.Synthesis of the polystyrene microgel grafting poly(ethyleneimine) (μ-PSt-g-PEI)
The balls of μ-PSt-g-PMOZ (2 g) were dispersed in 5 M HCl (30 mL) and stirred at 100 °C for 8 h. After being cooled down to room temperature, the resulting balls of μ-PSt-g-PEI were washed with methanol via centrifugation. Then, the separated particles were mixed with ammonium solution and stirred at room temperature for 24 h. The products were obtained by filtration (0.45 μm), washed with distilled water and acetone, and then dried under atmosphere. Yield: 1.6 g (white powders).Silica hybridization on μ-PSt-g-PEI
A mixture containing 1 mL of TMOS and 0.5 mL of distilled water was added to a dispersion of μ-PSt-g-PEI (50 mg) in methanol (20 mL) and stirred at room temperature for 3 h. After this, the precipitates of the silica hybrid (μ-PSt-g-PEI/SiO2) were obtained by centrifugation (4000 rpm, 5 min), washed with distilled water and acetone, and then dried under atmosphere.Moreover, for replacing methanol, other dispersion media such as ethanol, acetone, and water were used in the preparation of μ-PSt-g-PEI/SiO2 in the same manner.
Calcination of μ-PSt-g-PEI/SiO2
All the samples of μ-PSt-g-PEI/SiO2 mediated from different solvents were calcined at 700–900 °C for 2 h (rising time: 1 h, keeping time: 1 h) for removing the polymeric components from the hybrid balls. The resulting silica microballs were subjected to FT-IR, 29Si CP/MAS NMR, SEM, and nitrogen adsorption–desorption isotherm analyses.Characterization
Fourier transform infrared (FT-IR) spectra were obtained using a JASCO FT-IR-4600 spectrometer by diffusing the samples in KBr pellets. 13C cross polarization magic angle spinning (CP/MAS) NMR spectra and 29Si CP/MAS NMR spectra were obtained using a JEOL ECA-400 spectrometer operating at 400 MHz. Elemental analysis was conducted using Perkin Elmer 2400 II. Scanning electron microscopy (SEM) observations were performed using a Hitachi SU-8010 microscope at 5 kV after the samples were sputtered with Pt particles. X-ray diffraction (XRD) measurements were conducted via a Rigaku Rint-Ultima III X-ray diffractometer with Cu Kα radiation (λ = 0.1540 nm) operating at 40 kV and 40 mA. TGA was performed using a Seiko Instruments EXSTER 6000 at 30–900 °C at a heating rate of 10 °C min−1 under air condition. N2 adsorption/desorption isotherms were obtained using a Micrometrics Tristar-3000 instrument at liquid-nitrogen temperature, whereby all samples were degassed at 120 °C under vacuum for 3 h prior to analysis.Results and discussion
Synthesis and characterization of the μ-PStCl, μ-PSt-g-PMOZ, and μ-PSt-g-PEI microballs
The synthetic route to a series of polymeric microballs (microgels) is presented in Scheme 1. As abovementioned, the chloromethylene (–CH2Cl) group on the monomer unit of 4-chloromethylstyrene in the polymeric network would act as an initiator site for the polymerization of 2-oxazoline. Therefore, we tried the dispersion polymerization of 4-chloromethylstyrene and divinylbenzene (2 mol%) using different stabilizers, such as polyvinylpyrrolidone (PVP), hydroxypropyl cellulose (HPC), and poly(2-ethyloxazoline) (PEOZ), for the preparation of monodispersed gelated balls with the targeted sizes of 1–2 micrometers. Via comparison of the effect of the type of stabilizers (PVP K25 (Mw = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), PVP K60 (Mw = 160,000), PVP K90 (Mw = 600000), hydroxypropyl cellulose 3–5.9 (HPC 3–5.9, Mw = 160000), HPC 150–400 (Mw = 620000), poly(2-ethyl-2-oxazoline) (PEOZ, Mw = 50000), PEOZ (Mw = 500000)) on the size and yields of the microgels (see Fig. S1†), we found that polyvinylpyrrolidone with a molecular weight of 25000 is more suitable as a stabilizer than others in the preparation of mono-dispersed 1–2 micrometer spherical gels of μ-PStCl. Thus, we performed the preparation of μ-PStCl using CMS as a monomer, DVB as a cross-linker (2 mol%), PVP K25 as a stabilizer, and AIBN as an initiator in the mixed solvents of ethanol and DMAc under a N2 atmosphere, as described in the experimental section.
000), PVP K60 (Mw = 160,000), PVP K90 (Mw = 600000), hydroxypropyl cellulose 3–5.9 (HPC 3–5.9, Mw = 160000), HPC 150–400 (Mw = 620000), poly(2-ethyl-2-oxazoline) (PEOZ, Mw = 50000), PEOZ (Mw = 500000)) on the size and yields of the microgels (see Fig. S1†), we found that polyvinylpyrrolidone with a molecular weight of 25000 is more suitable as a stabilizer than others in the preparation of mono-dispersed 1–2 micrometer spherical gels of μ-PStCl. Thus, we performed the preparation of μ-PStCl using CMS as a monomer, DVB as a cross-linker (2 mol%), PVP K25 as a stabilizer, and AIBN as an initiator in the mixed solvents of ethanol and DMAc under a N2 atmosphere, as described in the experimental section.
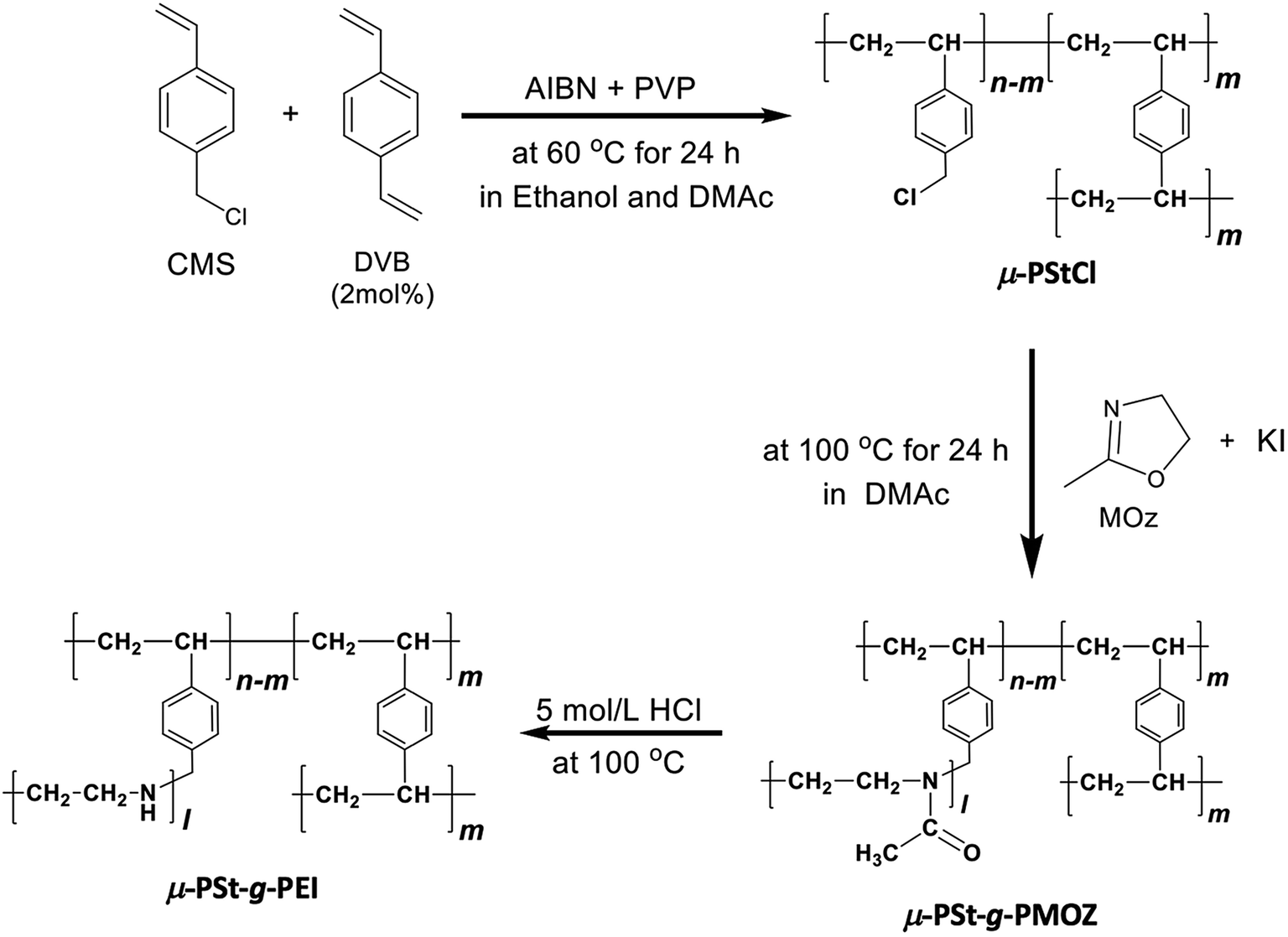 | ||
| Scheme 1 Synthetic route from μ-PStCl to μ-PSt-g-PEI. | ||
As shown in the FT-IR and 13C NMR spectra (Fig. 1), the characteristic vibration peaks at 676 cm−1 (C–Cl stretching) and 1265 cm−1 (C–Cl vending) due to –CH2Cl in μ-PStCl and the chemical shift at 50 ppm due to the –CH2Cl group in μ-PStCl can be observed. We also estimated from the elemental analysis that the content of Cl atom in μ-PStCl was 6 mmol g−1. Using the microgels of μ-PStCl as a solid-type initiator, we performed the polymerization of 2-methyl-2-oxazoline (MOZ) in the presence of potassium iodide (KI) in DMAc by changing the feeding ratio of Cl/MOZ (about 1/20, 1/50, and 1/100) at 100 °C for 24 h. The yields of the recovered solid μ-PSt-g-PMOZ were 0.9, 2.0, and 1.9 g, respectively, corresponding to the 1/20, 1/50, and 1/100 feeding ratios of Cl/MOZ. It was apparent that the polymerization of MOZ promoted by the solid initiator of μ-PStCl proceeded very well with different feeding ratios, but there was limited growth even if the monomer was used in large excess. This would be in relation to the container property of the microballs in which the volume to load the monomer unit will be restricted. The sample obtained by the 1/50 (Cl/MOZ) feeding ratio was subjected to FT-IR, 13C NMR, and elemental analyses. As shown in Fig. 1, the peaks of 676 cm−1 (C–Cl stretching) and 1265 cm−1 (C–Cl vending) due to the –CH2Cl group disappeared, and new signals at 1024 cm−1 (C–N) and 1639 cm−1 (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) due to the imide group were observed. Moreover, the 13-carbon chemical shift at 50 ppm due to –CH2Cl completely disappeared, and instead of it, new chemical shifts appeared at 20 and 175 ppm due to –CH3 and carbonyl N–CO groups, respectively. These results indicated the formation of poly(2-methyl-2-oxazoline) (PMOZ) chains in the micro spheres. From elemental analysis, the nitrogen content was determined to be 14.6%, which should be contributed by the MOZ unit in μ-PSt-g-PMOZ. Therefore, we estimated that the content of the MOZ unit in μ-PSt-g-PMOZ would be 75.3 wt% (equal to 10.4 mmol) for per gram microballs.
O stretching) due to the imide group were observed. Moreover, the 13-carbon chemical shift at 50 ppm due to –CH2Cl completely disappeared, and instead of it, new chemical shifts appeared at 20 and 175 ppm due to –CH3 and carbonyl N–CO groups, respectively. These results indicated the formation of poly(2-methyl-2-oxazoline) (PMOZ) chains in the micro spheres. From elemental analysis, the nitrogen content was determined to be 14.6%, which should be contributed by the MOZ unit in μ-PSt-g-PMOZ. Therefore, we estimated that the content of the MOZ unit in μ-PSt-g-PMOZ would be 75.3 wt% (equal to 10.4 mmol) for per gram microballs.
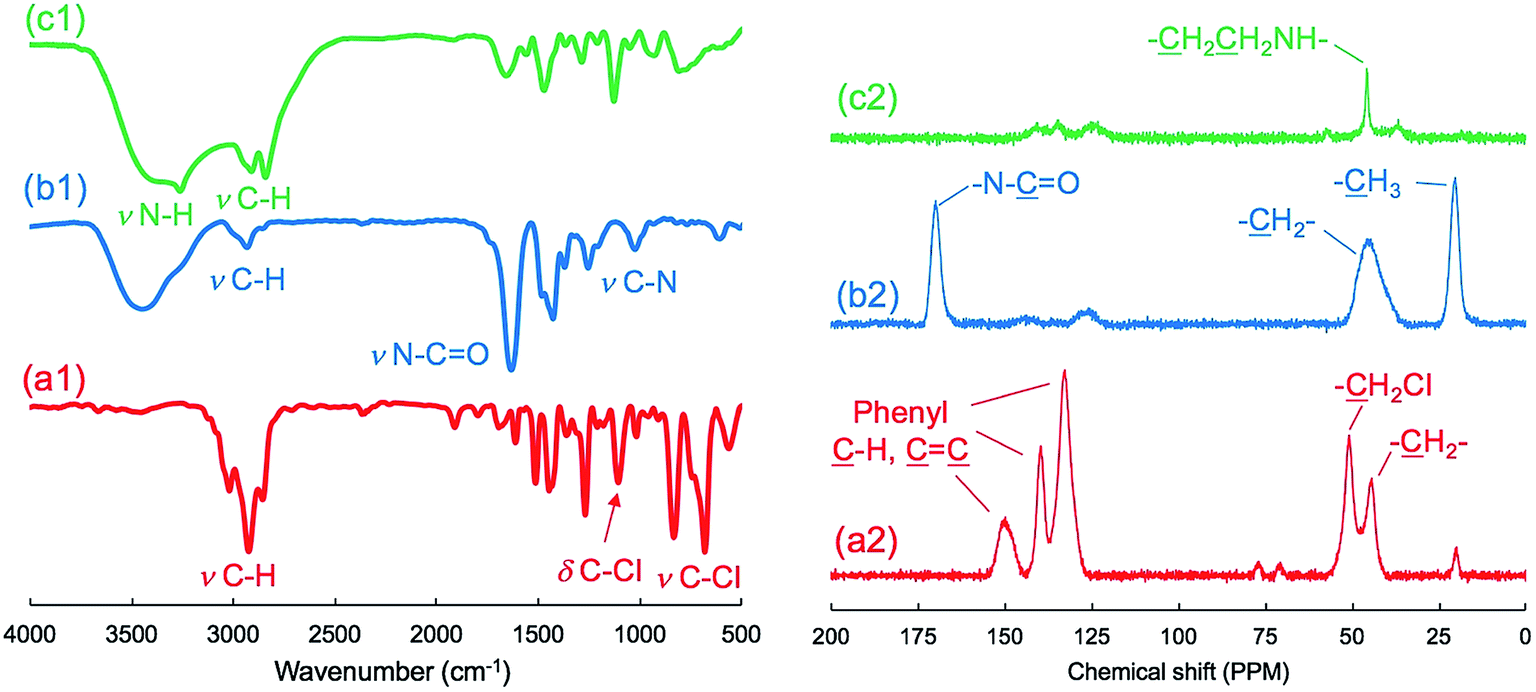 | ||
| Fig. 1 FT-IR (left) and solid state 13C NMR (right) spectra of (a) μ-PStCl, (b) μ-PSt-g-PMOZ, and (c) μ-PSt-g-PEI. | ||
Furthermore, we transformed the microballs of μ-PSt-g-PMOZ (10.4 mmol g−1) into μ-PSt-g-PEI via hydrolyzation of the PMOZ chains using 5 M HCl (aq) under reflux conditions. After treatment by ammonia solution, the resulting free-base form of μ-PSt-g-PEI was characterized by FT-IR and XRD. We can see from Fig. 1c that the vibration peaks at 1639 cm−1 (N–CO stretching) due to the imide group vanished and a new peak at 3277 (N–H stretching) appeared in the FT-IR spectrum. Moreover, from Fig. 2b and c, it was observed that the chemical shifts at 20 ppm due to the –CH3 group and at 175 ppm due to the carbonyl group (N–CO) of μ-PSt-g-PMOZ completely disappeared after hydrolysis. These results indicate that the transformation of μ-PSt-g-PMOZ into μ-PSt-g-PEI is very easy. Herein, note that the soluble polymers of linear poly(ethyleneimine)s with different topologies, such as straight or star/comb forms, always crystallize via association of two water molecules per ethyleneimine (EI) unit (EI/H2O = 1:2, dihydrated form) when the polymers are placed in the atmosphere or in water, and its crystalline packing is easily changed when the number of water molecules associated with the EI unit decreases.53 Unexpectedly, the crystalline feature also emerged even from extremely small isolated microballs possessing forest-like PEI chains, as seen in the XRD diffraction peaks at 2θ = 15.9, 17.9, 24.0, 25.5, and 32.0 degree (see Fig. 2). According to the classification rule of Chatani's model,54 we deduced that the crystal had a sesquihydrate form (EI/H2O = 1:1.5).
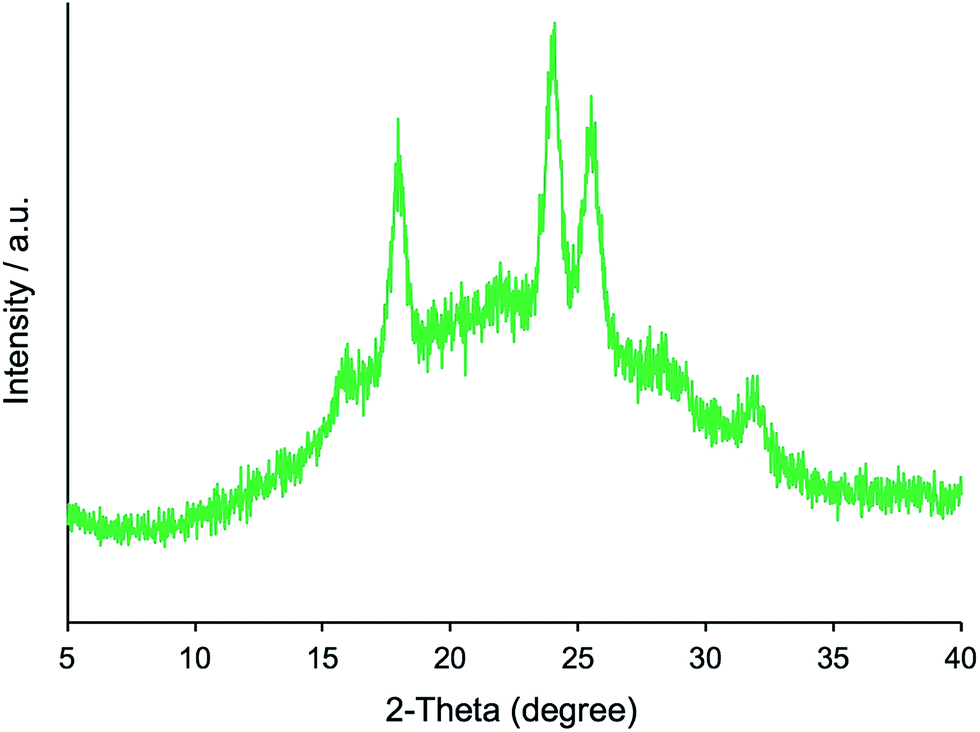 | ||
| Fig. 2 XRD pattern of μ-PSt-g-PEI. | ||
To evaluate the size change of the microballs, we further subjected the μ-PStCl, μ-PSt-g-PMOZ (1/50), and μ-PSt-g-PEI microballs to SEM visualization. Fig. 3 shows the SEM images of the three kinds of microballs. The initial microballs of μ-PStCl were 1.54 (±0.19) μm in size, with mono-dispersion form, but the size increased to 2.8 (±0.50) μm for μ-PSt-g-PMOZ generated from μ-PStCl. After the PMOZ chains were transformed into PEI, the size decreased to 2.2 (±0.21) μm. Probably, this diameter change was related to the mass of per ball. The dried balls of μ-PStCl with low cross-liking degree (2 mol%) must be in the contracted state, but would potentially have a large empty volume to load substances when the network frame undergoes expansion in the swollen state. The polymerization of MOZ by μ-PStCl just allowed the loading of the PMOZ fractions in the swollen state of μ-PStCl; thus, the empty volume decreased in the resulting μ-PSt-g-PMOZ to resist the contraction as compared to that in μ-PStCl. As a result, the diameters of the μ-PSt-g-PMOZ balls remarkably increased. However, the removal of the side chain of acetyl group from the μ-PSt-g-PMOZ balls via hydrolyzation led to the reduction of the mass of the balls by more than half, and thus, the size decreased. This size change means that the microballs would be like a flexible micro container and would be useful in hybridization with other components.
 | ||
| Fig. 3 SEM images of the three types microballs (a and d) μ-PStCl, (b and e) μ-PSt-g-PMOZ, and (c and f) μ-PSt-g-PEI. | ||
To understand the characteristic features of the three microballs in the dispersion state in different solvents, we tested the compatible behavior of the three microballs (2 mg) in a mixture of water/chloroform (1.5/1.5 in mL). Note that the polystyrene backbone is hydrophobic, being soluble only in chloroform. In contrast, PMOZ is an amphiphile, being highly soluble both in water and chloroform and is thus able to work as a surfactant to form oil-in-water droplets in water/chloroform mixtures.44 As seen in the image shown in Fig. 4, the μ-PStCl balls swell in the chloroform phase as they are put in a mixture of water/chloroform. However, in the case of μ-PSt-g-PMOZ, interestingly, when the amphiphilic microballs were put in the water/chloroform mixture, the whole chloroform phase became an accumulation of chloroform droplets, and the water/chloroform interface disappeared. This indicates that the μ-PSt-g-PMOZ microballs act as surfactants, entrapping chloroform to form droplets that sink and pile up in the bottom (see Fig. 4). Compared to the abovementioned two microballs, the μ-PSt-g-PEI microballs having PEI chains were neither in the swollen nor in the dispersion state, whereas they floated at the interface between water and chloroform (some precipitated in the bottom) when they were put in the mixture of water/chloroform. This is a reasonable phenomenon because linear PEI usually exists as crystals associating with water molecules and is not soluble in water and chloroform under ambient conditions. The μ-PSt-g-PEI microballs also possess crystalline domains and cannot be solvated by water and chloroform although they are wettable in water.
 | ||
| Fig. 4 (a) Images of wettability of (a) μ-PStCl (left), μ-PSt-g-PMOZ (center), and μ-PSt-g-PEI (right) (2 mg) in the mixture of water/chloroform (1.5 mL/1.5 mL). (b) magnified image of the center sample. | ||
The hybrid silica balls μ-PSt-g-PEI/SiO2 and calcined silica
We have established LPEI chemistry based on the crystallization-driven self-assembly of LPEI in an aqueous system for constructing hybrid nanomaterials via a biomimetic mineralization process. LPEI plays an important role as a catalytic template for spontaneous deposition of silica/titania on the surface of LPEI crystalline morphologies once mixed with silica or titania sources.50,51 Therefore, it was also expected that μ-PSt-g-PEI itself would effectively serve as a sub-5 μm type catalytic template for fabricating sub-5 μm inorganic oxide materials. Herein, we focused on silica deposition in the presence of μ-PSt-g-PEI. The process of silica deposition on μ-PSt-g-PEI and the subsequent calcination are shown in Scheme 2. In detail, first, TMOS (1 mL) mixed with a small amount of water (0.5 mL) was added to the dispersion solution of μ-PSt-g-PEI (50 mg) in various media (20 mL), such as water, methanol, ethanol, and acetone, and then stirred for 3 h at room temperature. This resulted in silica hybrid balls (μ-PSt-g-PEI/SiO2). Calcination of the as-prepared silica hybrids at a definite temperature under atmosphere finally yielded silica balls.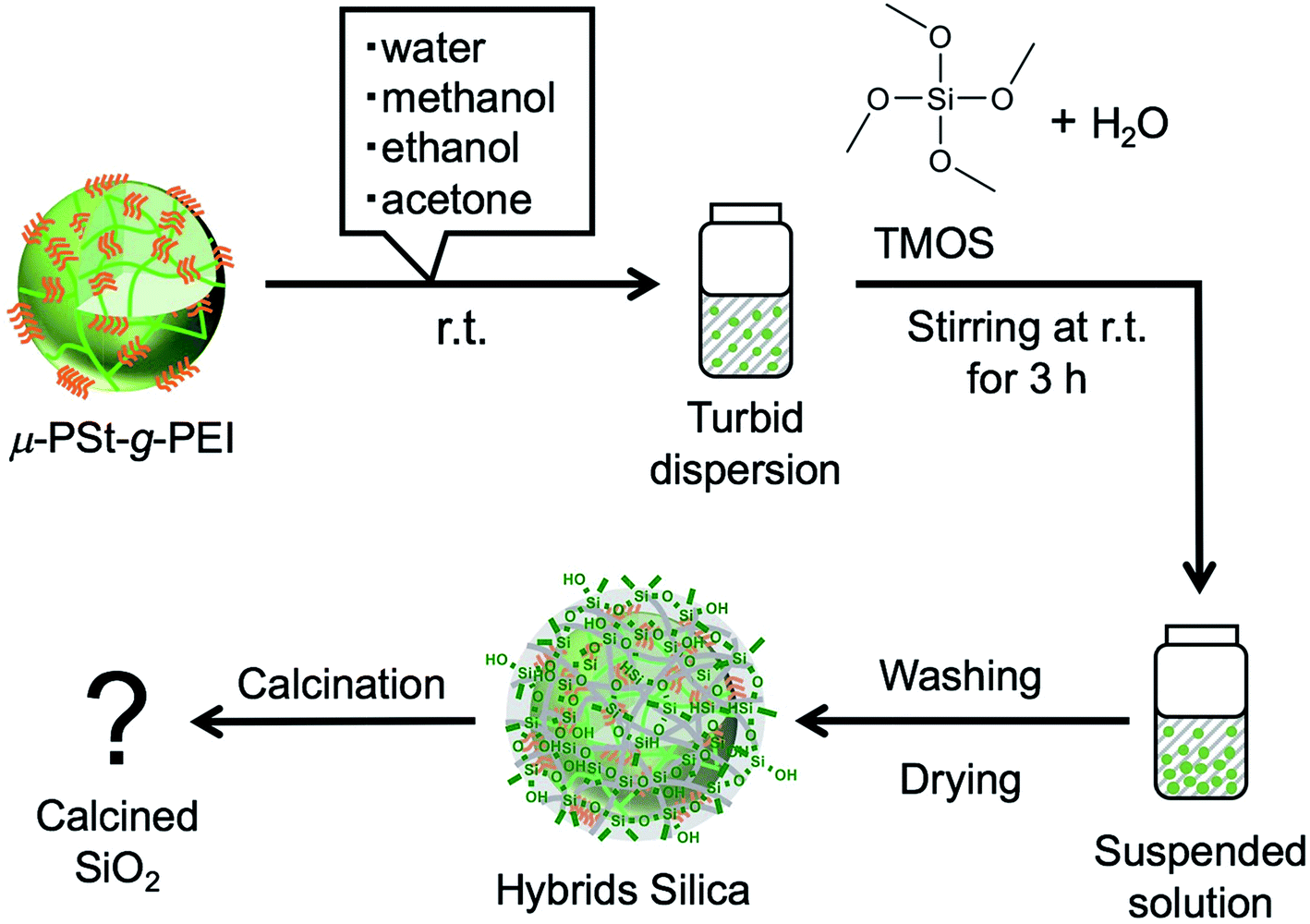 | ||
| Scheme 2 Schematic of the templated silica microballs. | ||
It can be seen from FT-IR spectra (Fig. 5, left) that all the as-prepared forms of μ-PSt-g-PEI/SiO2 show apparent vibration peaks at 1050 cm−1 (Si–O–Si, stretching) and 950 cm−1 (Si–OH stretching) indicating silica formation on the μ-PSt-g-PEI microballs. Fig. 6 shows the TGA traces of the hybrid μ-PSt-g-PEI/SiO2 balls prepared in different dispersion media such as methanol, ethanol, acetone, and water. The initial weight loss until 100 °C was due to the evaporation of water adsorbed on the silica networks. Herein, we assigned the remaining weight to the silica content after the removal of organic components at around 700 °C. It was clear that the silica content decreased in the order of 67, 61, 52, and 38 wt% with their corresponding mediation conditions using the dispersion media of methanol, ethanol, water, and acetone. This result means that the activity of μ-PSt-g-PEI for silica deposition is higher in methanol than that in acetone medium. It would be related to the solvation ability of the PEI chains. The μ-PSt-g-PEI microballs are in the fully swollen state in methanol, but are hardly swollen in acetone. The swollen PEI chains could act as a molecular-like basic catalyst to effectively promote the hydrolytic polycondensation of TMOS through the voids of the microballs μ-PSt-g-PEI. In contrast, in the case of the non-swollen state, the PEI chains distributed in both the exterior and the interior of the microballs would show different activities for silica deposition; the exterior is more active than the interior. All the hybrid silica microballs mediated from different solvents were calcined at 800 °C and subjected to 29Si CP/MAS NMR spectroscopy. As shown in Fig. 5 (right), the chemical shifts appeared at −90, −100, and −120 ppm were assigned to the signal of Q2, Q3, and Q4, respectively. It seems that the four types of silica microballs after calcination have almost no differences in their silica bonding (note: the samples before calcination could not provide reliable spectral results in the 29Si CP/MAS NMR measurements).
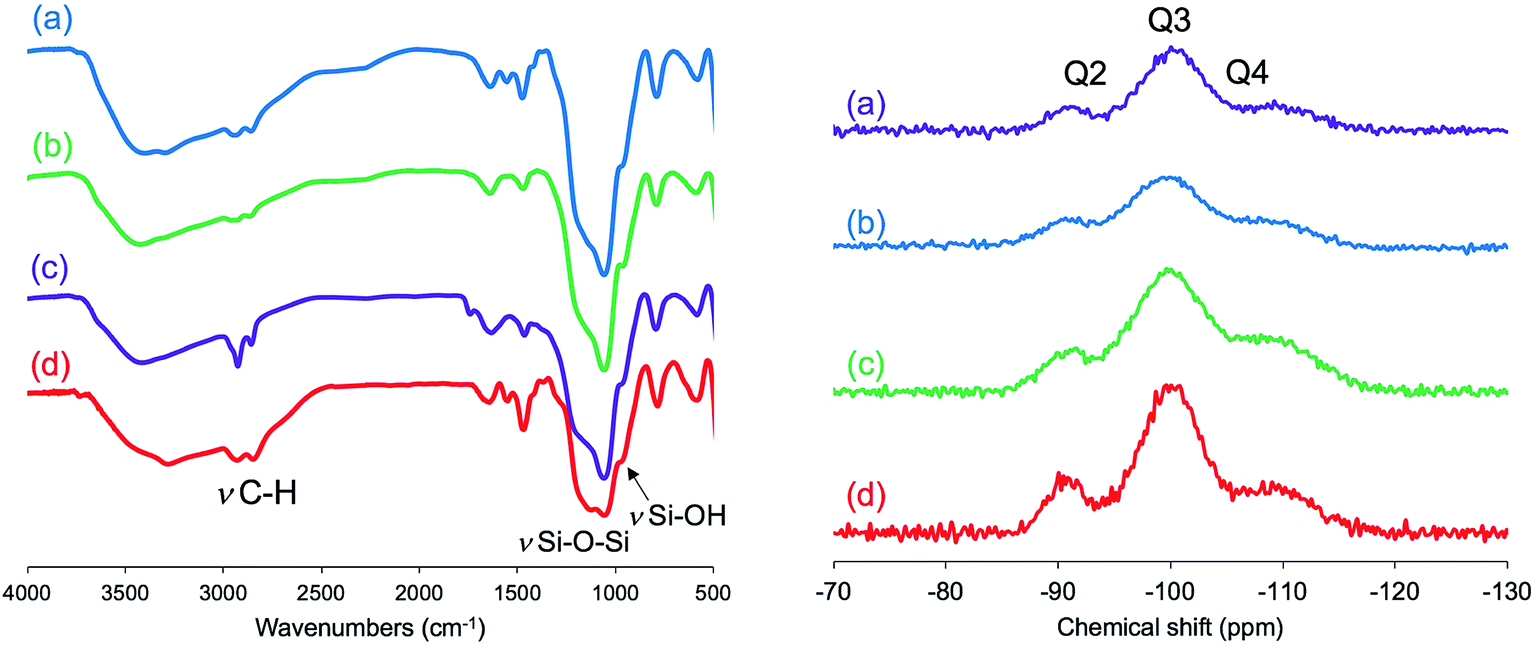 | ||
| Fig. 5 FT-IR spectra (left) of the as-prepared hybrid μ-PSt-g-PEI@SiO2 microballs obtained in different media: methanol (a), ethanol (b), water (c), and acetone (d). 29Si CP/MAS NMR (right) spectra after calcination at 700 °C of the μ-PSt-g-PEI@SiO2 microballs mediated by different solvents: water (a), methanol (b), ethanol (c), and acetone (d). | ||
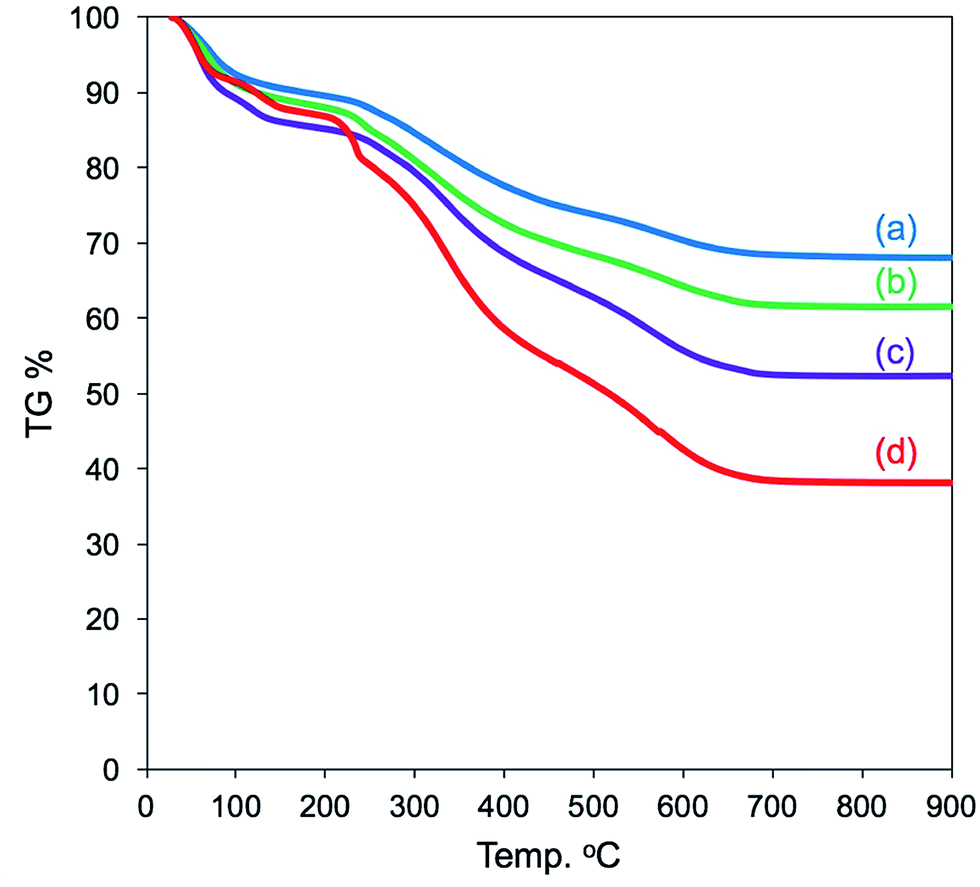 | ||
| Fig. 6 TGA curves of μ-PSt-g-PEI@SiO2 hybrids mediated from methanol (a), ethanol (b), water (c), and acetone (d). | ||
In Fig. S2,† the SEM images of the as-prepared hybrid silica microballs are shown. Similar to the spherical morphology of the catalytic template of μ-PSt-g-PEI, all the hybrid silica mediated under different conditions well retained their spherical shape, and their average particle sizes (in diameter) appeared to be about 3.2–3.5 μm for mediation in methanol and ethanol and about 2.2–2.5 μm for mediation in water. Interestingly, we found that the sizes of the μ-PSt-g-PEI@SiO2 hybrid microballs mediated in different solvents shrunk differently after calcination at 800 °C (see Fig. 7). The average particle sizes of the calcined silica decreased to about 2.5 (methanol), 2.3 (ethanol) > 1.8 (water) > 1.2 μm (acetone). The shrink degree estimated from the diameter reduction is about 65% for the silica microballs mediated from methanol and ethanol as compared to about 50% for the silica microballs mediated from water and acetone. This different shrinking behavior would be in relationship with the internal structures of the silica microballs (details have been discussed hereinafter). However, the particles after calcination could maintain their spherical shape without collapsing; this indicated the thermostability of the silica microballs even after the removal of organic components with remarkable shrinkage. From both the SEM images and TGA traces, it can be concluded that the sizes of the hybrid microballs and the deposition amount of silica on the hybrid microballs correlate with the swelling property of the μ-PSt-g-PEI microballs under the mediation conditions. It was found from the DLS measurements that the swelling property decreased in the following order: methanol > ethanol > water > acetone (see Fig. S3†). The good solvent (methanol) for μ-PSt-g-PEI afforded larger particles and higher amount of silica deposition, whereas the poor solvent (acetone) for μ-PSt-g-PEI yielded smaller particles and lower amount of silica deposition.
 | ||
| Fig. 7 SEM images of the calcined (at 800 °C) silica microballs mediated from different solvents: water (a and e), methanol (b and f), ethanol (c and g), and acetone (d and h). | ||
Furthermore, N2 adsorption–desorption isotherm measurement was used to investigate the feature of the internal structure of the calcined silica. As shown in Fig. 8 (left), there are remarkable differences for silica mediated in both methanol and ethanol and in both water and acetone. The former exhibited IV type hysteresis loop, whereas the latter exhibited II type hysteresis loop. In addition, the N2 quantity adsorbed on the calcined silica was lower for the former than that for the latter. The calculated BET surface areas for the calcined silica decreased in the following order: 575.5 (water) > 351.1 (acetone) > 251.1 (methanol) > 221.0 (ethanol) m2 g−1. Herein, note that the BET surface area is largest for silica mediated from water, being over two times those of the other silica microballs mediated from methanol and ethanol, indicating the formation of different internal structures on the silica microballs. We know that the PEI chains in the μ-PSt-g-PEI microballs tend to form cluster-like crystalline domains in water, which would be attributed to the formation of silica network frame with unusual pore volumes and cavities. Fig. 8 (right) unambiguously shows that the calcined silica mediated from both water and acetone are similar to each other with widely distributed mesopores in the range of 2–4 nm. In comparison, the calcined silica mediated from both methanol and ethanol showed relatively sharp mesopores distribution at 3.8 nm but without the pores below 3 nm. These differences would also be related to the internal structures of the silica microballs.
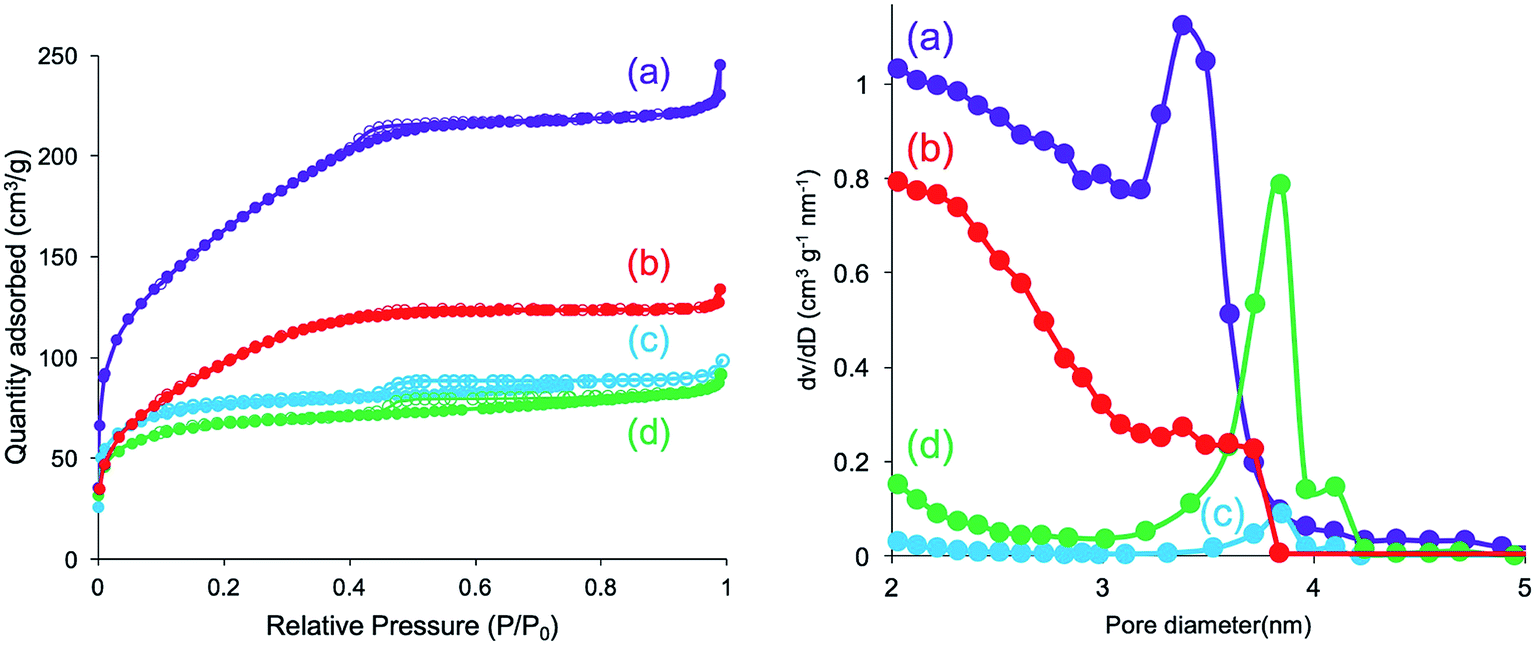 | ||
| Fig. 8 N2 adsorption desorption isotherm (left) and pore size distribution (right) of the calcined (at 800 °C) μ-PSt-g-PEI@SiO2 mediated from water (a), acetone (b), methanol (c), and ethanol (d). | ||
Recently, we developed a very unique silica pulverizing method55 that can be used for etching the surface of silica particles and thus for imaging their internal structures. Herein, we selected two kinds of the calcined (at 800 °C) silica microballs mediated from water and methanol and subjected them to a special treatment for slightly making a hole on their external surfaces using our etching method (see Schemes S1 and S2†). After the hole-making treatment, the silica microballs were subjected to SEM observation (see Fig. 9). Very interestingly, the silica microballs mediated from water and from methanol had remarkably different internal structures although the template was the same. The microballs mediated from water possess hollow structures with large inside cavity (Fig. 9a), while the balls mediated from methanol consist of co-continuous structures with dense silica skeletons and through-pores in which the silica skeletons appeared to be fibrous with about 30 nm diameter (Fig. 9b). From the abovementioned two types of silica microballs, we can conclude that the template microballs of μ-PSt-g-PEI can be adapted under mediation conditions to switch the catalytic functions of PEI in silica deposition around the external surface of the microballs or through the whole of the microballs. In water mediation, the PEI chains on the microballs tend to crystallize; this prevents the penetration of the silica sources; thus, the external surfaces of the microballs significantly act as catalysts in the deposition of silica to form tough silica shells. In contrast, in the case of methanol mediation, the PEI chains through the microballs are solvated very well; thus, the silica sources penetrate through the microballs and deposit here and there to form the continuous silica skeletons through the microballs.
 | ||
| Fig. 9 SEM images of the internal structures of the calcined (at 800 °C) silica microballs mediated from both water (a1 and a2) and methanol (b1 and b2). | ||
Considering the silica mediated from methanol, we investigated the effect of calcination temperatures on the nitrogen sorption isotherms (see Fig. 10, left). When the calcination temperature increased from 700, 800 to 900 °C, the corresponding silica showed remarkable reduction in nitrogen adsorption with accompanying reduction of BET surface areas in the order of 582.0 > 251.1 > 189.0 m2 g−1. This suggests that the lower calcination temperature is beneficial for producing larger surface areas. We also found that the calcination temperature markedly affects the size (diameters) of the silica balls, being in the order of 3.5 μm (700 °C), 2.5 μm (800 °C), and 1.6 μm (900 °C) (see SEM images in Fig. S4†). These large shrinkage with temperature increment indicates not only the presence of network-like silica skeletons and cavities through the balls but also the destruction and reconstruction of the silica skeletons through the internal cavities with the retention of the spherical shape; this caused the reduction of the surface areas, as seen in the sorption isotherms. In addition, the pore distributions shown in Fig. 10 (right) indicate the formation of mesopores in the silica microballs for all calcination temperatures. A pair of mesopores with the sizes of 3.5 (majority) and 4.2 nm (minority) and 3.8 (majority) and 4.2 nm (minority) appeared, respectively, for the silica calcined at 700 and 800 °C. In the case of 900 °C, silica showed mesopores with the size of 3.7 nm with the increment of the distribution-line towards the micropore field. The effect of calcination temperatures on the silica structures was also estimated by 29Si CP/MAS NMR spectra (see Fig. S5 and Table S1†). The silica structure slightly changed with the reducing integration ratio of Q3/Q4 (from 2.63 to 1.61) when the calcination temperature increased to 900 °C; this indicated that the silanol groups (Si–OH) on Q3 were partly transformed into Q4 via condensation at 900 °C. This result is well consistent with the severe shrinkage of the silica microballs at 900 °C.
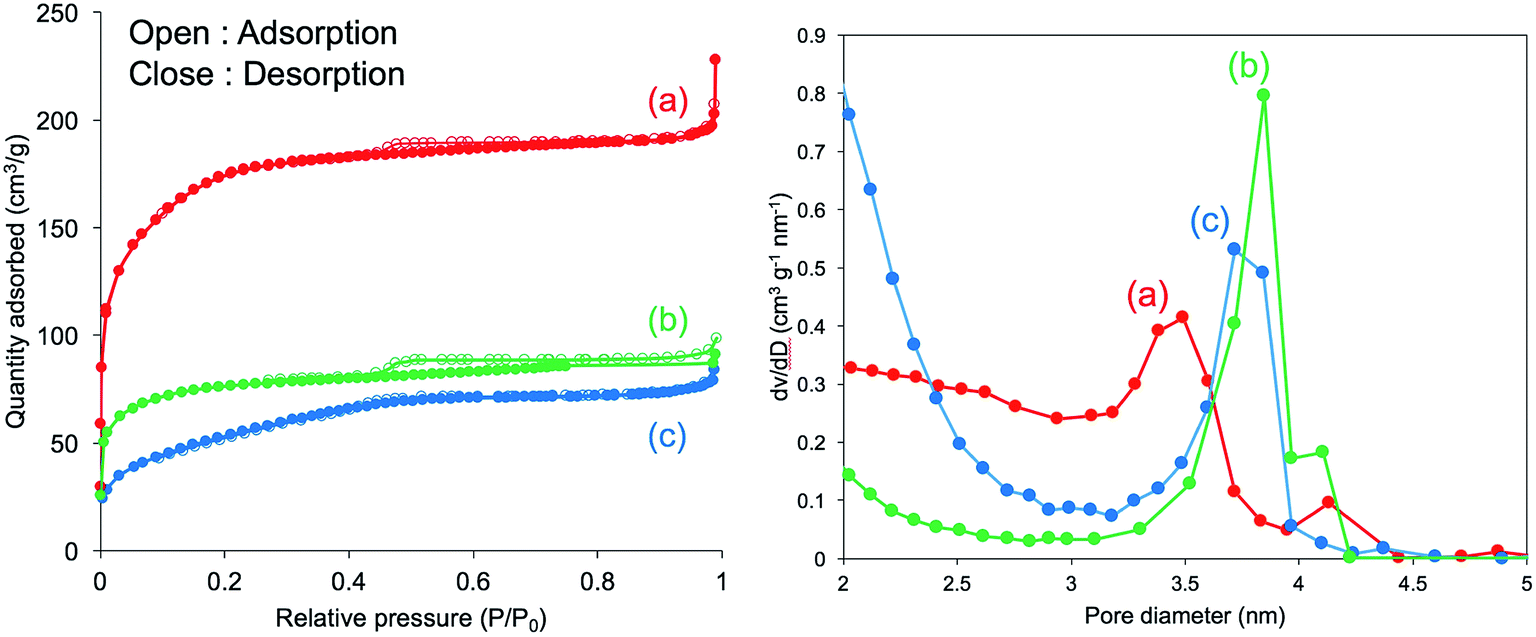 | ||
| Fig. 10 The influences of the calcination temperatures on N2 adsorption desorption isotherm (left) and pore size distribution (right). Sample of μ-PSt-g-PEI@SiO2 mediated from methanol. Calcination temperatures: 700 (a), 800 (b), and 900 °C (c). | ||
Conclusions
Herein, sub-5 μm polymeric balls of μ-PSt-g-PMOZ and μ-PSt-g-PEI with forest-like hydrophilic polymer chains were synthesized by a combination of dispersion polymerization of 4-chloromethylstyrene monomer as a starting material, cationic ring-opening polymerization of MOZ, and acidic hydrolysis reaction of PMOZ brushes. In this process, the lowly cross-linked solid μ-PStCl balls possessing 3-dimensionally and uniformly distributed reactive sites effectively act as solid initiators for the polymerization of MOZ to produce a controllable chain length of PMOZ to form the corresponding microballs of μ-PSt-g-PMOZ. These microballs are different, with the micellar-like spherical polymer aggregates having external brush layer and internal core in their structures. Both the parts of the exteriors and the interiors of the microballs are filled up by the PMOZ chains to form the PMOZ forest in and around the microballs. Therefore, the corresponding hydrolyzed microballs of μ-PSt-g-PEI have the same forest structures. Due to the characteristic features (crystallization or solvation) of the PEI chains in different media, the μ-PSt-g-PEI microballs can control silica deposition either around the microball surfaces, resulting in silica shells, or through the microballs to provide co-continuous silica skeletons. The two paths are easily switchable only by selecting mediation solvents during silicification. In addition, the calcined silica microballs mediated form different solvents showed different surface areas, different diameters, and slightly different sizes of mesopores. Even silica mediated from the same solvent also changed its structural characters when calcined at different temperatures. Especially, the sizes of the silica balls can shrink dramatically, but their spherical shape is retained without collapsing as the calcination temperature is increased.These sub-5 μm silica microballs are potentially applicable in the fields of separation, catalysis, carrier, and coating and optical materials. Further studies on the silica microballs with details of internal structures and their applications in catalytic supports are in progress.
Acknowledgements
This work was supported in part by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities: “Creation of new fusion materials by integration of highly ordered nano inorganic materials and ultra-precisely controlled organic polymers” (2013–2017).References
- A. Biffis and L. Minati, J. Catal., 2005, 236, 405–409 CrossRef CAS
.
- T. Terashima, M. Ouchi, T. Ando and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1061–1069 CrossRef CAS
- C. D. Sorrell, M. C. D. Carter and M. J. Serpe, ACS Appl. Mater. Interfaces, 2011, 3, 1140–1147 CAS
- Q. M. Zhang and M. J. Serpe, ACS Appl. Mater. Interfaces, 2015, 7, 27547–27553 CAS
- Y. Xia, X. He, M. Cao, C. Chen, H. Xu, F. Pan and J. R. Lu, Biomacromolecules, 2013, 14, 3615–3625 CrossRef CAS PubMed
- S. Schmidt, M. Zeiser, T. Hellweg, C. Duschl, A. Fery and H. Möhwald, Adv. Funct. Mater., 2010, 20, 3235–3243 CrossRef CAS
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC
- A. Imaz and J. forcada, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1173–1181 CrossRef CAS
- J. A. Bonham, M. A. Faers and J. S. van Duijneveldt, Soft Matter, 2014, 10, 9384–9398 RSC
- B. R. Saunders and U. B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–12 CrossRef CAS
- S. Baruch-Sharon and S. Margel, Colloid Polym. Sci., 2010, 288, 869–877 CAS
- C. M. Tseng, Y. Y. Lu, M. S. El-Aasser and J. W. Vanderhoff, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 2995–3007 CrossRef CAS
- T. Bahar and A. Tuncel, Polym. Eng. Sci., 1999, 39, 1849–1855 CAS
- J. S. Song and M. A. Winnik, Macromolecules, 2005, 38, 8300–8307 CrossRef CAS
- W. H. Li, K. Li and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2295–2303 CrossRef CAS
- S. F. Li, X. L. Yang and W. Q. Huang, Chin. J. Polym. Sci., 2005, 23, 197–202 CrossRef CAS
- Q.-Q. Liu, L. Wang, A.-G. Xiao, H. J. Yu and Q. H. Tan, Eur. Polym. J., 2008, 44, 2516–2522 CrossRef CAS
- S. Shen, X. Zhang and L. Fan, Mater. Lett., 2008, 62, 2392–2395 CrossRef CAS
- D. Wang, V. L. Dimonie, E. D. Sudol and M. S. El-Aasser, J. Appl. Polym. Sci., 2002, 84, 2710–2720 CrossRef CAS
- T. Gong and C. C. Wang, J. Mater. Sci., 2008, 43, 1926–1932 CrossRef CAS
- N. Kihara, Y. Adachi, K. Nakao and T. Fukutomi, J. Appl. Polym. Sci., 1998, 69, 1863–1873 CrossRef CAS
- S. Margel, E. Nov and I. Fisher, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 347–355 CrossRef CAS
- H. Schlaad, C. Diehl, A. Gress, M. Meyer, A. L. Demirel, Y. Nur and A. Bertin, Macromol. Rapid Commun., 2010, 31, 511–525 CrossRef CAS PubMed
- S. Kobayashi and H. Uyama, J. Polym. Sci., Part A: Polym. Chem., 2001, 40, 192–195 CrossRef
- M. Lobert, R. Hoogenboom, C.-A. Fustin, J.-F. Ghohy and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5859–5868 CrossRef CAS
- K. Kempe, M. Lobert, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3829–3838 CrossRef CAS
- P. V. Caeter, E. J. Goethals, V. Gancheva and R. Vellchkova, Polym. Bull., 1997, 39, 589–596 CrossRef
- C.-H. Wang and G.-H. Hsiue, Biomacromolecules, 2003, 4, 1487–1490 CrossRef CAS PubMed
- R. Hoogenboom, F. Wiesbrock, H. Huang, M. A. M. Leenen, H. M. L. Thijs, S. F. G. M. van Nispen, M. van der Loop, C.-A. Fustin, A. M. Jonas, J.-F. Gohy and U. S. Schubert, Macromolecules, 2006, 14, 4719–4725 CrossRef
- R. Weberskirch, R. Hettich, O. Nuyken, D. Schmaljohann and B. Voit, Macromol. Chem. Phys., 1999, 200, 863–873 CrossRef CAS
- R.-H. Jin and K. Motoyoshi, J. Porphyrins Phthalocyanines, 1999, 3, 60–64 CrossRef CAS
- O. Nuyken, J. Rueda-Sanchez and B. Voit, Polym. Bull., 1997, 38, 657–664 CrossRef CAS
- R.-H. Jin, J. Mater. Chem., 2003, 13, 672–675 RSC
- O. Nuyken, J. Rueda-Sanchez and B. Voit, Macromol. Rapid Commun., 1997, 18, 125–131 CrossRef CAS
- R. Hoogenboom, M. W. M. Fijiten, G. Kickebick and U. S. Schubert, Beilstein J. Org. Chem., 2010, 6, 773–783 CrossRef CAS PubMed
- J. Rueda-Sμnchez and M. C. Galloso, Macromol. Rapid Commun., 2001, 22, 859–863 CrossRef
- J. C. Rueda, S. Zschoche, H. Komber, F. Krahl, K. F. Arndt and B. Voit, Macromol. Chem. Phys., 2010, 211, 706–716 CrossRef CAS
- J. C. Rueda, H. Komber, J. C. Cedrón, B. Voit and G. Shevtsova, Macromol. Chem. Phys., 2003, 204, 947–953 CrossRef CAS
- J. N. Haigh, Y. M. Chuang, B. Farrugia, R. Hoogenboom, P. D. Dalton and T. R. Dargaville, Macromol. Rapid Commun., 2016, 37, 93–99 CrossRef CAS PubMed
- S. Kobayashi, T. Igarashi, Y. Moriuchi and T. Saegusa, Macromolecules, 1986, 19, 535–541 CrossRef CAS
- O. Nuyken, J. Rueda-Sanchez and B. Voit, Polym. Bull., 1997, 38, 657–664 CrossRef CAS
- R.-H. Jin, Chem. Commun., 2002, 198–199 RSC
- R.-H. Jin, Macromol. Chem. Phys., 2003, 204, 403–409 CrossRef CAS
- R.-H. Jin, J. Mater. Chem., 2004, 14, 320–327 RSC
- F. Wiesbrock, R. Hoogenboom, C. H. Abeln and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1895–1899 CrossRef CAS
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijis, B. M. van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659–671 CrossRef CAS
- R. Hoogenboom, M. W. M. Fijten, M. A. R. Meier and U. S. Schubert, Macromol. Rapid Commun., 2003, 24, 92–97 CrossRef CAS
- D. D. Yao and R.-H. Jin, Polym. Chem., 2015, 6, 2255–2263 RSC
- R. Tanaka, I. Ueoka, Y. Takaki, K. Kataoka and S. Saito, Macromolecules, 1983, 16, 849–853 CrossRef CAS
- R.-H. Jin and J.-J. Yuan, Macromol. Chem. Phys., 2005, 206, 2160–2170 CrossRef CAS
- R.-H. Jin and J.-J. Yuan, Chem. Mater., 2006, 18, 3390–3396 CrossRef CAS
- R. H. Jin, D. D. Yao and R. Levi, Chem.–Eur. J., 2014, 20, 7196–7214 CrossRef CAS PubMed
- J.-J. Yuan and R.-H. Jin, Langmuir, 2005, 21, 3136–3145 CrossRef CAS PubMed
- Y. Chatani, T. Kobatake and H. Tadokoro, Macromolecules, 1983, 16, 199–204 CrossRef CAS
- X.-L. Liu, S. Tsunega and R.-H. Jin, ACS Omega, 2017, 2, 1431–1440 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05329k |
| This journal is © The Royal Society of Chemistry 2017 |