
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA network similarity integration method for predicting microRNA-disease associations†
Xiaoying Li ,
Yaping Lin* and
Changlong Gu
,
Yaping Lin* and
Changlong Gu
College of Information Science and Engineer, Hunan University, Changsha, Hunan 410012, China
First published on 23rd June 2017
Abstract
Increasing evidence has indicated that microRNAs (miRNAs) regulate gene expression at the post-transcriptional level. Aberrant miRNA expression has been associated with many types of human disease, including cancers. Their associations can be used to understand the pathogenesis of diseases. However, using experimental methods to identify the associations between diseases and miRNAs is time consuming and costly. Computational methods could find the most promising miRNA-disease associations in a short time, thereby significantly reducing experimental time and cost. This paper presents a network similarity integration method (NSIM) for predicting potential miRNA-disease associations, considering that diseases associated with highly related miRNAs are more similar (and vice versa). The NSIM is based on 5425 experimentally verified human miRNA-disease associations, which consist of 495 miRNAs and 381 diseases. The NSIM integrates the disease similarity network, miRNA similarity network, and known miRNA-disease association network on the basis of cousin similarity to predict novel miRNA-disease associations. We evaluate the NSIM using leave-one-out cross validation. The area under the curve of the method is 0.9475, indicating its outstanding performance. Case studies on prostate, breast, and colon neoplasms further proved the outstanding performance of the NSIM to predict not only disease-related miRNAs but also isolated diseases (diseases without any related miRNAs).
Introduction
MicroRNAs (miRNAs) are small endogenous non-coding RNAs of about 22 nt long. MiRNAs are involved in many important biological processes, including cell development, proliferation, differentiation, apoptosis, and cellular signalling.1–6 Increasing evidence has indicated that miRNAs play important roles in the development and progression of human diseases.7–9 Aberrant miRNA expression has been associated with many types of human disease, including cancers, such as cardiovascular diseases,10 prostate neoplasms,11 and breast neoplasms.12 Therefore, prediction and identification of disease-related miRNAs are critical to understand the pathogenesis of diseases, and thereby improve disease prognosis, diagnosis, treatment, and prevention.In the last few years, many efforts have been exerted to identify potential miRNA-disease associations. Research using biological experimentation has determined a large number of miRNA-disease associations. Databases such as HMDD,13 miR2Disease,14 dbDEMC,15 miRCancer16 have been built to provide a platform for searching experimentally verified miRNA-disease associations. HMDD and miR2Disease are a collection of experimentally supported human miRNA-disease associations, manually retrieved on the basis of the literature. Database miRCancer stores miRNA-cancer associations, which are extracted using the rule-based text mining method. In addition, dbDEMC stores differentially expressed miRNAs in 14 human cancers by using significance analysis of microarrays to retrieve the miRNAs that have different expression levels in cancers when compared with normal tissues. These databases serve as a solid data foundation for predictive research of miRNAs in human diseases.
Considering that the experimental identification of disease-related miRNAs is time consuming and expensive, researchers proposed computational methods as important complementary ways to predict miRNA-disease associations. Computational methods mainly aim to select the most promising disease-related miRNAs for further experimental examination to reduce experimental time and cost. The key problem in miRNA-disease association inference is similarity calculation. These computational methods are divided into two categories:17 network-based methods18–26 and machine-learning-based methods.26–30
Network-based methods predict miRNA-disease associations in consideration of the hypothesis that functionally related miRNAs are usually associated with phenotypically similar diseases.13 This hypothesis was proposed by Lu et al.13 when they analyzed the human miRNA-disease association data in HMDD. Basing on this hypothesis, Jiang et al.18 constructed a functional association miRNA network, i.e., a human phenome-miRNAome network. For a given disease, they computed the similarity score of all human miRNAs in these networks and then prioritized all these miRNAs according to score. The top-ranked miRNAs were expected as the potential disease miRNAs. However, this model uses only the neighboring information of each miRNA and strongly relies on predicted miRNA-target interactions, thereby producing false-positive and false-negative results, that can influence the final prediction accuracy. Shi et al.21 presented a computational framework to identify miRNA-disease associations and further constructed a bipartite miRNA-disease network for systematically analyzing the global properties of miRNA regulation of disease genes. From these analyses, they found that most diseases in the same co-regulated module belong to the same category. Their work extended the previous hypothesis. However, this method is limited in application because of the low accuracy of target prediction and the fact that many disease–gene associations of miRNA-target interactions are unknown. On the basis of the weighted k most similar neighbours, HDMP22 was proposed to predict disease-related miRNAs. HDMP was used to evaluate the function similarity between miRNAs by considering disease terms and the phenotype similarity between diseases, as well as assigning higher weight to members of the miRNA family or cluster. However, HDMP only considers local network similarity measure and disregards diseases without any known related miRNA. Recently, Zou et al.26 have presented method KATZ, which uses the functional similarity score to denote the associations on the basis of the different lengths between the miRNA and disease nodes. However, the performance of KATZ is relatively poor on the spare known associations.
Machine-learning-based methods have been used to solve the problem by improving the classification accuracy and prediction performance. Jiang et al.29 proposed a Naïve Bayes model to rank candidate disease-related miRNAs through genomic data integration. This method strongly relies on datasets of disease–gene associations and miRNA-target interactions, but over half of human diseases are still unknown. To distinguish positive miRNA-disease associations from negative ones, Jiang et al.27 proposed a support vector machine approach by extracting the features based on miRNA-target data and phenotype similarity data. Considering the assumption that miRNAs implicated in a specific tumor phenotype show aberrant regulation of their target genes, Xu et al.30 prioritized novel disease miRNAs on the basis of the miRNA target-dysregulated network method. The common problem of the two aforementioned methods is that the negative training samples consisting of non-association between miRNAs and diseases do not demonstrate sufficient statistical confidence; the lack of a miRNA-disease association during observation in a biological experiment does not directly indicate absence of such an association. Chen et al.28 developed regularized least squares for miRNA-disease association (RLSMDA) to find potential miRNA candidates for a specific disease. RLSMDA is a semi-supervised method that integrates known disease-miRNA associations, disease–disease similarity dataset, and miRNA–miRNA functional similarity network. Despite its good prediction performance for diseases with or without related miRNAs, RLSMDA does not consider the topology information of the miRNA network.
The aforementioned methods have three main limitations. First, some methods are inefficient at cross-validation. Second, some approaches are unable to predict isolated disease-related miRNAs. Third, negative samples are difficult to obtain for some machine learning methods. Consequently, we propose a network similarity integration method (NSIM) to solve these limitations. The NSIM integrates miRNA similarities, diseases similarities, and known miRNA-disease association information to predict potential miRNA-disease associations. The advantages of the NSIM are as follows. First, this method is easy to understand and can effectively be implemented. Cross validations and global predictions for all 381 diseases are run simultaneously. Second, case studies about prostate, breast, and colon neoplasms demonstrate that the NSIM has good predictive performance. Third, the NSIM can also predict isolated diseases.
Materials
Dataset
The database we used in this study contains data on miRNA–miRNA functional similarities, disease semantic similarities, and known human miRNA-disease associations. Below is a brief description of these data.Known human miRNA-disease associations
Data on known human miRNA-disease were downloaded from HMDD 2.0 (ref. 13) (http://www.cuilab.cn/hmdd, Jun-14-2014 Version). We removed duplicated associations and those associations whose disease could not be mapped to the MeSH database or whose disease did not have a related MeSH tree number. After filtering, we finally received 5425 high-quality experimentally verified human miRNA-disease associations consisting of 495 miRNAs and 381 diseases in the dataset. Matrix AS denotes miRNA-disease associations and AS(i,j) = 1 means there exists a validated association between miRNA i and disease j; otherwise, AS(i,j) = 0.Disease directed acyclic graph
In our study, a functional similarity score for each disease pair was calculated based on the hypothesis that miRNAs with similar functions used to be associated with similar diseases. We improved the detailed description provided by ref. 19 about the calculated method. The diseases are mapped to the MeSH database (the website is http://www.ncbi.nlm.nih.gov/), and their MeSH headings (or called descriptors) are downloaded. Each MeSH heading shows a tree structure of a hierarchical organization. This tree structure of a disease is described as a directed acyclic graph (DAG). The nodes of the tree represent diseases while the edges represent the relationship between the parent node and their children nodes. The higher the hierarchy of a node is, the more general its meaning is. Otherwise, the lower the hierarchy is, the more specific its meaning is. The DAG of gastrointestinal neoplasms is shown in Fig. 1.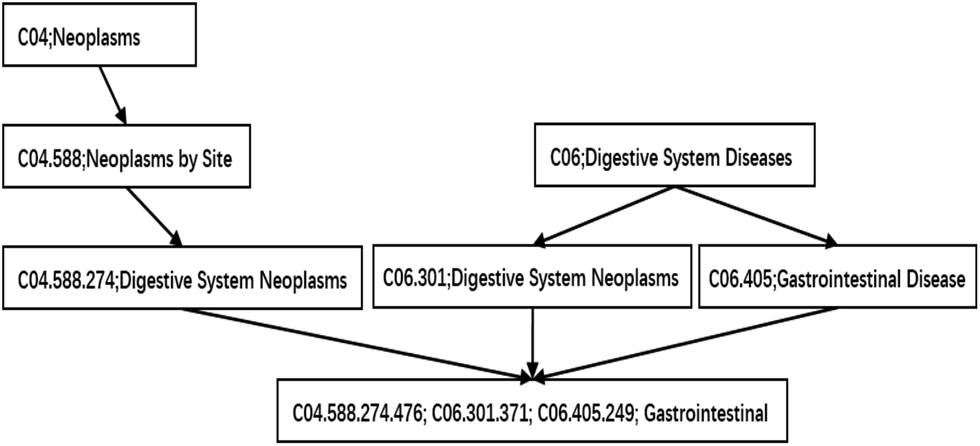 | ||
| Fig. 1 The disease DAG of gastrointestinal neoplasms. | ||
Methods
Based on the information of experimentally validated miRNA-disease association network and two common assumptions, we reconstructed miRNA and disease similarity networks, and employed the NSIM to predict potential miRNA-disease associations. One of the assumptions is miRNAs with similar functions are normally associated with phenotypically similar diseases and vice versa,13,31 and the other is diseases with similar functions are often having similar semantic descriptions and vice versa.19 The NSIM contains four processes. First, it calculates the semantic similarity score of diseases according to the semantic tree structure. Second, it calculates miRNA–miRNA functional similarities based on the semantic similarity score of diseases. A miRNA functional network was built on the basis of these calculations. Third, it calculates the similarity score of diseases to reconstruct a disease similarity network by considering the disease semantic similarities and disease similarities of known miRNA-disease associations. Fourth, it integrates the disease similarities, miRNA similarities, and known miRNA-disease associations to predict potential associations between miRNAs and diseases. The flowchart of the NSIM is shown in Fig. 2.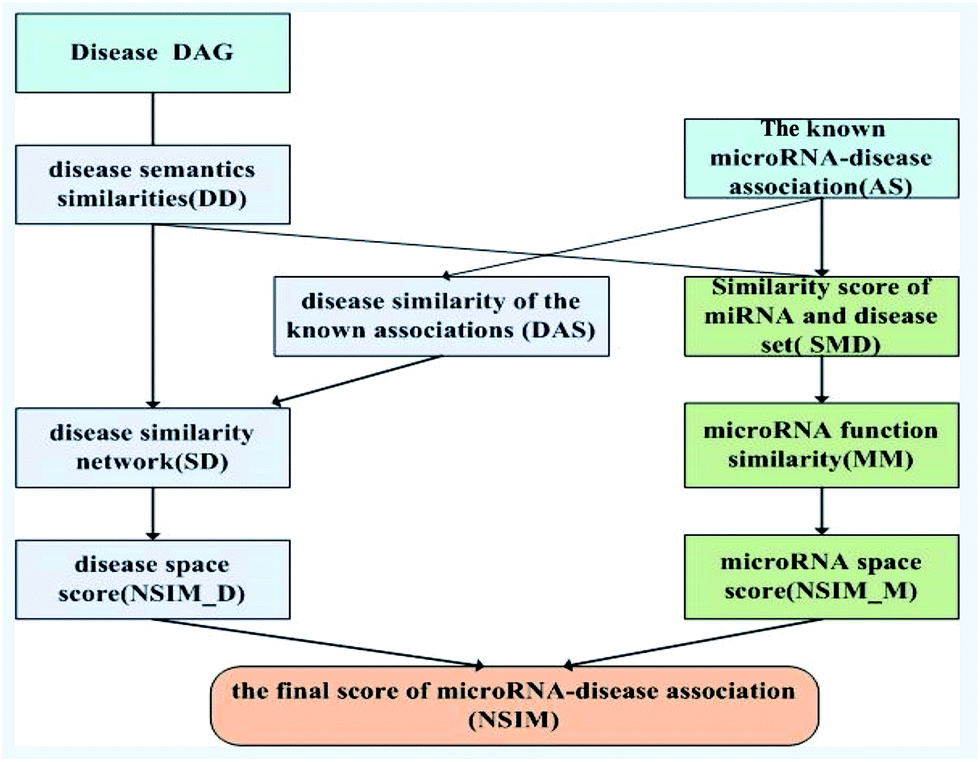 | ||
| Fig. 2 The flowchart of NSIM. | ||
Measurement of disease semantic similarities
Some researchers have measured the similarity of diseases by the hierarchical structure of disease semantics.19,32 In this work, the semantic similarity measure for disease is developed based on Wang et al.,19 but not the same as it.A disease A can be represented as a graph, DAG(A) = (A, TA, EA), where TA is the set of all ancestor nodes of A including A itself and EA is the set of corresponding links of A. The contribution of ancestor node t to A is defined as follows:
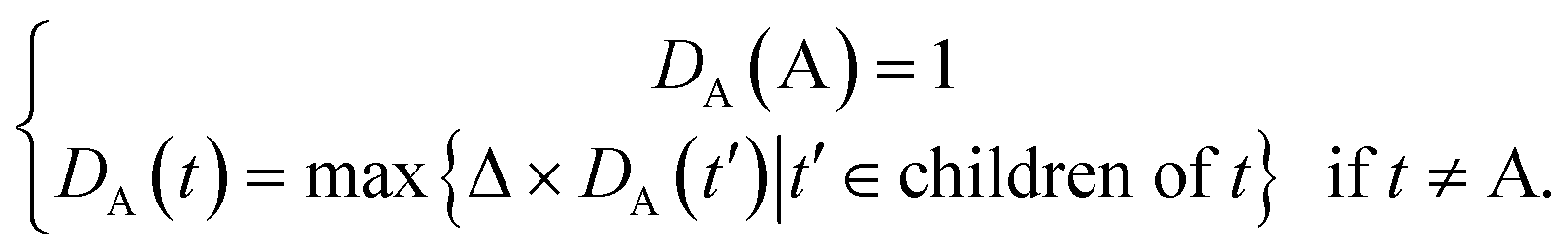 | (1) |
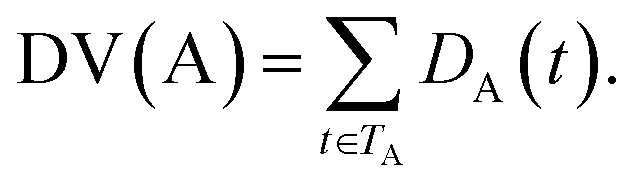 | (2) |
The semantic similarity score of disease A and disease B is defined as:
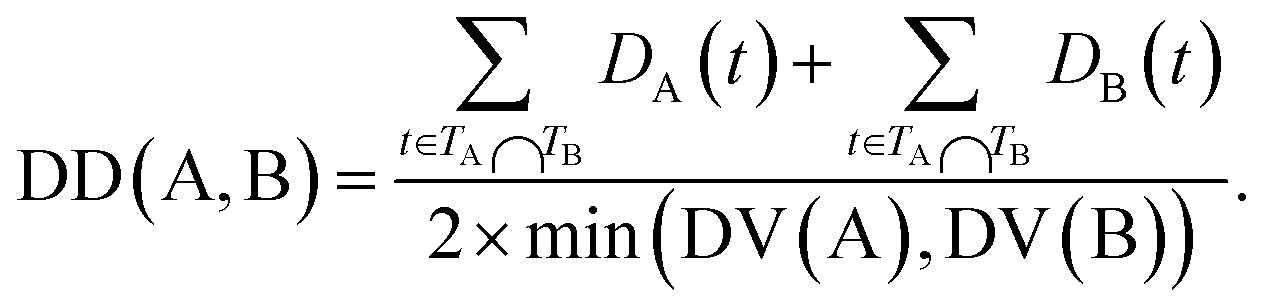 | (3) |
Measurement of miRNA functional similarity
We define DSj = {d1,d2,⋯,dn}, the disease set associated with miRNA j. The related score between disease d ∈ DSi and set DSj is defined as follows:
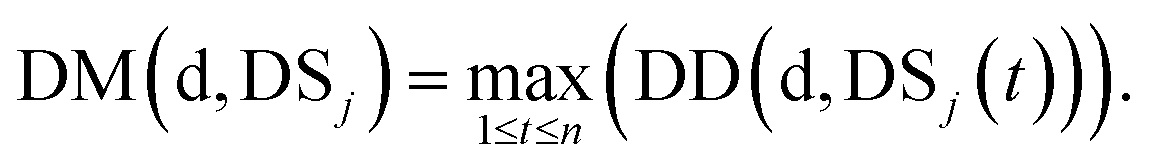 | (4) |
Here, we define the maximum similarity of disease d and diseases in DSj as the related score between disease d and miRNA j.
We define matrix MM as the miRNA–miRNA function similarity matrix, where MM(i,j) in row i and column j expresses the functional similarity score between miRNA i and miRNA j. By considering the contribution of the similarity diseases, the functional similarity of MM(i,j) is calculated as follows:
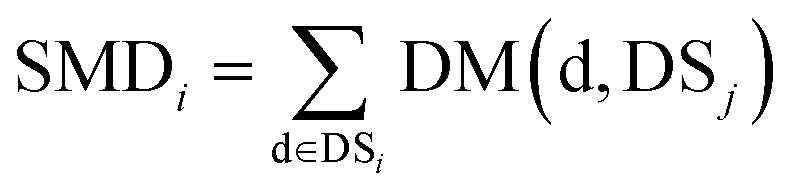
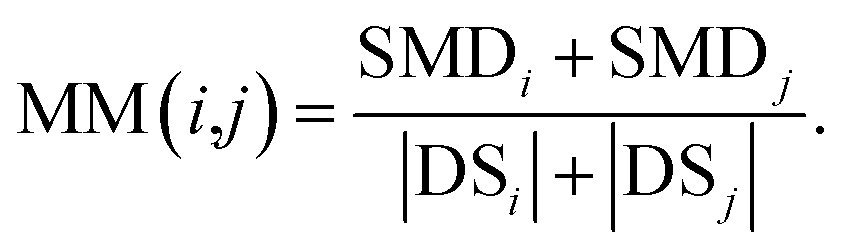 | (5) |
Reconstruction of a disease similarity network
A disease similarity network was reconstructed by considering the disease semantic similarities and disease similarities of known miRNA-disease associations. Considering the assumption that the more common miRNAs of a disease pair has, the more similar they are, we define the disease similarity value of a known disease-miRNA association on the basis of matrix AS and Jaccard similarity measurement as
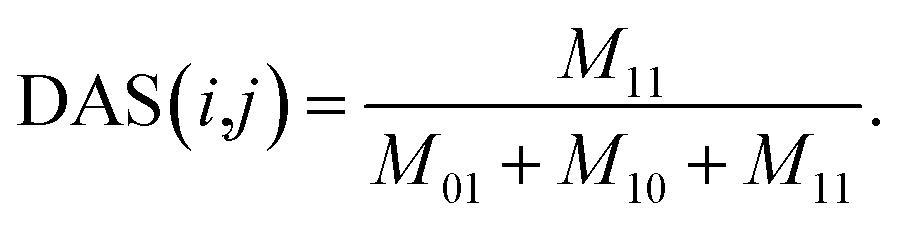 | (6) |
Considering disease i and disease j in matrix AS, we count the total number of commonly associated miRNAs of disease i and j, and then define it as M11. Similarly, M01 represents the total number of miRNAs that are only associated with disease i, M10 represents the total number of miRNAs that are only associated with disease j. The total number of miRNAs that are not associated with neither i nor j is disregarded. For a certain disease pair, the similarity value is set to 0 when the total number of miRNAs associated with these two diseases is zero.
We reconstruct the disease similarity network as:
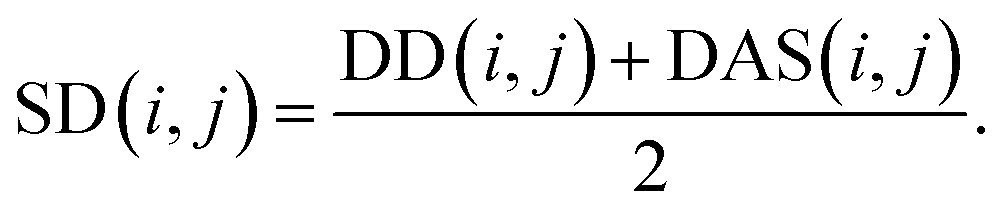 | (7) |
NSIM for miRNA-disease associations
The NSIM calculates the potential miRNA-disease association scores by integrating the miRNA and disease vector space score. Cosine similarity is employed to calculate the vector space score.In the miRNA vector space, the similarity between miRNA i and all miRNAs is described as a vector VMMi, and MMi, (the ith row of matrix MM) is used to represent it. Likewise, the similarity between the associations of disease j and all miRNAs is described as a vector VDj, and ASj (the jth column of matrix AS) is used to represent it.
| VMMi = MMi, |
| VDj = ASj |
The miRNA space score is defined as
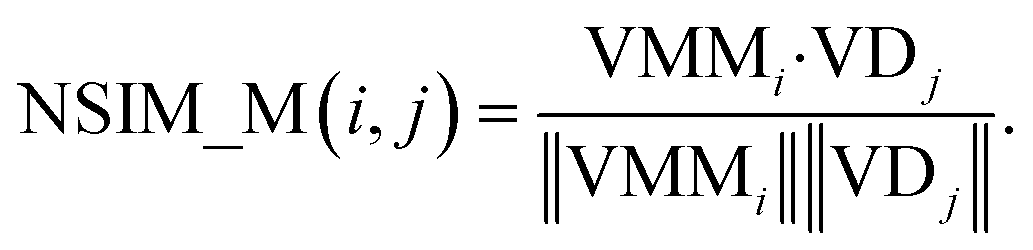 | (8) |
Obviously, the higher the spatial similarity of miRNA i-associated miRNAs in the miRNA–miRNA similarity network is, the greater the association between miRNA i and disease j is. Similarly, the higher the spatial similarity of disease j-associated miRNAs in the known miRNA-disease network is, the greater the association between miRNA i and disease j is.
In the disease vector space, the similarity between the associations of miRNA i and all diseases is described as a vector VMi. We could use ASi, (the ith row of matrix AS) to represent it. Similarly, the similarity between disease j and all diseases is described as vector VSDj, and we could use SDj (the jth column of matrix SD) to represent it.
| VMi = ASi, |
| VSDj = SDj |
The disease space score is defined as
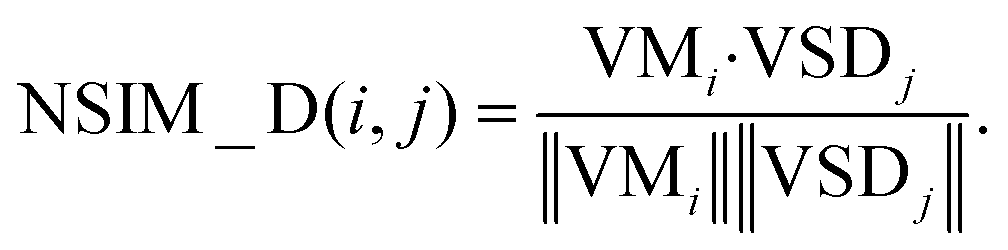 | (9) |
Obviously, that the higher the spatial similarity of miRNA i-associated diseases in the known miRNA-disease network is, the greater the association of miRNA i and disease j is. Likewise, the higher the spatial similarity of the disease j associated diseases in disease similarity network is, the greater the association of miRNA i and disease j is.
Finally, the miRNA space score and disease space score are integrated together as
| NSIM(i,j) = α × NSIM_M(i,j) + (1 − α) × NSIM_D(i,j) | (10) |
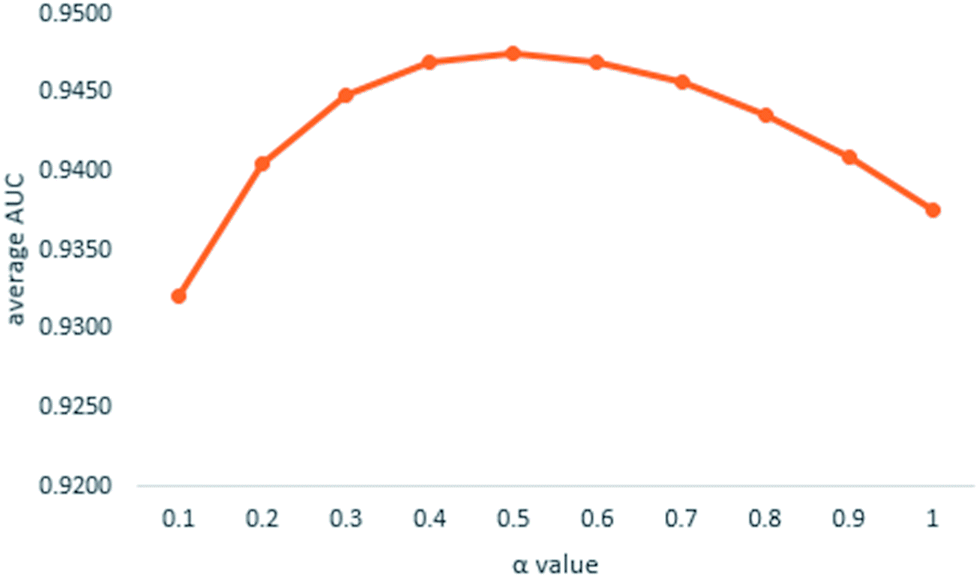 | ||
| Fig. 3 Average AUCs affected by α value. | ||
Results
Performance evaluation of the NSIM
In our study, we implemented leave-one-out cross validation (LOOCV) on experimentally verified miRNA-disease associations to evaluate the predictive performance of the NSIM. Each known miRNA-disease association was left out in turn as a test sample, and other known miRNA-disease associations were taken as a training set. A receiver operating characteristic (ROC) curve was plotted by varying the threshold, and the value of area under curve (AUC) was calculated. In the ROC, the vertical and horizontal axes are the true positive rate (TPR, sensitivity) and false positive rate (FPR, 1-specificity) at different thresholds, respectively. Sensitivity refers to the percentage of test miRNAs with ranking above a given threshold, whereas specificity refers to the percentage of associations below the threshold. When the AUC is closer to 1, the prediction performance is better.To our knowledge, HDMP,22 RLSMDA,28 KATZ,26 and the global network algorithm developed by Shi et al.21 are the-state-of-art computational approaches to predict miRNA-disease associations. We compared NSIM with RLSMDA and KATZ. HDMP could not predict disease without known associated miRNAs; the method developed by Shi et al. integrated the dataset from disease gene associations, miRNA-target interactions, and protein interactions, which were different from the dataset used in the NSIM.
We implemented a LOOCV for RLSMDA and KATZ. In the present study, the NSIM achieved an AUC value of 0.9475 when α is 0.5. For RLSMDA, when optimal parameters were selected as described in the literature, the AUC value was 0.8870. For KATZ, the AUC value was 0.9202. The comparison result of overall AUC between NSIM and RLSMDA, KATZ is shown in Fig. 4.
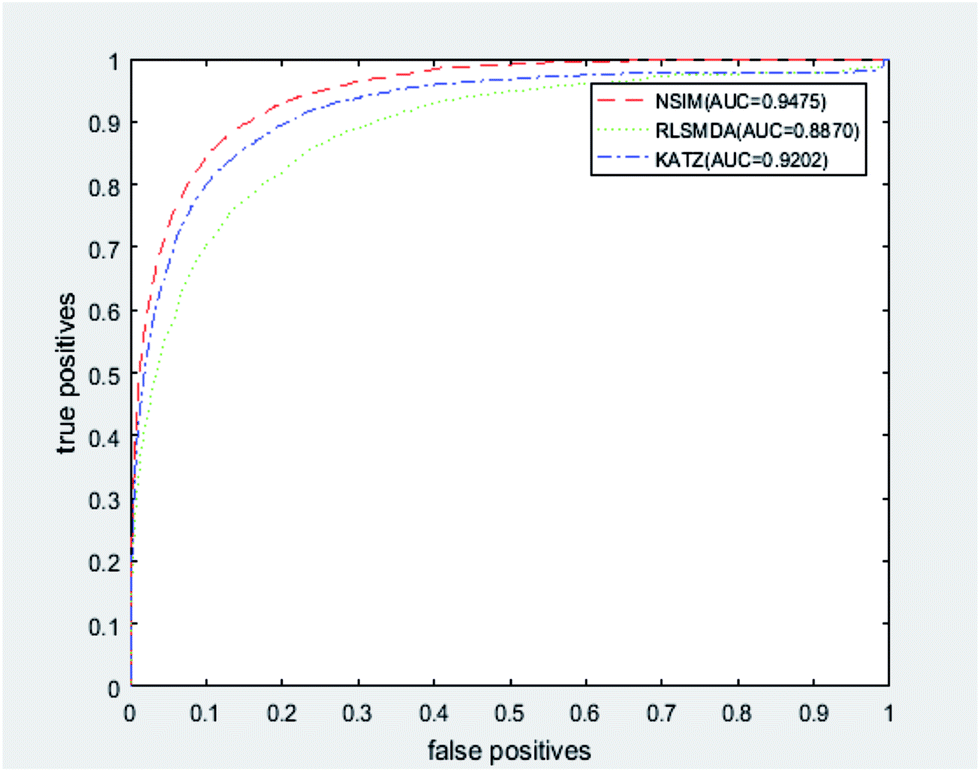 | ||
| Fig. 4 The comparison result between NSIM, RLSMDA and KATZ was shown, which demonstrated the superiority performance of NSIM to other two methods. | ||
To obtain reliable judgment, we tested 19 human diseases that are related to at least 70 microRNAs respectively. As shown in Table 1, the NSIM achieved the highest AUC of 0.9446 with lung neoplasms and the lowest AUC of 0.8813 with esophageal neoplasms. The average AUC value for the 19 diseases was 0.9125 (Table 1). For RLSMDA, the average AUC value for the 19 diseases was 0.8450. The average AUC value was increased by 6.75%. For KATZ, the average AUC value for the 19 diseases was 0.8945. The average AUC value of the NSIM was 1.8% higher than that of KATZ. The AUC values of the NSIM for of neoplasms and ovarian neoplasms were lower than those of RLSMDA and KATZ. The AUC values of the NSIM for the 17 other diseases were all higher than those of RLSMDA and KATZ. Obviously, the prediction performance of NSIM was more accurate than those of RLSMDA and KATA.
| Disease name | Number of associated microRNAs | AUC | ||
|---|---|---|---|---|
| NSIM | RLSMDA | KATZ | ||
| Breast neoplasms | 202 | 0.9353 | 0.8951 | 0.9296 |
| Carcinoma, hepatocellular | 214 | 0.9119 | 0.8631 | 0.9012 |
| Carcinoma, non-small-cell lung | 95 | 0.9031 | 0.8342 | 0.8800 |
| Carcinoma, renal cell | 107 | 0.8926 | 0.8172 | 0.875 |
| Carcinoma, squamous cell | 80 | 0.9048 | 0.8386 | 0.8895 |
| Colonic neoplasms | 78 | 0.8834 | 0.8232 | 0.8728 |
| Colorectal neoplasms | 147 | 0.8845 | 0.8461 | 0.8819 |
| Esophageal neoplasms | 74 | 0.8813 | 0.7747 | 0.8466 |
| Glioblastoma | 96 | 0.9006 | 0.7934 | 0.8595 |
| Glioma | 71 | 0.9131 | 0.8704 | 0.9114 |
| Heart failure | 120 | 0.9071 | 0.8454 | 0.8636 |
| Lung neoplasms | 132 | 0.9446 | 0.7844 | 0.9249 |
| Melanoma | 141 | 0.9185 | 0.8850 | 0.8903 |
| Neoplasms | 110 | 0.9436 | 0.8339 | 0.9751 |
| Ovarian neoplasms | 114 | 0.9286 | 0.9630 | 0.9271 |
| Pancreatic neoplasms | 99 | 0.9312 | 0.8991 | 0.9126 |
| Prostatic neoplasms | 118 | 0.9209 | 0.8665 | 0.883 |
| Stomach neoplasms | 174 | 0.9104 | 0.8217 | 0.8984 |
| Urinary bladder neoplasms | 92 | 0.9227 | 0.8493 | 0.8732 |
Comprehensive prediction of unknown associations
The NSIM was utilized to predict unknown microRNA-disease associations. Initially, the related score of each microRNA-disease pair was calculated by using all known and experimented microRNA-disease associations. Then, the unknown associations were ranked by their scores. Finally, the top 50 associations were manually verified through two databases: dbDEMC (the database is being upgraded, the experimental verified microRNA-disease associations are obtained from the author) and miRCancer. The predicted results are listed in Table S1 (ESI†), and their verified evidences is presented. For the top 50 predictive associations, all 50 had been confirmed in the aforementioned databases.Case studies
Many researchers have found more and more evidences that microRNAs are related with various human cancers.8,33343536 To further evaluate the performance of the NSIM to predict disease-associated miRNA candidates, we selected prostate, breast, and colon neoplasms as case studies.Prostate neoplasm is the most common cancer among males in 84 countries,35 especially in developed countries. Prostate neoplasm is the second most common type of cancer and the fifth leading cause of cancer-related death among men worldwide.37 MiRNAs are over expressed during the progression of prostate neoplasms. Thus, miRNAs are promising diagnostic or prognostic biomarkers. For example, miR-409-3p, miR-361-3p, miR-133b, miR-221, and miR-128 are under expressed and miR-375, miR-141, miR-378*, and miR-203 are upregulated in prostate cancer.38–40 Candidate miRNAs were ranked in terms of scores obtained from the NSIM. The top 20 potential miRNAs associated with prostate neoplasms and evidence for the associations with prostate are listed in Table 2. Among the top 20 predicted prostate-related miRNAs, 18 have been confirmed by dbDEMC or miRCancer. Unconfirmed potential miRNA with the highest rank is has-mir-17 (ranked 4th). However, we found in the literature41,42 that the miR-17 family is over expressed in prostate neoplasms by targeting the p300/CBP-associated factor and modulating androgen receptor transcriptional activity in cultured prostate neoplasms cells.
| Rank | miRNA | Evdances |
|---|---|---|
| 1 | Hsa-mir-182 | dbDEMC, miRCancer |
| 2 | Hsa-mir-143 | dbDEMC, miRCancer |
| 3 | Hsa-mir-21 | dbDEMC, miRCancer |
| 4 | Hsa-mir-17 | PMID: 27650539 |
| 5 | Hsa-mir-34a | dbDEMC, C |
| 6 | Hsa-mir-100 | dbDEMC, miRCancer |
| 7 | Hsa-mir-126 | dbDEMC |
| 8 | Hsa-mir-150 | dbDEMC |
| 9 | Hsa-mir-20a | miRCancer |
| 10 | Hsa-mir-142 | Unconfirmed |
| 11 | Hsa-mir-200a | dbDEMC |
| 12 | Hsa-mir-203 | miRCancer |
| 13 | Hsa-mir-141 | miRCancer |
| 14 | Hsa-mir-31 | dbDEMC, miRCancer |
| 15 | Hsa-mir-146a | miRCancer |
| 16 | Hsa-mir-96 | dbDEMC, miRCancer |
| 17 | Hsa-mir-200c | dbDEMC |
| 18 | Hsa-mir-200b | miRCancer |
| 19 | Hsa-mir-223 | dbDEMC, miRCancer |
| 20 | Hsa-mir-9 | dbDEMC |
Breast neoplasm is the most common invasive cancer among women especially in developed countries, accounting for 25% of cancer cases among women. MiRNAs play regulatory roles in the invasion and metastasis of breast neoplasms. For example, miR-182, miR-21 are over expressed in breast neoplasms,12,35 and miR-205, miR-200c, miR-141, and miR-429 are down regulated in breast cancer.43 The top 20 potential miRNAs associated with breast neoplasms and evidence for the associations with breast are listed in Table 3. Among these candidate miRNAs, only 4 were not confirmed in the dbDEMC or miRCancer dataset. However, the literature44 provided information that miRNA hsa-mir-542 induces angiogenic inhibition in breast neoplasms.
| Rank | miRNA | Evidences |
|---|---|---|
| 1 | Hsa-mir-99a | dbDEMC, miRCancer |
| 2 | Hsa-mir-138 | dbDEMC |
| 3 | Hsa-mir-142 | miRCancer |
| 4 | Hsa-mir-106a | dbDEMC |
| 5 | Hsa-mir-130a | dbDEMC, miRCancer |
| 6 | Hsa-mir-378a | Unconfirmed |
| 7 | Hsa-mir-150 | dbDEMC, miRCancer |
| 8 | Hsa-mir-185 | dbDEMC, miRCancer |
| 9 | Hsa-mir-15b | dbDEMC |
| 10 | Hsa-mir-98 | dbDEMC, miRCancer |
| 11 | Hsa-mir-192 | dbDEMC |
| 12 | Hsa-mir-542 | PMID: 26272182 |
| 13 | Hsa-mir-196b | dbDEMC |
| 14 | Hsa-mir-92b | dbDEMC |
| 15 | Hsa-mir-186 | dbDEMC |
| 16 | Hsa-mir-30e | Unconfirmed |
| 17 | Hsa-mir-372 | dbDEMC |
| 18 | Hsa-mir-130b | dbDEMC |
| 19 | Hsa-mir-370 | dbDEMC |
| 20 | Hsa-mir-449a | Unconfirmed |
Colon neoplasm is the third most common cancer in the digestive tract worldwide. MiRNAs can be accurately diagnosed as biomarkers of colon neoplasms and can help predict colon neoplasms.45,46 MiRNA differential expression provides a promising application for early diagnosis and screening of colon neoplasms. For example, miR-21, miR-155, miR-31, miR-92a, and miR-17 are involved in the development of colon neoplasms.47 The top 20 potential miRNAs associated with colon neoplasms and evidence for the associations with colon neoplasms are listed in Table 4. Among these candidate miRNAs, 5 were not confirmed by the dbDEMC or miRCancer dataset. Nevertheless, they all have been identified in the literature. The PMID of the literature is shown in the tables.
| Rank | miRNA | Evidences |
|---|---|---|
| 1 | Hsa-mir-20a | dbDEMC |
| 2 | Hsa-mir-18a | dbDEMC, miRCancer |
| 3 | Hsa-mir-19b | dbDEMC |
| 4 | Hsa-mir-21 | dbDEMC, miRCancer |
| 5 | Hsa-mir-143 | dbDEMC, miRCancer |
| 6 | Hsa-mir-19a | dbDEMC |
| 7 | Hsa-mir-155 | dbDEMC, miRCancer |
| 8 | Hsa-mir-92a | PMID: 26463716 |
| 9 | Hsa-mir-125b | PMID: 24774301 |
| 10 | Hsa-mir-29b | PMID: 26466603 |
| 11 | Hsa-mir-34a | dbDEMC, miRCancer |
| 12 | Hsa-mir-146a | dbDEMC |
| 13 | Hsa-mir-16 | PMID: 22049153 |
| 14 | Hsa-mir-106b | dbDEMC |
| 15 | Hsa-let-7a | miRCancer |
| 16 | Hsa-mir-181a | dbDEMC, miRCancer |
| 17 | Hsa-mir-31 | dbDEMC, miRCancer |
| 18 | Hsa-mir-15a | dbDEMC |
| 19 | Hsa-mir-150 | PMID: 24705249 |
| 20 | Hsa-mir-221 | dbDEMC |
The above results demonstrate that the NSIM performs well in predicting potential disease-associated miRNA candidates.
Application of NSIM to predict isolated diseases
An isolated disease refers to a disease without any known related miRNAs. To demonstrate the predictive ability of NSIM on diseases without any known related miRNA, we removed the known verified miRNA-disease associations related to predictive diseases. This operation ensured that we only used known miRNA-disease association and similarity information of other diseases to predict candidate miRNAs related to the given disease while prioritizing these candidate miRNAs.We take isolated disease j as an example, VDj = ASj = null vector and NSIM_M(i,j) = 0. The predictor score between miRNA i and disease j is calculated by NSIM_D(i,j). The disease similarity consists of disease semantic similarities (eqn (3) DD) and disease similarities of known miRNA-disease associations (eqn (6) DAS). When disease j is an isolated disease, DAS(i,j) = null vector; and DD calculated by disease MeSH DAG, do not depend on the associated miRNAs. What we call isolated disease refers to a disease without any known related miRNAs, and the associations between the disease and other diseases exists. So we use DD(i,j) as SD(i,j) to calculate NSIM_D(i,j). Therefore, our method can be applied to predict isolated disease-related miRNAs.
The average AUC of NSIM to predict isolated diseases is 0.8146. The predicted results of breast neoplasms are listed in Table 5.
| Rank | miRNA name | Evidences |
|---|---|---|
| 1 | Hsa-mir-99a | dbDEMC, miRCancer |
| 2 | Hsa-mir-663b | Unconfirmed |
| 3 | Hsa-mir-138 | dbDEMC |
| 4 | Hsa-mir-331 | dbDEMC |
| 5 | Hsa-mir-185 | dbDEMC, miRCancer |
| 6 | Hsa-mir-372 | dbDEMC |
| 7 | Hsa-mir-378a | Unconfirmed |
| 8 | Hsa-mir-1224 | Unconfirmed |
| 9 | Hsa-mir-130a | dbDEMC, miRCancer |
| 10 | Hsa-mir-98 | dbDEMC, miRCancer |
| 11 | Hsa-mir-532 | dbDEMC |
| 12 | Hsa-mir-370 | dbDEMC |
| 13 | Hsa-mir-542 | Unconfirmed |
| 14 | Hsa-mir-498 | dbDEMC |
| 15 | Hsa-mir-371a | Unconfirmed |
| 16 | Hsa-mir-142 | miRCancer |
| 17 | Hsa-mir-130b | dbDEMC |
| 18 | Hsa-mir-150 | dbDEMC, miRCancer |
| 19 | Hsa-mir-449a | Unconfirmed |
| 20 | Hsa-mir-15b | dbDEMC |
Discussion
The recommendable performance of NSIM could be mainly attributed to the several factors. First, NSIM is a prediction method based on experimentally confirmed microRNA-disease associations. It integrates scores from disease space and microRNA space to construct a global network, which improves prediction accuracy. Second, the NSIM is an understandable method involving only one parameter, which is easy to adjust. Furthermore, new diseases (isolated diseases without any known related microRNA) are constantly being discovered. Thus, computational methods are used to predict isolated diseases. The NSIM performs well in predicting isolated diseases.The current version of NSIM has limitations. Despite its good performance, the NSIM was constructed on basis of miRNA-disease associations. The number of associations affected the prediction accuracy. The more the number of associations, the more accurate the prediction is. Hence, the performance of the NSIM could be improved by obtaining more miRNA-disease associations. Furthermore, this method only considers the semantic relation in calculating the disease similarity score. Information on gene-disease, miRNA-lncRNA, and miRNA-target gene interactions could further improve the similarity measure between miRNAs and diseases.
Conclusions
Predicting potential microRNA-disease associations through computational methods can provide support for experimental studies on microRNAs. In this study, we proposed the NSIM to predict miRNA-disease associations by integrating miRNAs similarities, disease similarities, and known miRNA-disease associations. The NSIM obtained a high AUC of 0.9475 in LOOCV. Furthermore, case studies of prostate, breast, and colon neoplasms were implemented, and 19, 17, and 20 miRNAs in the top 20 prediction list were confirmed, respectively. These results demonstrate that NSIM can effectively identify potential disease-related miRNAs. NSIM also performs well in predicting isolated diseases. The results demonstrated that the performance of the NSIM is superior to that of other existing prediction methods. The NSIM could be an effective biological tool that can be extended to research on drug-disease and environmental factor–disease associations.References
- A. M. Krichevsky, K. S. King, C. P. Donahue, K. Khrapko and K. S. Kosik, RNA, 2003, 9, 1274–1281 CrossRef CAS PubMed.
- Q. H. Cui, Z. B. Yu, E. O. Purisima and E. Wang, Mol. Syst. Biol., 2006, 2 Search PubMed.
- T. Du and P. D. Zamore, Cell Res., 2007, 17, 661–663 CrossRef CAS PubMed.
- E. Berezikov, E. Cuppen and R. H. Plasterk, Nat. Genet., 2006,(38), S2–S7 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2009, 136 Search PubMed.
- E. A. Miska, Curr. Opin. Genet. Dev., 2005, 15, 563–568 CrossRef CAS PubMed.
- J. Kim, K. Inoue, J. Ishii, W. B. Vanti, S. V. Voronov, E. Murchison, G. Hannon and A. Abeliovich, Science, 2007, 317 Search PubMed.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS PubMed.
- J. P. Cogswell, J. Ward, I. A. Taylor, M. Waters, Y. Shi, B. Cannon, K. Kelnar, J. Kemppainen, D. Brown and C. Chen, J. Alzheimer's Dis., 2008, 14 Search PubMed.
- P. K. Mishra, N. Tyagi, M. Kumar and S. C. Tyagi, J. Cell. Mol. Med., 2009, 13, 778–789 CrossRef CAS PubMed.
- J. Ribas, X. H. Ni, M. Haffner, E. A. Wentzel, A. H. Salmasi, W. H. Chowdhury, T. A. Kudrolli, S. Yegnasubramanian, J. Luo, R. Rodriguez, J. T. Mendell and S. E. Lupold, Cancer Res., 2009, 69, 7165–7169 CrossRef CAS PubMed.
- M. Han, Y. Wang, M. Liu, X. Bi, J. Bao, N. Zeng, Z. Zhu, Z. Mo, C. Wu and X. Chen, Cancer Sci., 2012, 103, 1058–1064 CrossRef CAS PubMed.
- M. Lu, Q. Zhang, M. Deng, J. Miao, Y. Guo, W. Gao and Q. Cui, PLoS One, 2008, 3 CAS.
- Q. Jiang, Y. Wang, Y. Hao, L. Juan, M. Teng, X. Zhang, M. Li, G. Wang and Y. Liu, Nucleic Acids Res., 2009, 37 Search PubMed.
- Z. Yang, F. Ren, C. N. Liu, S. M. He, G. Sun, Q. A. Gao, L. Yao, Y. D. Zhang, R. Y. Miao, Y. Cao, Y. Zhao, Y. Zhong and H. T. Zhao, BMC genomics, 2010, 11 CAS.
- B. Xie, Q. Ding, H. Han and D. Wu, Bioinformatics, 2013, 29, 638–644 CrossRef CAS PubMed.
- X. Zeng, X. Zhang and Q. Zou, Briefings Bioinf., 2016, 17, 193–203 CrossRef PubMed.
- Q. H. Jiang, Y. Y. Hao, G. H. Wang, L. R. Juan, T. J. Zhang, M. X. Teng, Y. L. Liu and Y. D. Wang, BMC Syst. Biol., 2010, 4 Search PubMed.
- D. Wang, J. A. Wang, M. Lu, F. Song and Q. H. Cui, Bioinformatics, 2010, 26, 1644–1650 CrossRef CAS PubMed.
- X. Chen, M. X. Liu and G. Y. Yan, Mol. BioSyst., 2012, 8 Search PubMed.
- H. Shi, J. Xu, G. Zhang, L. Xu, C. Li, L. Wang, Z. Zhao, W. Jiang, Z. Guo and X. Li, BMC Syst. Biol., 2013, 7, 1–12 CrossRef PubMed.
- P. Xuan, K. Han, M. Guo, Y. Guo, J. Li, J. Ding, Y. Liu, Q. Dai, J. Li, Z. Teng and Y. Huang, PLoS One, 2013, 8, e70204 CAS.
- H. Shi, G. Zhang, M. Zhou, L. Cheng, H. Yang, J. Wang, J. Sun and Z. Wang, PLoS One, 2016, 11, e0148521 Search PubMed.
- X. Chen, C. C. Yan, X. Zhang, Z. H. You, L. X. Deng, Y. Liu, Y. D. Zhang and Q. H. Dai, Sci. Rep., 2016, 6 Search PubMed.
- J. Luo, P. Ding, C. Liang, B. Cao and X. Chen, IEEE/ACM Trans. Comput. Biol. Bioinf., 2016 DOI:10.1109/TCBB.2016.2599866.
- Q. Zou, J. Li, Q. Hong, Z. Lin, Y. Wu, H. Shi and Y. Ju, BioMed Res. Int., 2015, 2015, 810514 Search PubMed.
- Q. Jiang, G. Wang, S. Jin, Y. Li and Y. Wang, Int. J. Data Min. Bioinform., 2013, 8, 282–293 CrossRef PubMed.
- X. Chen and G. Y. Yan, Sci. Rep., 2014, 4, 5501 CrossRef CAS PubMed.
- Q. Jiang, G. Wang and Y. Wang, An approach for prioritizing disease-related microRNAs based on genomic data integration, Biomedical Engineering and Informatics (BMEI), 2010 3rd International Conference On, IEEE, 2010, vol. 6, pp. 2270–2274 Search PubMed.
- J. Xu, C. X. Li, J. Y. Lv, Y. S. Li, Y. Xiao, T. T. Shao, X. Huo, X. Li, Y. Zou, Q. L. Han, X. Li, L. H. Wang and H. Ren, Mol. Cancer Ther., 2011, 10, 1857–1866 CrossRef CAS PubMed.
- S. Bandyopadhyay, R. Mitra, U. Maulik and M. Q. Zhang, Silence, 2010, 1, 6 CrossRef PubMed.
- A. Schlicker, T. Lengauer and M. Albrecht, Bioinformatics, 2010, 26, i561–i567 CrossRef CAS PubMed.
- J. Jiang, E. J. Lee, Y. Gusev and T. D. Schmittgen, Nucleic Acids Res., 2005, 33 Search PubMed.
- K. Musilova and M. Mraz, Leukemia, 2015, 29, 1004–1017 CrossRef CAS PubMed.
- S. McGuire, Adv. Nutr., 2016, 7, 418–419 CrossRef PubMed.
- J. Weidhaas, Lancet Oncol., 2010, 11 Search PubMed.
- P. D. Baade, D. R. Youlden and L. J. Krnjacki, Mol. Nutr. Food Res., 2009, 53, 171–184 CAS.
- M. Alshalalfa, G. D. Bader, A. Goldenberg, Q. Morris and R. Alhajj, BMC Syst. Biol., 2012, 6, 112 CrossRef CAS PubMed.
- M. Jin, T. Zhang, C. Liu, M. A. Badeaux, B. Liu, R. Liu, C. Jeter, X. Chen, A. V. Vlassov and D. G. Tang, Cancer Res., 2014, 74, 4183–4195 CrossRef CAS PubMed.
- E. Guzel, O. F. Karatas, A. Semercioz, S. Ekici, S. Aykan, S. Yentur, C. J. Creighton, M. Ittmann and M. Ozen, Int. J. Cancer, 2015, 136, 875–879 CrossRef CAS PubMed.
- A. Y. Gong, A. N. Eischeid, J. Xiao, J. Zhao, D. Chen, Z. Y. Wang, C. Y. Young and X. M. Chen, BMC Cancer, 2012, 12, 492 CrossRef CAS PubMed.
- R. Ottman, J. Levy, W. E. Grizzle and R. Chakrabarti, OncoTargets Ther., 2016, 7, 73739–73753 Search PubMed.
- H. L. Wu and Y. Y. Mo, Expert Opin. Ther. Targets, 2009, 13, 1439–1448 CrossRef CAS PubMed.
- T. He, F. Qi, L. Jia, S. Wang, C. Wang, N. Song, Y. Fu, L. Li and Y. Luo, Cancer Lett., 2015, 368, 115–125 CrossRef CAS PubMed.
- A. Drusco, G. J. Nuovo, N. Zanesi, G. Di Leva, F. Pichiorri, S. Volinia, C. Fernandez, A. Antenucci, S. Costinean, A. Bottoni, I. A. Rosito, C. G. Liu, A. Burch, M. Acunzo, Y. Pekarsky, H. Alder, A. Ciardi and C. M. Croce, Plos One, 2014, 9 Search PubMed.
- R. Siegel, C. DeSantis and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 104–117 CrossRef PubMed.
- J. J. Ye and J. Cao, World J. Gastroenterol., 2014, 20, 4288–4299 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A supplemental table is available as a single excel file. See DOI: 10.1039/c7ra05348g |
| This journal is © The Royal Society of Chemistry 2017 |