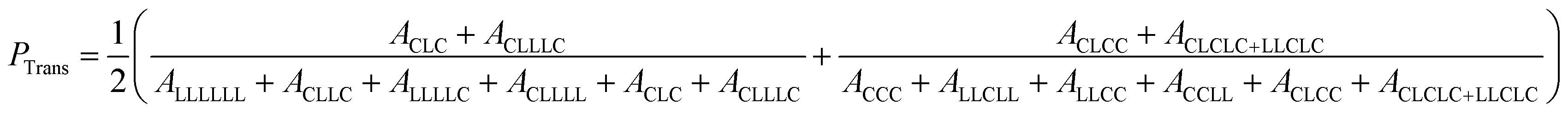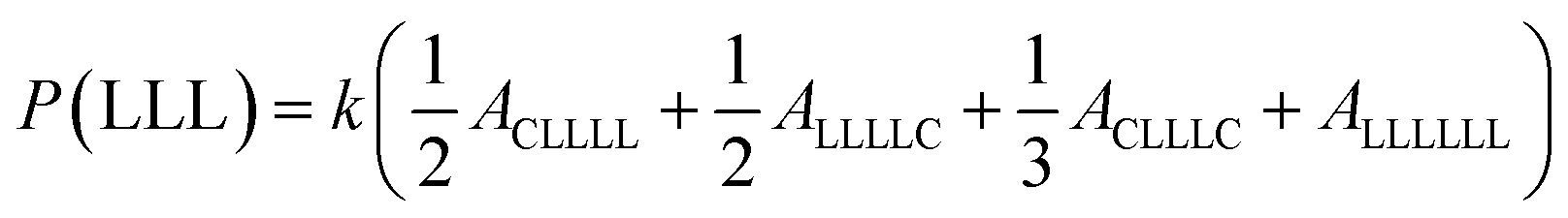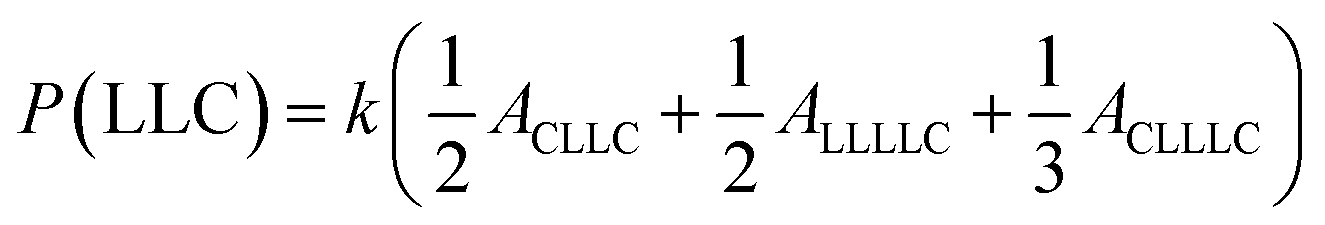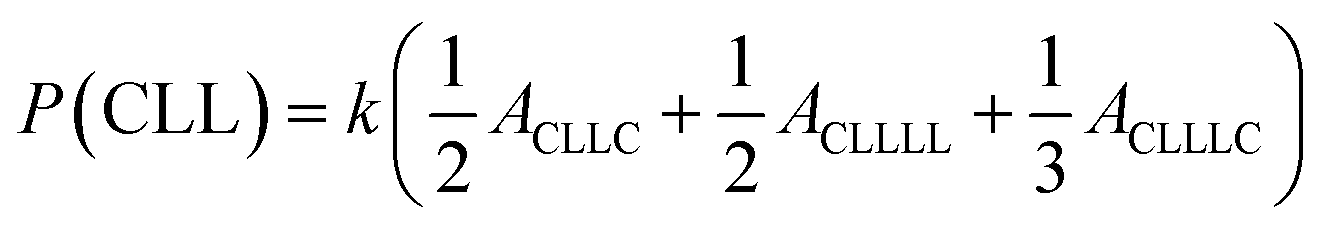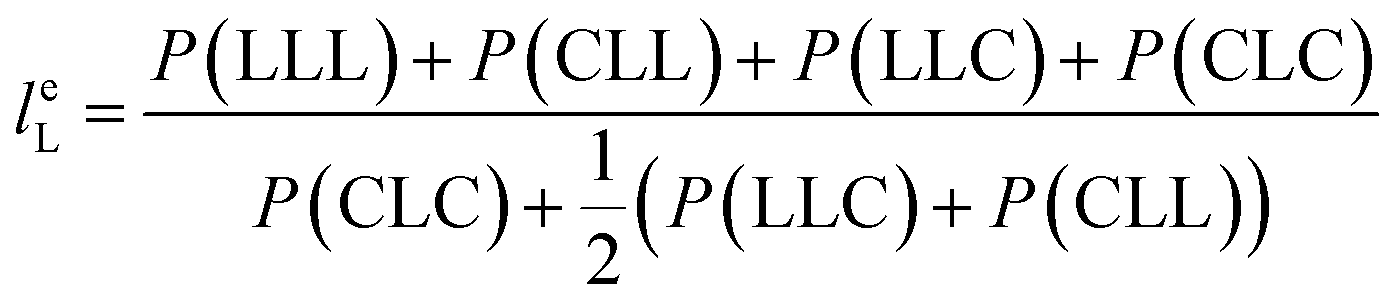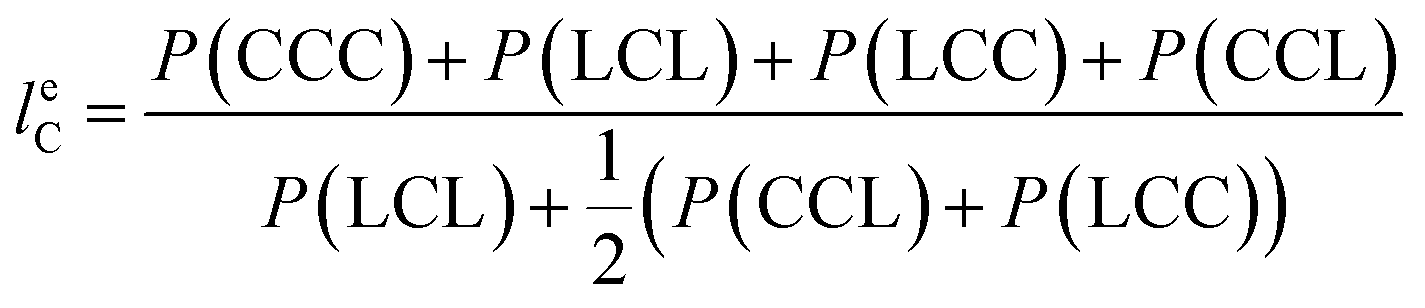DOI:
10.1039/C7RA05531E
(Paper)
RSC Adv., 2017,
7, 28661-28669
Tailoring chain structures of L-lactide and ε-caprolactone copolyester macromonomers using rac-binaphthyl-diyl hydrogen phosphate-catalyzed ring-opening copolymerization with monomer addition strategy†
Received
16th May 2017
, Accepted 19th May 2017
First published on 30th May 2017
Abstract
Copolymerizations involving polyester macromonomers (MMs) generated from biomass provide a new route for introducing high biomass content into existing polymeric products. Stannous 2-ethylhexanoate-catalyzed ring-opening copolymerization (ROcoP) is commonly utilized to synthesize MMs, but this approach generates polymer chains containing terminal metal residues and limits control of MM chain structures due to the presence of transesterification side reactions (TSRs). Here, rac-1,1′-binaphthyl-2,2′-diyl-hydrogenphosphate (rac-BNPH) was used for the 2-hydroxylethyl methacrylate (HEMA) initiated ROcoP of L-lactide (L-LA) and ε-caprolactone (ε-CL) to produce the well-defined MMs. The copolymerization kinetics and monomer feeding strategies, batch and semibatch, were studied, and the influence on MM chain structures was investigated using both 1H and 13C NMR analysis. The rac-BNPH was identified as an effective catalyst for the ROcoP of L-LA and ε-CL, producing narrowly dispersed MMs with a 96% retention of terminal vinyl groups associated with HEMA. The ε-CL was more reactive than L-LA, and the reactions exhibited characteristics of living polymerization. The TSRs could be significantly suppressed using batch operation or semibatch with fast ε-CL addition. It was found that slowing the ε-CL addition generated more randomly and uniformly distributed comonomers along MM chains. In general, it is demonstrated here that the rac-BNPH catalyzed semibatch ROcoPs is an effective means for tailoring the chain structures of MMs.
Introduction
Polyesters have been used in a wide variety of products due to their excellent mechanical properties, weather and corrosion resistance, and ease of processing. Polyesters generated from biomass such as lactide are renewable and biodegradable making them well-suited for disposable packaging and garments.1,2 They are also nontoxic and biocompatible which qualifies them for biomedical applications.3,4 More recently, an approach was introduced for increasing the biomass content in polymeric products by incorporating polyester grafts into polymers composed of acrylic backbones.5–8 This is achieved most commonly by growing polyester chains on initiators possessing a reactive terminal vinyl group through catalyzed ring-opening polymerizations (ROPs) to generate macromonomers, MMs. The MMs are then polymerized with other monomers to produce the polymers possessing polyester side chains. This approach opens a new route to convert a number of existing commodity products into more sustainable forms.
Previous work on these hybrid materials demonstrates their promise. Ishimoto et al.5 used 2-hydroxylethyl methacrylate, HEMA, as initiator to synthesize MMs with L-lactide, L-LA, containing approximately six lactidyl units. The MMs were then copolymerized with n-butyl methacrylate (BMA) using miniemulsion polymerizations to generate PBMA-g-PLLA. Films cast from the formed latexes were reported to demonstrate good elastic properties. In their subsequent work, Ishimoto et al. utilized renewable itaconic anhydride (IAn) to generate PLLA graft copolymers using the MM approach as well as a copolymer approach in which PLLA chains are generated on a polymer containing IAn repeat units. Both approaches resulted in materials that could be further optimized for plastic applications. The synthesis of PLLA MMs having 2 to 20 repeat units with various hydroxyalkyl methacrylates (HAA) initiators including HEMA, 2-hydroxyethyl acrylate (HEA), hydroxypropyl methacrylate (HPMA), and 4-hydroxybutyl acrylate (HBA) were also reported.9 The MMs were further copolymerized with HAA in solution to produce PHAA-g-PLLA copolymers, which were applied as scaffolding materials for tissue engineering applications. A more immediate application of these materials was demonstrated by Severtson et al.10,11 In order to tailor the thermal properties of the polyester MMs, ε-caprolactone, ε-CL, was copolymerized with L-LA to manipulate glass transition temperatures (Tgs) of formed MMs with HEMA as initiator. The MMs having low Tg values were then used to replace the acrylic soft monomer in miniemulsion polymerizations to generate pressure-sensitive adhesive (PSA) latexes containing 50 wt% MMs, which provided equivalent and often superior adhesive properties to commercial latex products. It was also demonstrated that the MM composition and chain length could be used to engineer adhesive properties. The use of the MMs approach was further extended for the production of thermoplastic adhesive materials. Much of the 2-ethylhexylacrylate (EHA) was replaced in a commercial acrylic hot-melt formulation with MMs synthesized from the hydroxyl ethylacrylamide initiated copolymerization of L-LA and ε-CL. The hot-melt adhesives containing 50 wt% MM possessed excellent adhesive properties. In general, the MM approach provides a path forward for synthesizing a broad array of polymer materials with high biomass contents.
The MMs discussed above were synthesized using stannous 2-ethylhexanoate, Sn(Oct)2, catalyzed ROP. Sn(Oct)2 is suitable for L-LA homopolymerizations and copolymerizations.12–15 Although widely used in ROP to prepare polyesters, metal-based catalysts possess complex structures and become bound to generated polymers.16,17 This inhibits or prevents their removal from polymer products, limiting applications for the generated polymers. Furthermore, metal-catalyzed ring-opening copolymerizations, ROcoPs, are typically accompanied by transesterification side reactions, TSRs.18 In generating MMs, such reactions limit the control of chain structure and chain length.13 They also promote the formation of cyclic units13 and MMs containing two vinyl initiator heads, which act as a crosslinker during the subsequent copolymerization of MM with vinyl monomers. Thus, the preparation of well-defined MMs, with predictable chain structures and chain lengths, requires that TSRs be minimized.
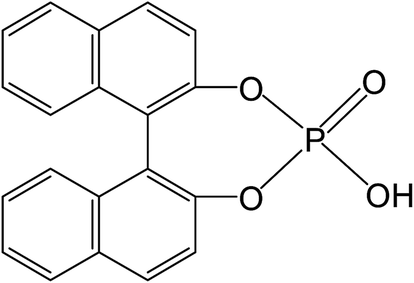
Recently, there has been significant interest in the use of metal-free organocatalysts for making polyesters. Such catalysts are relatively easy to generate and remove from end-products.19–22 The organocatalysts include organic acid,23–25 phosphazene,26,27 pyridine and carbene bases,28–31 thiourea/amine complex,32,33 and amidine/guanidine.34 Hedrick et al.35 was the first to report the successful catalysis of LA ROPs using strongly basic amines, 4-(dimethylamino)pyridine and 4-pyrrolidinopyridine. The nucleophilic alkali catalysts, in particular carbene bases, have been found to be effective in polymerizing lactone, epoxide, and carbonic anhydride.17,29 Carbene bases were found to catalyze zwitterionic ROP of cyclic esters in the absence of an alcohol initiator.36 The guanidine/amidine based catalysts, such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD),37–40 N-methyl-TBD,41 and 1,8-diazabicyclo[5.4.0]-undec-7-ene32 were also effective in catalyzing ROP of LA, δ-valerolactone, and ε-CL, and they demonstrated high activity in catalyzing LA polymerization in nonpolar solvents. Among the organocatalysts, simple organic acids such as 1,1′-binaphthyl-2,2′-diyl-hydrogenphosphate, BNPH, were found to possess high activities in catalyzing the ROP of cyclic monoesters.19–21 It was reported that the chiral (R)-BNPH was used for ROcoP of ε-CL and LA, but with very low LA incorporation in the copolymer.21
Although various organocatalysts have been found to promote ROPs, most of them are ineffective when it comes to catalyzing copolymerizations of ester monomers.42 Here, efforts to identify alternative catalyst for producing the HEMA initiated L-LA/ε-CL copolymer MMs through catalyzed ROcoPs are reviewed. The focus is on finding an organocatalyst that is easier to produce, use, and remove once the reaction is complete. As will be discussed, rac-BNPH appears to be a promising candidate for the generation of MMs containing L-LA and ε-CL. To the best of our knowledge, this is the first report of successful reactions for this system. The copolymerization kinetics including transesterification using different monomer feed strategies were studied. The effect of feeding strategies on the control of MM chain structures was also demonstrated.
Experimental section
Materials
The L-LA was purchased from Shenzhen Bright China Industrial Co. Ltd. The ε-CL, HEMA, Sn(Oct)2, rac-BNPH, hexane, and hydroquinone were obtained from Sigma Aldrich (St. Louis, MO) and used as received. Deuterated chloroform (CDCl3) and tetrahydrofuran (THF) were both purchased from J&K China Chemical, Ltd.
Synthesis of macromonomers
Macromonomers were synthesized with a fixed molar ratio of HEMA/L-LA/ε-CL = 1/5/4 at a variety of temperatures and polymerization times. Prior to the bulk ROcoPs, reactants were heated and stirred continuously in a round-bottom flask, and the contents were purged with nitrogen gas for approximately 10 min. The flask was then sealed and lowered into a hot oil bath and the contents were heated to the polymerization temperature prior to the addition of 0.5 mol% catalyst using a syringe. Temperature was maintained during the copolymerization under constant mechanical stirring. For copolymerizations involving semibatch operation with monomer addition, ε-CL (neat) was continuously introduced to the flask at rates of 0.0187 or 0.0468 mol h−1, respectively, using a syringe pump. Subsequent to copolymerizations, the flask and contents were cooled and washed with hexane twice to eliminate residual reactants, and the MMs were dried at room temperature in a vacuum oven for 24 h.
Characterization of macromonomers
Monomer conversions were determined with 1H nuclear magnetic resonance (NMR) analysis using a Bruker Advance III operated at 500 MHz. The MMs were dissolved in CDCl3 and tetramethylsilane (TMS) was added as the internal standard. All 1H NMR spectra were obtained with 512 scans and a 1 s acquisition time. In order to analyze the chain structures, the 13C NMR spectra of MMs were recorded at 125 MHz with the same spectrometer as used to conduct proton NMR measurements. The carbonyl carbon signals located between 165 and 175 ppm of the 13C NMR spectra were used for quantitative analysis. Spin-lattice relaxation times (T1) of carbonyl carbons in this range were determined (see ESI†). As T1 values for all carbonyl carbons used for sequential structure analysis are less than 3.628 s at present conditions, a delay time between pulses of 30 s was used to eliminate the saturation effect. Data were acquired using an acquisition time of 0.865 s, pulse width of 10.5 μs, and a spectral width of 300 ppm. Molecular weights and polydispersity indexes (Đ) of macromonomers were characterized using a Waters 1525/2414 Gel Permeation Chromatograph (GPC) equipped with a Waters 2414 differential refractive index detector. THF was used as the eluent at 1.0 mL min−1. The reported molecular weights and Đ values are relative to polystyrene standards.
Results and discussion
Batch copolymerization of L-LA/ε-CL using rac-BNPH
Along with Sn(Oct)2, which was used in previous studies,6,7,12,13 rac-BNPH (Scheme 1) was used for copolymerization initiated by HEMA at 100 °C with the HEMA/L-LA/ε-CL molar ratio of 1/5/4. The conversions of L-LA (xLA) and ε-CL (xCL) at polymerization times of 1, 3, and 7 hours are summarized in Table 1.
 |
| | Scheme 1 rac-BNPH as organocatalyst for ROcoP of L-LA and ε-CL. | |
Table 1 Monomer conversions for bulk ROcoP of L-LA and ε-CL initiated by HEMA in batch operation using rac-BNPH and Sn(Oct)2a
| Run |
Catalyst |
t = 1 h |
t = 3 h |
t = 7 h |
| xLA (%) |
xCL (%) |
xLA (%) |
xCL (%) |
xLA (%) |
xCL (%) |
| Molar ratio of HEMA/L-LA/ε-CL = 1/5/4, polymerization temperature (T) = 100 °C, 0.5 mol% (based on the monomer) catalyst, t denotes reaction time. |
| B1 |
rac-BNPH |
28.6 |
88.2 |
40.6 |
94.9 |
48.9 |
98.5 |
| B2 |
Sn(Oct)2 |
77.0 |
11.0 |
97.0 |
37.5 |
∼100 |
78.7 |
The results show significant differences between the catalysts, not only in the general activity they induce but also in their ability to catalyze the ROcoPs of L-LA and ε-CL. In copolymerization catalyzed by rac-BNPH, ε-CL initially reacted much faster than the L-LA. As the polymerization time was extended, the L-LA was converted. The observation that in rac-BNPH catalyzed ROcoPs, L-LA reactivity is less than that of ε-CL is opposite of that found when the same copolymerization is catalyzed with Sn(Oct)2. But the reactivity of L-LA is much higher than when solution ROcoPs are catalyzed with (R)-BNPH in toluene.21 It was reported that a little L-LA participated in copolymerizations with ε-CL, even with extended copolymerization times.21 Our results indicate that rac-BNPH shows promise for controlling the ROcoP of L-LA and ε-CL.
To explore the possibility of enhancing L-LA incorporation rates in rac-BNPH catalyzed copolymerizations, ROcoPs were conducted at increasing temperatures up to 160 °C. Table 2 lists characterization data for MMs generated in 7 h reactions including comonomer compositions, % HEMA vinyl group retentions (Rv), number-average molecular weights (Mn), and polydispersity index values (Đ). Raising temperature significantly increased monomer conversions, especially for L-LA. At 160 °C, the L-LA reached nearly full conversion in the 7 h polymerization. During the preparation of the MMs, a certain amount of HEMA is expected to polymerize. For copolymerization temperatures greater than 140 °C, the preserved MM terminal vinyl groups from HEMA, Rv, dropped to about 90%. Reactions between formed MMs are expected to form structures that are much larger than target structures based on molar ratios. Furthermore, it is expected that TSRs, which increase the chain disparity, are greatly enhanced at the higher temperatures. The influence of these processes is apparent from values for Đ with increasing temperature. For example, comparing polymerizations carried out at 160 and 100 °C, Đ increased from 1.33 to 2.63, respectively. This increase can be limited through the use of paradioxybenzene inhibitor, which protects the HEMA double bond. The addition of 0.1 mol% (based on monomers) to copolymerizations carried out at 160 °C lowered the Đ from 2.63 to 1.85, and 96% of terminal vinyl groups in HEMA were preserved in the MM as opposed to 91% in the absence of the inhibitor.
Table 2 Characterization data for rac-BNPH catalyzed bulk ROcoP of L-LA and ε-CL at various temperaturesa
| Run |
T (°C) |
xLA (%) |
xCL (%) |
Rvb (%) |
HEMA/L-LA/ε-CLc |
MNMRn (kg mol−1) |
MGPCn (kg mol−1) |
ĐGPC |
| Molar ratio of HEMA/L-LA/ε-CL = 1/5/4, 0.5 mol% (based on the monomer) rac-BNPH as catalyst, t = 7 h. The retention percentage of HEMA terminal vinyl groups in MMs. Molar fraction of monomers in macromonomers determined from 1H NMR spectra. 0.1 mol% (based on the monomers) paradioxybenzene added as inhibitor. |
| B1 |
100 |
48.9 |
98.5 |
98 |
1/4.67/4.06 |
0.89 |
1.12 |
1.33 |
| B3 |
120 |
73.6 |
∼100 |
96 |
1/7.38/4.34 |
1.11 |
1.39 |
1.45 |
| B4 |
140 |
92.4 |
∼100 |
93 |
1/11.75/5.39 |
1.59 |
1.70 |
1.46 |
| B5d |
140 |
92.7 |
∼100 |
96 |
1/9.20/4.28 |
1.28 |
1.44 |
1.43 |
| B6 |
160 |
∼100 |
∼100 |
91 |
1/14.01/5.98 |
1.95 |
1.71 |
2.63 |
| B7d |
160 |
∼100 |
∼100 |
96 |
1/10.67/4.41 |
1.36 |
1.56 |
1.85 |
rac-BNPH catalyzed semibatch bulk copolymerization of L-LA/ε-CL
The HEMA initiated ROcoP of L-LA and ε-CL is effectively catalyzed with rac-BNPH, but the balance between polymerization rates and the desired chain length parity requires elevated temperatures, e.g., 140 °C, along with use of an inhibitor. The rapid incorporation of ε-CL relative to L-LA, can be countered through effective monomer feed strategies in semibatch operations. The ε-CL is better suited for continuous feed operations because it is a liquid at room temperature, while L-LA is in its solid form. Two rac-BNPH catalyzed semibatch runs with ε-CL feeding rates of 0.0468 mol h−1 (SB1) and 0.0187 mol h−1 (SB2) were conducted at 140 °C, and the results are summarized in Table 3. Results for batch runs catalyzed with both rac-BNPH and Sn(Oct)2 at the same polymerization temperature are also reported in the table for comparison. The conversion of L-LA is increased significantly with the semibatch process involving the continuous feeding of ε-CL. Nearly complete conversion of the L-LA was obtained for the 0.0187 mol h−1 feed rate. This occurs without the large increase in Đ observed under semibatch operations.
Table 3 Data for bulk ROcoP of L-LA and ε-CL initiated with HEMA using batch and semibatch processesa
| Run |
Catalyst |
vCL (mol h−1) |
xLA (%) |
xCL (%) |
Rvb (%) |
HEMA/L-LA/ε-CLc |
MNMRn (kg mol−1) |
MGPCn (kg mol−1) |
ĐGPC |
| Molar ratio of HEMA/L-LA/ε-CL = 1/5/4, 0.5 mol% (based on the monomer) catalyst, t = 7 h, T = 140 °C. Retention percentage of terminal vinyl groups in MMs. Molar fraction of monomers in macromonomers determined from 1H NMR spectra. 0.1 mol% (based on monomers) paradioxybenzene added as inhibitor. |
| B8 |
Sn(Oct)2 |
— |
∼100 |
∼100 |
97 |
1/10.07/4.31 |
1.35 |
2.01 |
1.60 |
| B5d |
rac-BNPH |
— |
92.7 |
∼100 |
96 |
1/9.20/4.28 |
1.28 |
1.44 |
1.43 |
| SB1d |
rac-BNPH |
0.0468 |
96.0 |
∼100 |
97 |
1/9.74/4.62 |
1.35 |
1.83 |
1.38 |
| SB2d |
rac-BNPH |
0.0187 |
98.0 |
∼100 |
∼100 |
1/9.46/4.33 |
1.30 |
1.79 |
1.41 |
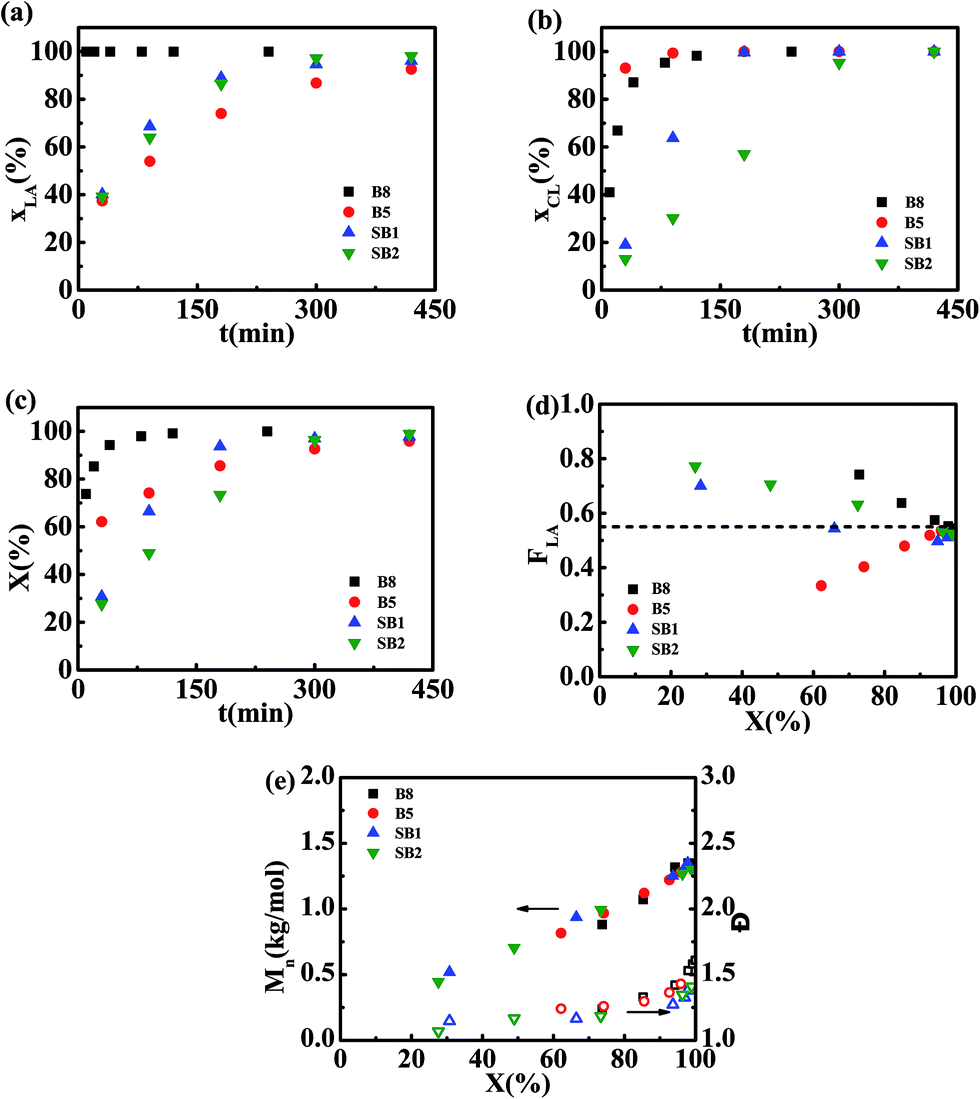
Conversions for L-LA, ε-CL and overall monomers (X) as a function of polymerization time are plotted in Fig. 1a–c. The information on MMs including cumulative L-LA composition (FL), Mn, and Đ are plotted as a function of overall monomer conversion in Fig. 1d and e. For batch reactions promoted with Sn(Oct)2, the L-LA is converted at a higher rate compared with the ε-CL comonomer. Batch polymerizations catalyzed with rac-BNPH demonstrate a more rapid conversion of ε-CL relative to L-LA. Regulating the ε-CL feed rates (at 0.0468 and 0.0187 mol h−1) in the rac-BNPH-catalyzed ROcoPs slows down the ε-CL polymerization rate in favor of more L-LA incorporated into MM chains. The benefits of this approach are evident in the composition of generated MMs. For batch processes, the FL in MM deviated substantially from the design value regardless of which catalyst, Sn(Oct)2 or rac-BNPH, was used (see Fig. 1d). Even at overall monomer conversions of greater than 60%, large deviations in comonomer composition exist. The MMs generated using the semibatch process are more homogeneous. The MM FL values are close to design values with regulated ε-CL feeding, even at the low monomer conversions. The Mn values of the MMs prepared using all of the various approaches catalyzed with rac-BNPH increased linearly with X, and all Đ values were held below 1.4, showing living polymerization characteristics. The slightly higher Đ values at high conversions are attributed to the increase of bulk viscosity inhibiting diffusion of monomers.43
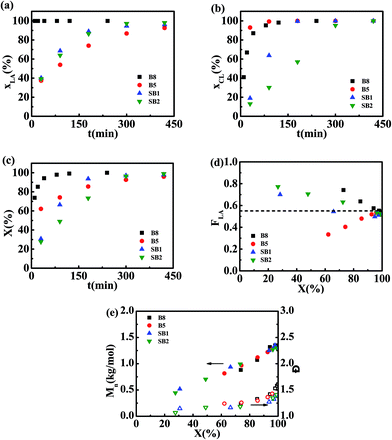 |
| | Fig. 1 Bulk ROcoP of L-LA and ε-CL carried out at 140 °C using a molar ratio of HEMA/L-LA/ε-CL = 1/5/4 with different catalysts and monomer addition modes (B8: Sn(Oct)2, batch; B5: rac-BNPH, batch; SB1: rac-BNPH, semibatch with vCL = 0.0468 mol h−1; and SB2: rac-BNPH, semibatch with vCL = 0.0187 mol h−1). Shown are results for (a) conversion of L-LA (xLA) versus polymerization time (t), (b) conversion of ε-CL (xCL) versus t, (c) total conversion of monomers (X) versus t, (d) cumulative molar fraction of L-LA in copolymer (FLA) versus X, and (e) Mn,NMR and Đ versus X. | |
Analysis of MM chain structure
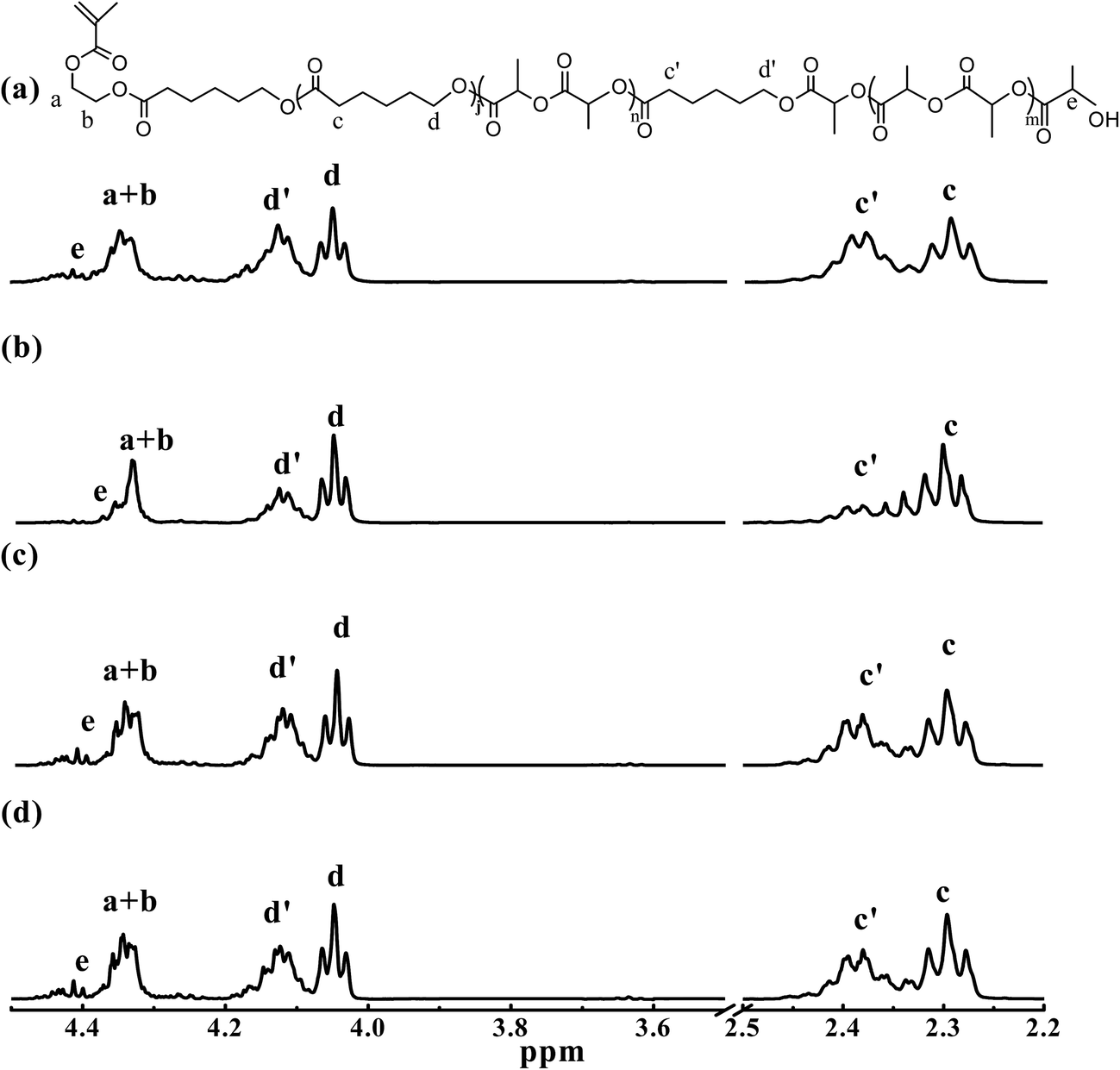
Intuitively, the polymerization kinetic behavior described above would be expected to provide MMs possessing a L-LA rich block near the HEMA head group when Sn(Oct)2 is used as the catalyst in batch operations, while a block of L-LA would be expected to terminate the MM chain for batch ROcoPs catalyzed with rac-BNPH. The 1H NMR spectra of the ε- and α-methylenes and protons adjacent to the terminal hydroxyl group in MMs generated using rac-BNPH under batch and semibatch conditions as well as spectra generated using Sn(Oct)2 under batch conditions are shown in Fig. 2 along with peak assignments for MMs. Full spectra for all of the generated MMs are provided as part of the ESI (Fig. S1†). The reference MMs, L-LA homo-macromonomer with five lactidyl units (LA5-HEMA), ε-CL homo-macromonomer having four ε-CL units (CL4-HEMA), and a block macromonomer of ε-CL and L-LA using HEMA as initiator (LA5-b-CL4-HEMA) were generated using Sn(Oct)2 as catalyst. These acted as references to establish chemical shifts that provide the local sequence for the LA and CL units in the formed copolymers. The details for their preparation and peak assignments of 1H NMR spectra are provided in the ESI (Fig. S2†).
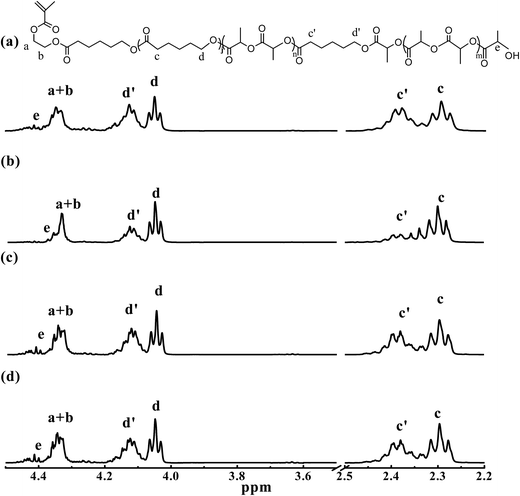 |
| | Fig. 2 1H NMR spectra of the ε- and α-methylenes and protons adjacent to terminal hydroxyl group in macromonomers. Shown are 1H NMR spectra for (a) B8: Sn(Oct)2, batch; (b) B5: rac-BNPH, batch; (c) SB1: rac-BNPH, semibatch with vCL = 0.0468 mol h−1; and (d) SB2: rac-BNPH, semibatch with vCL = 0.0187 mol h−1. | |
From all four spectra shown in Fig. 2, the triple peak of terminal HO-C around 3.61–3.69 ppm could not be observed clearly, but peaks around 4.4 ppm associated the HO-L end-group are easily distinguishable. The results are consistent with what is expected for the MMs catalyzed with rac-BNPH, but it is not statistically consistent with the ROcoP catalyzed with Sn(Oct)2 in which the ε-CL is incorporated slowly. The results suggest the presence of TSRs in which the terminal active caproyl end from a MM chain acts as the nucleophile and attacks a carbonyl group located on an adjacent MM chain in the Sn(Oct)2 catalyzed ROcoP.44–47 It has been reported that the HO-C end is significantly more active in such reactions compared with a HO-L end-group.4 Thus, when the MM is synthesized using the rac-BNPH catalyst in the batch mode, a long blocky L-LA would be expected at the end of the MM. This is confirmed from the chemical shift of HO-L changing from 4.39–4.48 ppm in Runs B8, SB1 and SB2 to 4.35–4.41 ppm in Run B5, which is consistent with the chemical shifts shown for sample LA5-HEMA in the ESI (Fig. S2†).
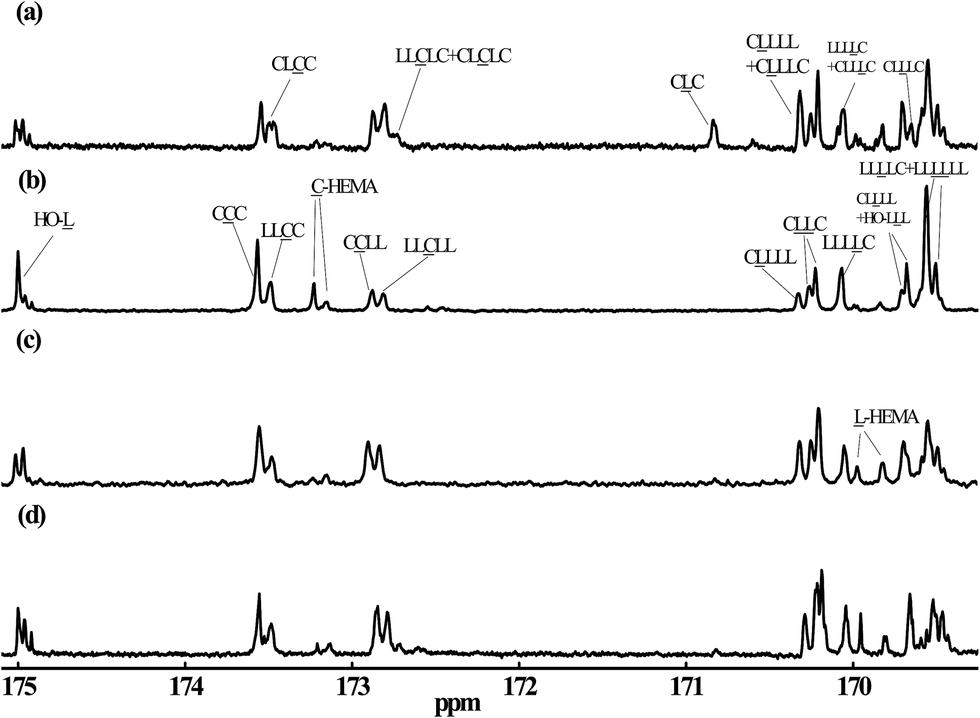
To further parse the MM chain structure for the various reaction conditions, 13C NMR analysis was employed. The carbonyl carbon peaks between 165 and 175 ppm of the 13C NMR spectra were the main focus of this analysis. These regions of the spectra and sequential assignments of each peak for the MMs generated via the different synthesis approaches are shown in Fig. 3. The 13C NMR analysis for MMs LA5-HEMA, CL4-HEMA, and LA5-b-CL4-HEMA was also carried out. Their 13C NMR spectra and triad assignments are given in Fig. S3 in the ESI.† Here LL is used to denote the lactidyl unit while C is for carproyl unit. All peak assignments in the figure are consistent with those reported by Kasperczyk and Bero,48 with the exception of chemical shifts 170.32 and 170.08 ppm, which were assigned to sequences LLLLC and CLLLL, respectively. This is the opposite of the previously reported assignments in ref. 48, and is confirmed by the results of the reference MMs presented in the ESI.†
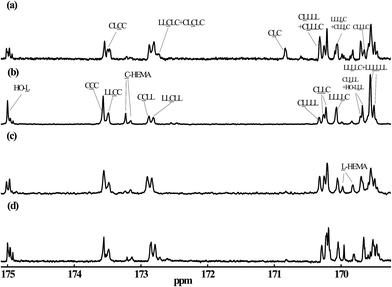 |
| | Fig. 3 Carbonyl carbon regions of 13C NMR spectra for MMs produced through different synthesis approaches. Shown are 13C NMR spectra for (a) B8, batch and Sn(Oct)2 catalyzed, (b) B5, batch and rac-BNPH catalyzed, (c) SB1, semibatch with vCL = 0.0468 mol h−1 and rac-BNPH catalyzed, and (d) SB2, semibatch with vCL = 0.0187 mol h−1 and rac-BNPH catalyzed. | |
The peaks at 173.13 and 173.21 ppm from MMs generated in Run B5 (Fig. 3) correspond to the triad of CC-HEMA head groups of MMs prepared by rac-BNPH catalyzed batch ROcoP of L-LA and ε-CL with HEMA initiator. Approximately 75% of formed MMs were initiated via reaction between HEMA and ε-CL in the rac-BNPH catalyzed batch ROcoP, while 25% involved reactions between HEMA and L-LA, which is indicated by the peaks at peaks of 169.79 and 169.98 ppm corresponding to the triad of LL-HEMA. These results along with proton NMR spectra strongly suggest the rapid initial addition of ε-CL for these reaction conditions. By changing the operation from a batch to semibatch process through ε-CL metering, the percentages of HEMA initially incorporated with ε-CL dropped to approximately 30% in both Runs SB1 and SB2 corresponding to ε-CL feed rates of 0.0468 and 0.0187 mol h−1, respectively. In comparison, almost all of the HEMA was initially reacted with L-LA in Sn(Oct)2 catalyzed batch ROcoP of L-LA and ε-CL. The absence of peak corresponding to HO-CC at 173.75 ppm in all MMs further verifies the observation of the MMs possessing terminal HO-LL groups in the 1H NMR measurement.
The presence of TSRs during Sn(Oct)2 catalyzed ROcoP of L-LA and ε-CL has been reported previously.18 The resonances of CLC triad at 170.83 ppm and CLLLC at 169.65 ppm for Run B8 shown in Fig. 3, indicating the presence of an odd number of latidyl units (L), are evidence of the presence of such reactions. For polymerizations catalyzed with rac-BNPH these peaks become indistinguishable for reaction runs under batch mode (Run B5) and semibatch mode using the higher ε-CL feed rate (Run SB1), but becomes slightly noticeable at the lower ε-CL feed rate (Run SB2). For a clearer assessment of the extent to which TSRs are present during ROcoPs, the percentage of lactyl units (L) present in the MMs, denoted here as PTrans, was determined by comparing the 13C NMR spectral area A associated with sequences possessing these units with the total area associated with sequences having both lactyl and lactidyl units (eqn (1)). The PTrans value is 20.2% when polymerizations are catalyzed with Sn(Oct)2 under batch reaction conditions. This value sharply declines to 0% for rac-BNPH catalyzed batch and semibatch reactions using the higher ε-CL feed rate in Run SB1, and 2.6% for rac-BNPH catalyzed semibatch reaction conditions used in Run SB2. This suggests the TSRs can be suppressed in the rac-BNPH catalyzed ROcoP by controlling the ε-CL concentration in the system.
| |
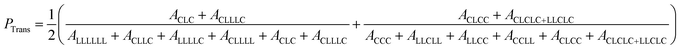 | (1) |
The sequential distribution in the MMs is further investigated through the triad distribution and average sequence length of each monomer unit. Since there exist lactyl units in some MM samples, the triads and average sequence lengths on the basis of lactyl unit L are considered. The triad ijk distributions (P(ijk)) in the MMs can be estimated from Fig. 3. The relationships between the triad distributions and the integral areas in the 13C NMR spectra are49
| |
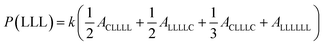 | (2) |
| |
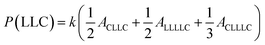 | (3) |
| |
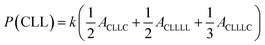 | (4) |
| | |
P(LCL) = k(ALLCLL + ALLCLC + ACLCLL + ACLCLC)
| (6) |
| | |
P(CCL) = k(ACCLC + ACCLL)
| (7) |
| | |
P(LCC) = k(ACLCC + ALLCC)
| (8) |
where
k is the NMR signal proportionality constant.
The triad distributions for the MMs are summarized in Table 4. For the batch copolymerization Run B5 using rac-BNPH as the catalyst, more CCC and LLL triads were identified compared to that found in Run B8 with Sn(Oct)2 as the catalyst. In comparing Run B5 with rac-BNPH catalyzed semibatch reactions at the lowest ε-CL feed rate (SB2), the CCC sequence distributions decrease from 0.231 to 0.129, respectively, while the LLL sequences decrease from 0.297 to 0.194, respectively. This is accompanied by an increase in the CCL, LCL, LLC, and CLL sequences, suggesting more uniformly distributed comonomer sequential structures in the MMs due to slow addition of ε-CL. The triad distributions except CLC, which is contributed by the TSR, in Run SB2 are similar.
Table 4 Triad distributions in macromonomers synthesized in HEMA initiated bulk ROcoP of L-LA and ε-CL catalyzed by either rac-BNPH or Sn(Oct)2
| Triad distribution |
B8 |
B5 |
SB1 |
SB2 |
| P(CCC) |
0.090 |
0.231 |
0.186 |
0.129 |
| P(CCL) |
0.087 |
0.088 |
0.148 |
0.150 |
| P(LCC) |
0.101 |
0.125 |
0.083 |
0.114 |
| P(LCL) |
0.187 |
0.076 |
0.116 |
0.129 |
| P(CLC) |
0.076 |
0.000 |
0.000 |
0.010 |
| P(LLC) |
0.099 |
0.116 |
0.120 |
0.149 |
| P(CLL) |
0.099 |
0.068 |
0.123 |
0.126 |
| P(LLL) |
0.261 |
0.297 |
0.224 |
0.194 |
From the triad distribution data, the average sequence lengths of lactyl and caproyl units, leL and leC, respectively, in the MMs can be estimated using,
| |
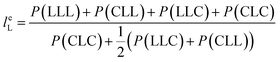 | (10) |
| |
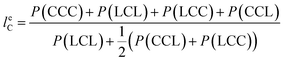 | (11) |
The results of average sequence lengths of lactyl and caproyl units are summarized in Table 5. The evidence of leL and leC becoming shorter with the change of ε-CL feed strategy from batch mode to slow addition of ε-CL is further observed. Run SB2 by rac-BNPH catalyzed semibatch ROcoP with slowest ε-CL addition has leL and leC values close to those in Run B8 by Sn(Oct)2 catalyzed batch reaction, suggesting the MM sequential structures become more random. The semibatch operation with ε-CL addition is an effective means for tailoring the MMs with more uniformly distributed comonomer sequential structures.
Table 5 Average sequence lengths of lactyl and caproyl units in macromonomers synthesized in HEMA initiated bulk ROcoP of L-LA and ε-CL catalyzed by either rac-BNPH or Sn(Oct)2 using batch and semibatch processes
| Run |
leL |
leC |
| B8 |
3.06 |
1.65 |
| B5 |
5.22 |
2.85 |
| SB1 |
3.84 |
2.30 |
| SB2 |
3.25 |
2.00 |
Conclusions
The first successful catalysis of HEMA initiated ROcoPs of L-LA and ε-CL using rac-BNPH is reported. Narrowly dispersed MMs with a 96% retention of HEMA terminal vinyl groups were generated at 140 °C using 0.5 mol% catalyst and 0.1 mol% inhibitor and a HEMA/L-LA/ε-CL molar ratio of 1/5/4. It was observed that when rac-BNPH is applied, ε-CL is more reactive than L-LA, which is the opposite to the reactivity order observed when using Sn(Oct)2 as the catalyst. The ROcoP showed living characteristics. Using semibatch operation involving the regulation of ε-CL feed rates to 0.0468 and 0.0187 mol h−1 generated MMs with more homogeneous comonomer compositions. The 1H and 13C NMR characterization results showed that the MMs prepared with rac-BNPH possess terminal OH-LA units. It was also found that 75% of MMs generated via batch reactions have initial CL-HEMA groups, which dropped to 30% for semibatch reactions. The prevalence of TSRs was greatly reduced when these reactions are catalyzed with rac-BNPH and ε-CL feed rates are controlled. No TSRs were observed in rac-BNPH catalyzed batch and semibatch reactions using the higher ε-CL feed rate. Approximately 2.6% lactyl units participated in the TSRs during rac-BNPH catalyzed semibatch ROcoPs using the lower feed rate of 0.0187 mol h−1, which is significantly lower than the 20.2% lactyl unit participation measured during batch copolymerization using Sn(Oct)2 as the catalyst. By moving from batch mode to slow addition of ε-CL in rac-BNPH catalyzed ROcoPs, both CCC and LLL sequence distributions in the MMs were decreased, and the comonomer sequential structures became more uniformly distributed and random in the MMs. In summary, it was shown that rac-BNPH catalyzed ROcoPs with ε-CL addition is an effective means for tailoring the comonomer sequential structures of the MMs.
Author contributions
The manuscript was written with contributions of all authors. All authors have given approval to the final version of the manuscript.
Acknowledgements
The authors thank National Key Research and Development Program of China (2016YFB0302402 and 2016YFB0302405), National Natural Science Foundation of China (21420102008), and Chinese State Key Laboratory of Chemical Engineering at Zhejiang University (SKL-ChE-15D03) for financial supports.
References
- L. Shen, E. Worrell and M. Patel, Present and Future Development in Plastics from Biomass, Biofuels, Bioprod. Biorefin., 2010, 4, 25–40 CrossRef CAS.
- T. Iwata, Biodegradable and Bio-Based Polymers: Future Prospects of Eco-Friendly Plastics, Angew. Chem., Int. Ed., 2015, 54, 3210–3215 CrossRef CAS PubMed.
- K. Sudesh and T. Iwata, Review: Sustainability of Biobased and Biodegradable Plastics, Clean, 2008, 36, 433–442 CAS.
- Z. Y. Wei, L. Liu, C. Qu and M. Qi, Microstructure Analysis and Thermal Properties of L-Lactide/ε-Caprolactone Copolymers Obtained with Magnesium Octoate, Polymer, 2009, 50, 1423–1429 CrossRef CAS.
- K. Ishimoto, M. Arimoto, H. Ohara, S. Kobayashi, M. Ishii, K. Morita, H. Yamashita and N. Yabuuchi, Biobased Polymer System: Miniemulsion of Poly(alkyl methacrylate-graft-lactic acid)s, Biomacromolecules, 2009, 10, 2719–2723 CrossRef CAS PubMed.
- G. Pu, D. A. Hauge, C. Gu, J. Zhang, S. J. Severtson, W. Wang and C. J. Houtman, Influence of Acrylated Lactide-Caprolactone Macromonomers on the Performance of High Biomass Content Pressure-Sensitive Adhesives, Macromol. React. Eng., 2013, 7, 515–526 CrossRef CAS.
- K. Ishimoto, M. Arimoto, T. Okuda, S. Yamaguchi, Y. Aso, H. Ohara, S. Kobayashi, M. Ishii, K. Morita, H. Yamashita and N. Yabuuchi, Biobased Polymers: Synthesis of Graft Copolymers and Comb Polymers Using Lactic Acid Macromonomer and Properties of the Product Polymers, Biomacromolecules, 2012, 13, 3757–3768 CrossRef CAS PubMed.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Renewable Biobased Polymeric Materials: Facile Synthesis of Itaconic Anhydride-Based Copolymers with Poly(L-lactic acid) Grafts, Macromolecules, 2012, 45, 4166–4174 CrossRef CAS.
- X. Liu and P. X. Ma, The Nanofibrous Architecture of Poly(L-lactic acid)-Based Functional Copolymers, Biomaterials, 2010, 31, 259–269 CrossRef CAS PubMed.
- G. Pu, M. R. Dubay, J. Zhang and S. J. Severtson, Polyacrylates with High Biomass Contents for Pressure-Sensitive Adhesives Prepared via Mini-emulsion Polymerization, Ind. Eng. Chem. Res., 2012, 51, 12145–12149 CrossRef CAS.
- C. Gu, M. R. Dubay and S. J. Severtson, Hot-Melt Pressure-Sensitive Adhesives Containing High Biomass Contents, Ind. Eng. Chem. Res., 2014, 53, 11000–11006 CrossRef CAS.
- P. Kurock, M. Kowalczuk, K. Hennek and Z. Jedlinski, Anionic Polymerization of Lactones Initiated with Alkali-Metal Alkoxides: Reinvestigation of the Polymerization Mechanism, Macromolecules, 1992, 25, 2017–2020 CrossRef.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Controlled Ring-Opening Polymerization of Lactide and Glycolide, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- C. M. Thomas, Stereocontrolled Ring-Opening Polymerization of Cyclic Esters: Synthesis of New Polyester Microstructures, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- P. J. Dijkstra, H. Z. Du and F. J. Jan, Single Site Catalysts for Stereoselective Ring-Opening Polymerization of Lactides, Polym. Chem., 2011, 2, 520–527 RSC.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Phosphazene Bases: A New Category of Organocatalysts for the Living Ring-Opening Polymerization of Cyclic Esters, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, First Example of N-Heterocyclic Carbenes as Catalysts for Living Polymerization: Organocatalytic Ring-Opening Polymerization of Cyclic Esters, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed.
- H. R. Kricheldorf, M. Berl and N. Scharnagl, Poly(lactones). 9. Polymerization Mechanism of Metal Alkoxide Initiated Polymerizations of Lactide and Various Lactones, Macromolecules, 1988, 21, 286–293 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Organocatalysis: Opportunities and Challenges for Polymer Synthesis, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- Y. Miao, Y. Phuphuak, C. Rousseau, T. Bousquet, A. Mortreux, S. Chirachanchai and P. Zinck, Ring-Opening Polymerization of Lactones using Binaphthyl-diyl Hydrogen Phosphate as Organocatalyst and Resulting Monosaccharide Functionalization of Polylactones, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2279–2287 CrossRef CAS.
- X. Zhou and L. Z. Hong, Controlled Ring-Opening Polymerization of Cyclic Esters with Phosphoric Acid as Catalysts, Colloid Polym. Sci., 2013, 291, 2155–2162 CAS.
- S. Wang, P. Liu, W.-J. Wang, Z. Zhang and B.-G. Li, Hyperbranched Polyethylene-Supported L-proline: A Highly Selective and Recyclable Organocatalyst for Asymmetric Aldol Reactions, Catal. Sci. Technol., 2015, 5, 3798–3805 CAS.
- T. Endo, Y. Shibasaki and F. Sanda, Controlled Ring-Opening Polymerization of Cyclic Carbonates and Lactones by An Activated Monomer Mechanism, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2190–2198 CrossRef CAS.
- G.-B. Stéphanie, D. Damien, M.-V. Blanca, B. Didier, N. Christophe and M. Stéphanie, Organo-Catalyzed ROP of ε-Caprolactone: Methanesulfonic Acid Competes with Trifluoromethanesulfonic Acid, Macromolecules, 2008, 41, 3782–3784 CrossRef.
- N. Susperregui, D. Delcroix, B. Martinvaca, D. Bourissou and L. Maron, Ring-Opening Polymerization of ε-Caprolactone Catalyzed by Sulfonic Acids: Computational Evidence for Bifunctional Activation, J. Org. Chem., 2010, 75, 6581–6587 CrossRef CAS PubMed.
- S. Boileau and N. Illy, Activation in Anionic Polymerization: Why Phosphazene Bases Are Very Exciting Promoters, Prog. Polym. Sci., 2011, 36, 1132–1151 CrossRef CAS.
- G. Mountrichas, C. Mantzaridis and S. Pispas, Well-Defined Flexible Polyelectrolytes with Two Cationic Sites per Monomeric Unit, Macromol. Rapid Commun., 2006, 27, 289–294 CrossRef CAS.
- C. G. Wade and J. L. Hedrick, Organocatalytic Stereoselective Ring-Opening Polymerization of Lactide with Dimeric Phosphazene Bases, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef PubMed.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, N-Heterocyclic Carbenes (NHCs) as Organocatalysts and Structural Components in Metal-free Polymer Synthesis, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, N-Heterocyclic Carbene-Organocatalyzed Ring-Opening Polymerization of Ethylene Oxide in the Presence of Alcohols or Trimethylsilyl Nucleophiles as Chain Moderators for the Synthesis of α,ω-Heterodifunctionalized Poly(ethylene oxide)s, Macromolecules, 2010, 43, 2814–2823 CrossRef CAS.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, N-Heterocyclic Carbene-Induced Zwitterionic Ring-Opening Polymerization of Ethylene Oxide and Direct Synthesis of α,ω-Difunctionalized Poly(ethylene oxide)s and Poly(ethylene oxide)-b-poly(ε-caprolactone) Block Copolymers, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, Thiourea-Based Bifunctional Organocatalysis:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Supramolecular Recognition for Living Polymerization, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS PubMed.
Supramolecular Recognition for Living Polymerization, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS PubMed. - M. Oshimura, T. Tang and A. Takasu, Ring-opening Polymerization of ε-Caprolactone Using Perfluoroalkanesulfonates and Perfluoroalkanesulfonimides as Organic Catalysts, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1210–1218 CrossRef CAS.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, New Paradigms for Organic Catalysts: The First Organocatalytic Living Polymerization, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS PubMed.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Zwitterionic Polymerization of Lactide to Cyclic Poly(Lactide) by Using N-Heterocyclic Carbene Organocatalysts, Angew. Chem., Int. Ed., 2007, 119, 2681–2684 CrossRef.
- M. L. Foresti and M. L. Ferreira, Synthesis of Polycaprolactone Using Free/Supported Enzymatic and Non-Enzymatic Catalysts, Macromol. Rapid Commun., 2004, 25, 2025–2028 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, Triazabicyclodecene: A Simple Bifunctional Organocatalyst for Acyl Transfer and Ring-Opening Polymerization of Cyclic Esters, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- G. Theryo, F. Jing, L. M. Pitet and M. A. Hillmyer, Tough Polylactide Graft Copolymers, Macromolecules, 2010, 43, 7394–7397 CrossRef CAS.
- S. Jaenicke, G. K. Chuah, X. H. Lin and X. C. Hu, Organic-inorganic Hybrid Catalysts for Acid- and Base-catalyzed Reactions, Microporous Mesoporous Mater., 2000, 99, 143–153 CrossRef.
- F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Functionalized Cyclic Carbonates: from Synthesis and Metal-free Catalyzed Ring-opening Polymerization to Applications, Polym. Chem., 2011, 2, 528–533 RSC.
- G. W. Nyce, T. Glauser, E. F. Connor, A. Möck, R. M. Waymouth and J. L. Hedrick, In Situ Generation of Carbenes: A General and Versatile Platform for Organocatalytic Living Polymerization, J. Am. Chem. Soc., 2003, 125, 3046–3056 CrossRef CAS PubMed.
- F. Y. Weng, X. H. Li, Y. J. Wang, W.-J. Wang and S. J. Severtson, Kinetics and Modeling of Ring-Opening Copolymerization of L-Lactide and ε-Caprolactone, Macromol. React. Eng., 2015, 9, 535–544 CrossRef CAS.
- M. Bero and J. Kusperczyk, Coordination Polymerization of Lactides, 5. Influence of Lactide Structure on the Transesterification Processes in the Copolymerization with ε-Caprolactone, Macromol. Chem. Phys., 1996, 197, 3251–3258 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Kinetics and Mechanism of Cyclic Esters Polymerization Initiated with Tin(II) Octoate, 1. Polymerization of ε-Caprolactone, Macromol. Rapid Commun., 1998, 19, 567–572 CAS.
- A. Kowalski, A. Duda and S. Penczek, Mechanism of Cyclic Ester Polymerization Initiated with Tin(II) Octoate. 2. Macromolecules Fitted with Tin(II) Alkoxide Species Observed Directly in MALDI–TOF Spectra, Macromolecules, 2000, 33, 689–695 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Kinetics and Mechanism of Cyclic Esters Polymerization Initiated with Tin(II) Octoate. 3. Polymerization of L,L-Dilactide, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- J. Kasperczyk and M. Bero, Coordination Polymerization of Lactides, 2. Microstructure Determination of Poly[(L,L-lactide)-co-(ε-caprolactone)] with 13C Nuclear Magnetic Resonance Spectroscopy, Makromol. Chem., 1991, 192, 1777–1787 CrossRef CAS.
- J. Kasperczyk and M. Bero, Coordination Polymerization of Lactides, 4. The Role of Transesterification in the Copolymerization of L,L-lactide and ε-Caprolactone, Makromol. Chem., 1993, 194, 913–925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details about synthesis procedure of L-LA and ε-CL homo- and block macromonomers and their 1H and 13C NMR spectra and peak assignments, 1H NMR spectra for macromonomers of Runs B8, B5, SB1, and SB2, chemical shift assignments of carbonyl carbon sequences in macromonomers, and spin-lattice relaxation time (T1) for carbonyl carbons in 13C NMR spectrum of macromonomer. See DOI: 10.1039/c7ra05531e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a