
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New cyclometalated Ir(III) complexes with NCN pincer and meso-phenylcyanamide BODIPY ligands as efficient photodynamic therapy agents†
Leila Tabrizi *ab and
Hossein Chiniforoshan*b
*ab and
Hossein Chiniforoshan*b
aSchool of Chemistry, National University of Ireland, Galway, University Road, Galway, Ireland. E-mail: LEILA.TABRIZI@nuigalway.ie
bDepartment of Chemistry, Isfahan University of Technology, Isfahan, Iran. E-mail: Chinif@cc.iut.ac.ir
First published on 7th July 2017
Abstract
A new class of cyclometalated iridium(III) with NCN pincer and meso-phenylcyanamide BODIPY ligands of the formula [Ir(L)(PPY)(Pcyd-BODIPY)], 1, [Ir(L)(PIQ)(Pcyd-BODIPY)], 2, in which HL: 5-methoxy-1,3-bis (1-methyl-1H-benzo[d]imidazol-2-yl)benzene, Pcyd-BODIPY: 8-(4-phenylcyanamide) BODIPY, PPY: 2-phenylpyridine, PIQ: 1-phenylisoquinoline have been synthesized and studied for photodynamic therapy (PDT). The photophysical properties, DNA binding, DNA photocleavage, cellular uptake, thioredoxin reductase activity, reactive oxygen species (ROS) generation, and cellular apoptosis of the complexes have also been studied. Therefore, we present a conceivable biologically applicable PDT modality that develops persuasively designed photoactivatable Ir(III) complexes.
1. Introduction
Photodynamic therapy (PDT) has been successfully used for cancer treatment and involves the interaction of the photosensitizer, light, and molecular oxygen.1–5 Recently, a ruthenium-based photosensitizer (TLD-1433) has been approved for phase Ib clinical trials in Canada for non-muscle invasive bladder cancer.6–8Some cyclometalated Ru(II), Ir(III), and Pt(II) complexes, Ru(II) arene complexes, and Re(I) tricarbonyl complexes have proven themselves for photocytotoxicity studies.9–15 Cyclometalated IrIII complexes have recently received great interest for PDT agents due to their exceptional properties such as simple color tuning, energy-level control, high quantum yields, large Stokes shifts, long-lived phosphorescence, and ROS generation under hypoxia conditions via electron (type I) or energy transfer (type II).16–27
BODIPY (boron dipyrromethene) derivatives have found to be applicable as potential applications for biological labeling, cellular imaging, and fluorescence sensing, unique narrow emission with high quantum yield in biological and medical fields. Furthermore, it has been found that the heavy atom substituted BODIPY dyes containing an electron-withdrawing acetoxymethyl moiety in the meso-position could be enhanced photostability for the PDT agents.28–32 Recently, our groups focused attention in the development Ni(II), Zn(II), Cd(II), Co(II), Ru(II), Ir(III), Pt(II), Pd(II), and triorganotin(IV) complexes containing phenylcyanamide derivatives as monodentate ligands in regard to traditional chemotherapy and PDT applications.33–39 With the aim of enhancing the photophysical and photobiological properties of BODIPY ligands in cyclometalated Ir(III) complexes, we have designed new monodentate ligand by linking meso-BODIPY ligands to phenylcyanamide ligands.
Here we present the synthesis, characterization, photophysical properties, and photocytotoxicity explorations of two new cyclometalated Ir(III) complexes of the type [Ir(L)(PPY)(Pcyd-BODIPY)], 1, [Ir(L)(PIQ)(Pcyd-BODIPY)], 2, in which HL: 5-methoxy-1,3-bis (1-methyl-1H-benzo[d]imidazol-2-yl)benzene, Pcyd-BODIPY: 8-(4-phenylcyanamide) BODIPY, PPY: 2-phenylpyridine, PIQ: 1-phenylisoquinoline.
2. Results and discussion
2.1. Synthesis and characterization
The ligand 8-(4-phenylcyanamide) BODIPY (Pcyd-BODIPY) was synthesized from 8-(4-anilino) BODIPY as previously reported.40 The ligand HL was synthesized as previously reported by us.41 Reaction of IrCl3·xH2O with pincer ligand HL in MeOH/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and then PPY (2-phenylpyridine) or PIQ (1-phenylisoquinoline) and Pcyd-BODIPY in MeOH/CH2Cl2 (1:2, v/v) in pH ∼7 gave the complexes 1 and 2 (Fig. 1).
:1, v/v) and then PPY (2-phenylpyridine) or PIQ (1-phenylisoquinoline) and Pcyd-BODIPY in MeOH/CH2Cl2 (1:2, v/v) in pH ∼7 gave the complexes 1 and 2 (Fig. 1).
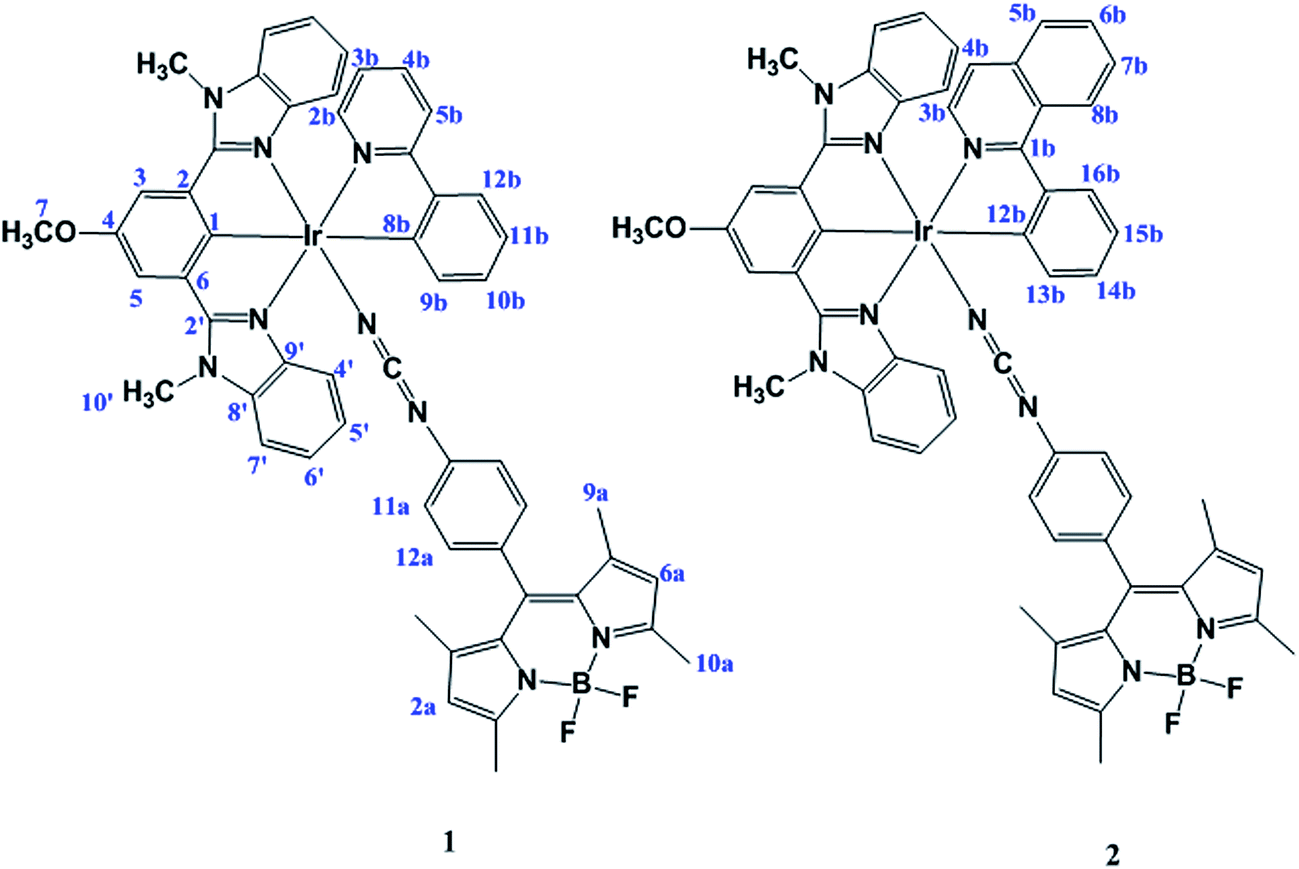 | ||
| Fig. 1 Structure of complexes 1 and 2. | ||
The ligand Pcyd-BODIPY and complexes were prepared in excellent yield and in high purity as described in the experimental section. Complexes were characterized by elemental analysis, IR, 1H NMR, 13C NMR spectroscopy (Fig. S1–S6†) and ESI-TOF mass spectrometry (Fig. S7–S9†).
The formation of Pcyd-BODIPY, 1, and 2 is clearly evidenced by the parent peaks in the mass spectra. The mass spectra of Pcyd-BODIPY, 1, and 2 show peaks of (M + H)+ centered at m/z = 365, 1078, and 1128, respectively (Fig. S7–S9†).
The disappearance of the H1 signal (7.54 ppm in ligand HL) and the upfield shift of the signal of H3/H5 of the central phenyl ring (7.29–7.35 ppm in ligand HL and 6.75 and 6.79 ppm in 1 and 2, respectively) in the 1H NMR spectra of 1 and 2 are consistent with the coordination of Ir to the deprotonated pincer ligand. The H4′ resonance is shifted downfield due to the ring current effect of the second ligand in complexes 1 and 2 (7.84–7.79 ppm in ligand HL and 8.31 and 8.80 ppm in 1 and 2, respectively).41
The proton chemical shifts of Pcyd-BODIPY decrease with respect to the free ligand due to coordination of ligand Pcyd-BODIPY to Ir. Also, the NH proton of ligand Pcyd-BODIPY disappeared due to coordination of the cyanamide group to Ir in complexes 1 and 2.
The ν (NCN) band of ligand Pcyd-BODIPY is observed at 2225 cm−1, when the Pcyd-BODIPY ligand coordinates to Ir, ν (NCN) of complexes 1 and 2 is shifted to lower energies (2092 and 2088 cm−1, respectively) representing the coordination of cyanamide through the cyano nitrogen.
2.2. Photophysical properties
The photophysical properties of Ir(III) complexes in acetonitrile and phosphate buffer solution (PBS) were considered at 298 K (Fig. S10, S11, and Table S1†). The complexes 1 and 2 display the absorption bands of lower intensity in the 300–480 nm region due to intraligand (IL) π → π* and n → π* transitions and singlet and triplex metal-to-ligand charge transfer (1MLCT and 3MLCT).42 In addition, the absorption spectra of complexes 1 and 2 are dominated by the intense bands at 541 and 526 nm in acetonitrile, respectively, due to the S0 → S1 (π–π*) transition localized on the BODIPY unit.43 The complexes 1 and 2 exhibited red emissions in acetonitrile and PBS upon excitation at 500 nm.The luminescence quantum yields (Φem) and luminescence lifetimes were evaluated in acetonitrile at room temperature (Table S1†). The luminescence lifetimes of complexes 1 and 2 are around 700 ns. These complexes have higher quantum yields and longer lifetimes than [Ru(bpy)3]2+ and Ru(II) pincer complexes.37,42
2.3. Singlet oxygen sensitization
The major cytotoxic species in PDT is the singlet oxygen to induce cancer cell death.44 Therefore, the determination of 1O2 quantum yields of complexes 1 and 2 was made upon irradiation at 500 nm (Table S2†). The 1O2 photosensitizing properties of 1 and 2 were investigated by direct and indirect measurement.37The complexes 1 and 2 have Φ (1O2) values of 79% and 92% in acetonitrile (500 nm, direct method), respectively. The obtained values for 1 and 2 in acetonitrile are comparable with 1O2 quantum yields reported for related compounds.37,45–47 These data display that the complexes 1 and 2 have a great efficacy in the photosensitization of molecular oxygen. Moreover, the quantum yields of 1O2 production in PBS are low (Table S2†). Thus, the effect of solvent on the efficiency of 1O2 production is obvious. In acetonitrile, the photosensitization of molecular oxygen is effective due to the absence of quenching mechanism on the triplet excited state.48
As a result, these complexes can produce 1O2 when they accumulate in hydrophobic compartments and complex 2 has the highest quantum yield of singlet oxygen production, emission and fluorescence lifetime.
2.4. Interaction of 1–3 with DNA
Electronic absorption spectroscopy is an efficient method to study the binding mode of metal complexes to DNA. The binding ability of complexes 1 and 2 with CT DNA was considered by considering their effects on UV absorption. The Kb values were measured in Tris–HCl buffer (pH 7.4) by changing the concentration of DNA from 0 to 100 μM while keeping the complex concentration constant (5 μM). The absorption spectra of 1 and 2 in the absence and presence of increasing amounts of CT DNA are shown in Fig. 2.
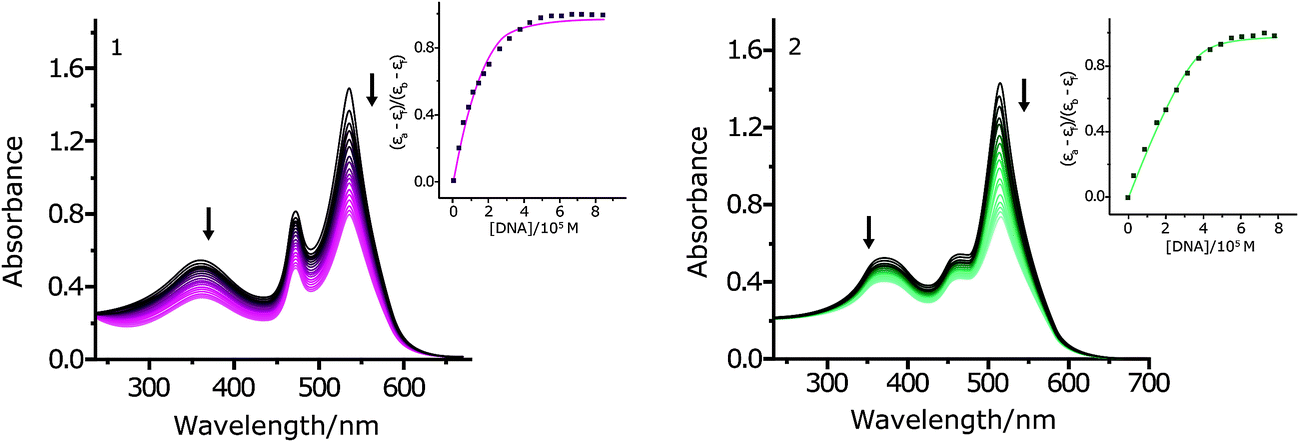 | ||
| Fig. 2 Absorption spectra of complexes 1 and 2 in Tris–HCl, pH 7.2, buffer upon addition of CT DNA. [Complex] = 5.0 μM, [DNA] = 0–100 μM. The arrows show the direction of increasing DNA concentration. | ||
The complexes 1 and 2 exhibited hypochromicity upon adding CT DNA. Such marked changes in the intensity of spectral bands confirm an intimate association of the complex with CT DNA. The Kb values of complexes 1 and 2 are (4.39 ± 0.2) × 105 M−1 and 1.31 × 106 M−1, respectively (Fig. 2, inset). Moreover, the s values of complexes 1 and 2 are (0.51 ± 0.03) and (0.91 ± 0.03), respectively. A low value of s (<1) could be due to an aggregation of hydrophobic molecules on the DNA surface by π stacking.51
This result suggests that the two complexes maybe bind to DNA by partial intercalation, involving a strong stacking interaction between the aromatic chromophore and the base pairs of the DNA. In addition, complex 2 binds with DNA more strongly than the complex 1 due to the stronger electrostatic interaction between DNA and complex 2.
The binding of the complexes 1 and 2 to CT DNA was further considered by a competitive binding fluorescence assay using ethidium bromide (EB). The EB–DNA system usually displays a major increase in the fluorescence emission when EB binds to DNA, and a decrease is observed when EB is displaced by another DNA intercalate molecule.52,53
Fig. 3 shows the fluorescence spectra of the EB–DNA system on addition of increasing amounts of complexes 1 and 2. The intensity of the emission band at 601 nm of the EB–DNA system decreases by increasing concentrations of 1 and 2.
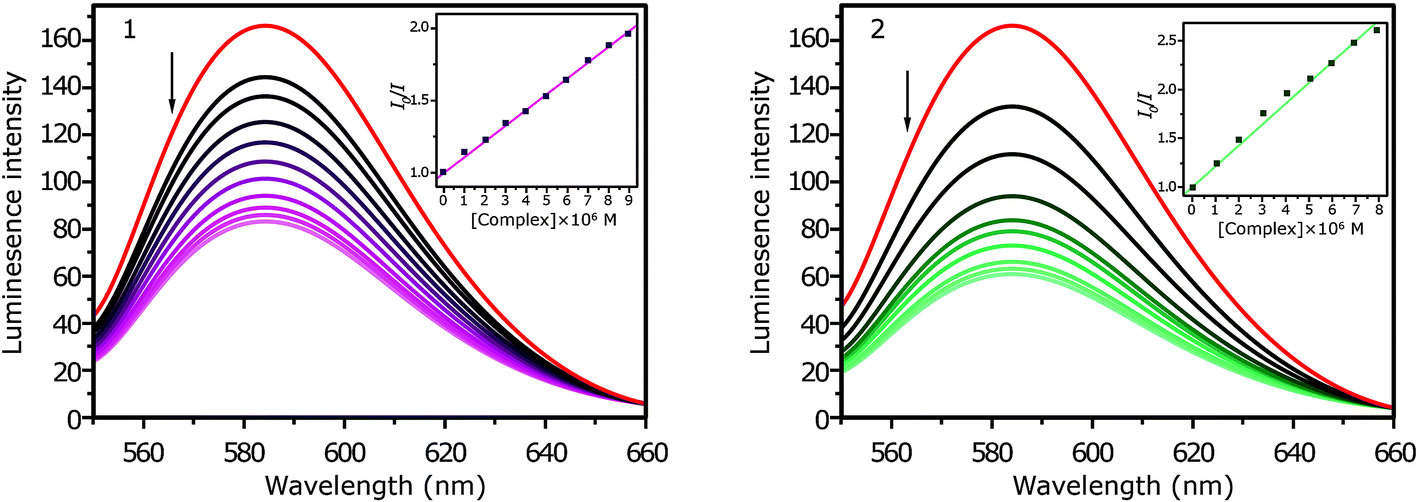 | ||
| Fig. 3 Fluorescence quenching curves of EtBr bound to DNA: 1 and 2. [DNA] = 10 μM, [EB] = 10 μM, [complex] = 0–100 μM (inset: plot of I0/I vs. [Q]). | ||
The Ksv values that were obtained from the experiments are (1.02 ± 0.01) × 105 M−1 and (2.23 ± 0.03) × 105 M−1 for complexes 1 and 2, respectively (Fig. 3, inset). Kapp was found to be (1.15 ± 0.02) × 105 M−1 and (2.62 ± 0.02) × 105 M−1 for complexes 1 and 2, respectively.
These results show the same magnitude of the intrinsic binding constants determined from the absorption spectra titration, further demonstrating the absorption spectral observations that the complex 2 with the ligand PIQ can intercalate to DNA more strongly than the complex 1.
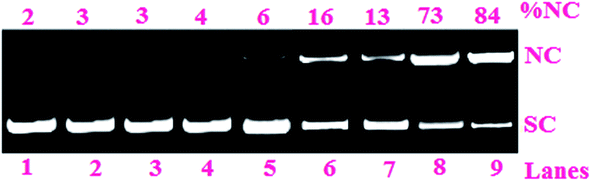 | ||
| Fig. 4 Cleavage of SC pUC19 DNA (0.2 μg, 30 μM) by the complexes 1 and 2 (100 μM) in 50 mM Tris–HCl/NaCl buffer (pH, 7.2) on photoirradiation at 500 nm for 1 h: (lane 1) DNA control; (lane 2) DNA + HL; (lane 3) DNA + PPY; (lane 4) DNA + PIQ; (lane 5) DNA + Pcyd-BODIPY; (lane 6) DNA + 1 (in dark); (lane 7), DNA + 2 (in dark); (lane 8) DNA + 1; (lane 9) DNA + 2. | ||
Addition of hydroxyl radical scavengers like DMSO, KI, catalase and superoxide radical scavengers such as superoxide dismutase, SOD did not inhibit the DNA photocleavage reaction whereas singlet oxygen quenchers such as 2,2,6,6-tetramethyl-4-piperidone (TEMP), 1,4-diazabicyclo-[2.2.2]octane (DABCO), and NaN3 inhibited the DNA photocleavage activity significantly (Fig. 5). These results suggested 1O2 are likely to be the reactive species that contribute to the cleavage process.
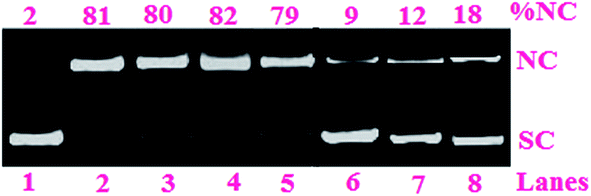 | ||
| Fig. 5 Photo-induced DNA cleavage activity of complex 2 (100 μM) in the presence of different additives at 500 nm for an exposure time of 1 h: (lane 1) DNA control; (lane 2) DNA + DMSO (2 μL) + 2; (lane 3) DNA + KI (0.2 mM) + 2; (lane 4) DNA + catalase (4 U) + 2; (lane 5) DNA + SOD (4 U) + 2; (lane 6) DNA + TEMP (0.2 mM) + 2; (lane 7) DNA + DABCO (0.2 mM) + 2; (lane 8) DNA + NaN3 (0.2 mM) + 2. | ||
2.5. Biological activity
| IC50 μM | ||||
|---|---|---|---|---|
| Pcyd-BODIPY | 1 | 2 | Cisplatin | |
| a 24 h incubation.b Irradiation with light at 500 nm (10 J cm−2). | ||||
| HeLa (dark)a | 78.52 ± 0.2 | 59.31 ± 0.3 | 60.11 ± 0.2 | 9.2 ± 1.7 |
| HeLa (light)b | 11.34 ± 0.4 | 0.78 ± 0.1 | 0.53 ± 0.1 | 27.5 ± 1.1 |
| PI (HeLa) | 6.92 | 76.04 | 113.41 | — |
| MRC-5 (dark)a | 80.15 ± 0.2 | 60.58 ± 0.1 | 61.03 ± 0.1 | 17.2 ± 1.3 |
| MRC-5 (light)b | 53.41 ± 0.1 | 23.82 ± 0.1 | 31.41 ± 0.2 | 16.4 ± 1.1 |
| PI (MRC-5) | 1.50 | 2.54 | 1.94 | — |
The phototoxicity index (PI = IC50 (dark)/IC50 (light)) values of complexes 1 and 2 in HeLa cells are 76.04 and 113.41, respectively. These values are auspicious considering that the commonly used photosensitizer available on the market (photofrin) with PI > 10.55
The phototoxicity of complexes 1 and 2 on the MRC-5 cell line was also considered (Table 1), which followed the same trend observed for HeLa cells. Notably, 1 and 2 are only moderately photocytotoxic against the nontumorigenic MRC-5 cell line (IC50 = 23.82 and 31.41 μM).
The ligand Pcyd-BODIPY is less toxic in the dark with IC50 values of ∼78 μM, but was significantly photocytotoxic in HeLa cells when irradiated with visible light of 500 nm giving IC50 value of 11.34 ± 0.4 μM (Table 1). Also, Pcyd-BODIPY is moderately photocytotoxic against the nontumorigenic MRC-5 cell line with IC50 = 53.41 μM.
These complexes are comparable with cyclometalated Ir(III) complexes of styryl-BODIPY ligands with IC50 values 2.58–6.18 μM and 8.16–9.81 μM on photoirradiation and in the dark for LLC cells (lung cancer cells) cell lines, respectively.56
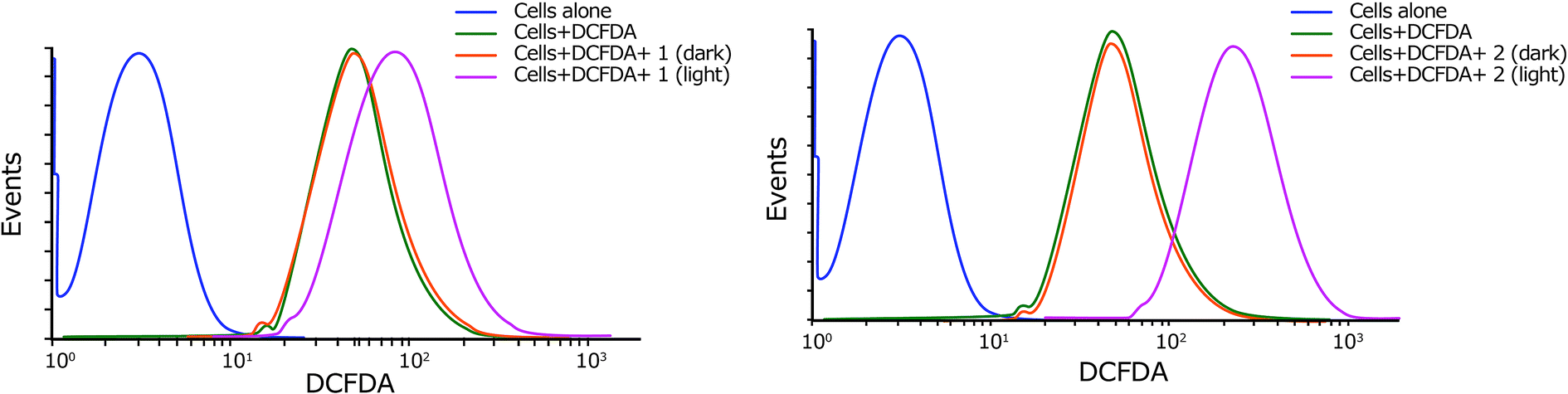 | ||
| Fig. 6 Cellular ROS generation by complexes 1 and 2 in HeLa cells in dark or light conditions as estimated by DCFDA assay (pre-incubation: 4 h in dark, light exposure: 500 nm, 1 h). | ||
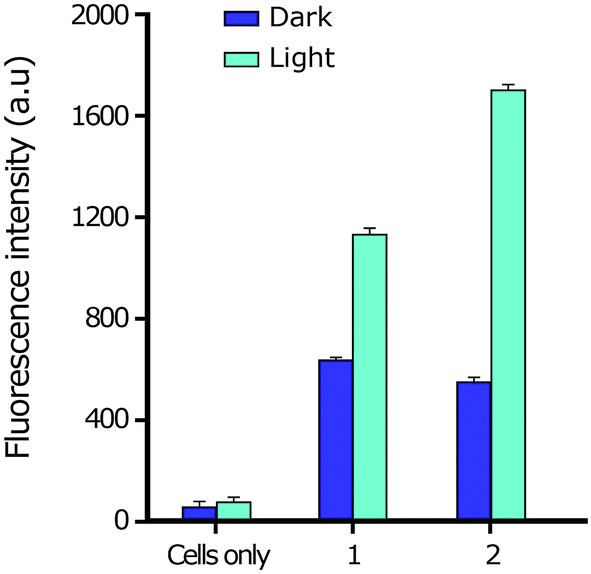 | ||
| Fig. 7 DCFH-DA assay showing generation of ROS in HeLa cells treated with complexes 1 and 2 for 4 h in the dark and exposed to light (500 nm). | ||
To evaluate the number of cells undergoing apoptosis, the Annexin-V-FITC/PI assay was performed by complexes 1 and 2 in HeLa cells. Cells were incubated with the complexes for 4 h in dark followed by light irradiation (500 nm). Complex 2 induced features of early apoptosis in ∼86% of the cells upon light (500 nm) exposure in comparison with ∼64% for complex 1 under identical experimental conditions. The observed early apoptotic cell population in the dark was 16.46% and 16.17% for 1 and 2, respectively, demonstrating the importance of light exposure to observe apoptotic cell death (Fig. 8).
 | ||
| Fig. 8 Dot plots of annexinV-FITC/PI assay on HeLa cells treated with complexes 1 and 2 either kept in dark or exposed to photoreactor (500 nm) as labeled showing % cell populations in respective quadrants: FITC−/PI− lower left (Q3), viable cells; FITC+/PI− lower right (Q4), early apoptotic cells; FITC+/PI+ upper right (Q2), late apoptotic cells; FITC−/PI+ upper left (Q1), necrotic cells. Representative data from three independent experiments are shown (pre-incubation: 4 h in dark, light exposure: 500 nm, 1 h). | ||
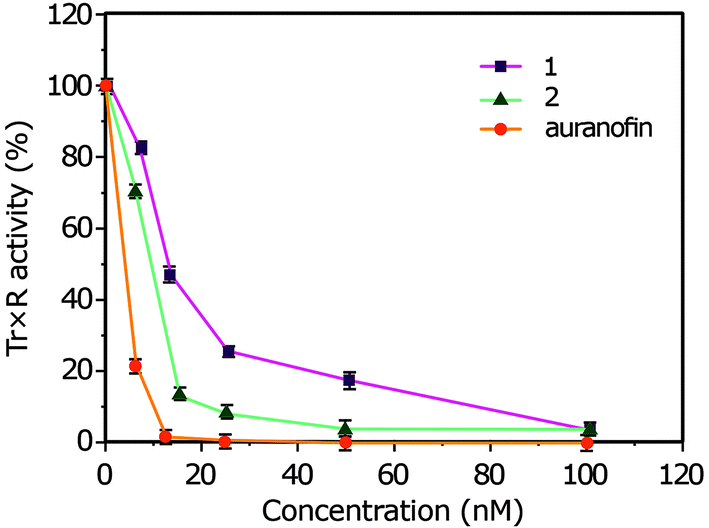 | ||
| Fig. 9 Inhibitory effects of complexes 1 and 2 on TrxR. Error bars indicate the standard deviation. | ||
3. Conclusions
Two new cyclometalated Ir(III) complexes bearing NCN pincer, meso-phenylcyanamide BODIPY, and 2-phenylpyridine (1) or 1-phenylisoquinoline (2) ligands have been demonstrated as photodynamic therapy agents (PDT). The DNA-binding interactions of the complexes with CT DNA display that the binding mode is mainly non-covalent via intercalation. The complexes 1 and 2 show cleavage SC-DNA efficiently on photoactivation at 500 nm with the formation of singlet oxygen (1O2) as reactive species to contribute in the cleavage process. The complexes 1 and 2 are significantly less toxic in the dark with IC50 values of ∼60 μM, while, upon light irradiation (500 nm), 1 and 2 were significantly photocytotoxic in HeLa cells with IC50 values of 0.78 ± 0.1 μM and 0.53 ± 0.1 μM, respectively. These complexes showed very good uptake and efficient ROS generation in HeLa cell lines upon photoexposure to light of 500 nm. Moreover, the complexes 1 and 2 revealed exceptional TrxR-inhibitory activities in comparison with auranofin. Therefore, the selenoenzyme TrxR may be as a protein target for cyclometalated Ir(III) complexes 1 and 2. Overall, these designing of cyclometalated iridium complexes can open new opportunities for metal-based PDT agents that may be used for a wide variety of applications as potential anticancer drugs.4. Experimental
4.1. General procedures, materials and physical measurements
All manipulations were carried out under nitrogen atmosphere using standard Schlenk techniques and vacuum-line systems. Work-up procedures were performed in air. All materials used were received from commercial sources unless stated otherwise. The pincer ligand HL was synthesized as previously reported by us.41 8-(4-Anilino) BODIPY (boron dipyrromethene difluoro) was prepared according to literature method.60 All solvents were dried by standard procedures and freshly distilled prior to use. Human cervical cancer (HeLa), human fibroblast (MRC-5) nontumorigenic cells, and human plasma were supplied by the National Cell Bank of Pasteur Institute, Tehran, Iran. All reagents and cell culture media were purchased from Gibco Company (Germany). CT DNA was obtained from Sigma-Aldrich.Fourier transform infrared spectra were recorded on a PerkinElmer Spectrum 400 (FT-IR/FT-NIR spectrometer). 1H, 13C {1H} nuclear magnetic resonance (NMR) spectra were recorded on a Bruker-400 MHz spectrometer at ambient temperature in DMSO-d6. Elemental analyses were performed with an EA 3000 CHNS. Electronic absorption spectra were recorded on a JASCO 7580 UV-vis-NIR double-beam spectrophotometer. Emission intensity measurements were carried out using a Hitachi F 4500 spectrofluorometer. Luminescence quantum yields were measured in degassed acetonitrile using [Ru(bpy)3]Cl2 as the reference.
4.2. Synthesis of ligands and the complexes 1 and 2
:1) produced analytically pure orange solid (304 mg, 83.4%, and 1 mmol). Anal. calc. (%) for C20H19BF2N4 (364.19): C, 65.96; H, 5.26; N, 15.38; found (%): C, 65.92; H, 5.22; N, 15.33. IR (KBr): 2225 (vs., NCN), 1548, 1512 (m, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN), 1192 (m, B–F) cm−1. TOF-MS: 365 [M + H]+. 1H NMR (DMSO-d6): δ 8.132 (s, 1H, H–NH), 6.95 (d, 2H, H-11a or 12a, 3J 7.6), 6.77 (d, 2H, H-11a or 12a, 3J 7.6), 6.09 (s, 2H, H-2a and H-6a), 2.28 (s, 6H, H-9a), 1.60 (s, 6H, H-10a). 13C NMR (DMSO-d6): δ 150.0 (Ar), 140.1 (Ar), 138.0 (Ar), 136.0 (Ar), 133.1 (Ar), 131.6 (Ar), 129.9 (Ar), 127.1 (Ar), 116.8 (Ar), 112.0 (NCN), 12.5 (C–CH3), 10.2 (C–CH3).:1, v/v) was refluxed under an inert atmosphere of nitrogen for 12 h. To this solution, PPY (2-phenylpyridine) (155 mg, 1 mmol) and Pcyd-BODIPY (8-(4-phenylcyanamide) BODIPY) (364 mg, 1 mmol) in MeOH/CH2Cl2 (30 mL, 1:2, v/v) was added. The pH of the solution was adjusted to ∼7 using triethylamine. The solution was refluxed again under an inert atmosphere for 24 h. The reaction mixture was then cooled to room temperature and then evaporated under vacuum. The residual orange solid was dissolved in CH2Cl2 and purified by column chromatography on silica gel. The desired product was eluted with CH2Cl2/MeOH (10:1, v/v) and subsequent recrystallized from CH2Cl2/diethyl ether to afford an orange solid (954 mg, 88.6% yield, and 1 mmol). Anal. calc. (%) for C54H45BF2IrN9O (1077.02): C, 60.22; H, 4.21; N, 11.70; found (%): C, 60.18; H, 4.18; N, 11.65. IR (KBr): 2092 (vs., NCN), 1542, 1507 (m, CC, CN), 1187 (m, B–F) cm−1. TOF-MS: 1078 [M + H]+. 1H NMR (DMSO-d6): δ 8.31 (d, 2H, H-4′, 3J 8.0), 8.24 (d, 1H, H-5b, 3J 7.6), 7.96 (t, 1H, H-4b, 3J 7.6), 7.86 (d, 1H, H-12b, 3J 7.6), 7.68 (d, 1H, H-2b, 3J 7.6), 7.25 (t, 2H, H-6′, 3J 7.6), 6.94–7.01 (m, 9H, H-phenyl), 6.75 (s, 2H, H-3,5), 6.61 (d, 2H, H-11a or 12a, 3J 7.6), 6.35 (d, 1H, H-9b, 3J 7.6), 6.00 (s, 2H, H-2a,6a), 3.80 (s, 3H, H-7), 3.49 (s, 6H, H-10′), 2.09 (s, 6H, H-9a), 1.60 (s, 6H, H-10a). 13C NMR (DMSO-d6): δ 162.9 (Ar), 159.9 (Ar), 153.1 (Ar), 149.9 (Ar), 147.9 (Ar), 144.1 (Ar), 141.0 (Ar), 138.5 (Ar), 136.6 (Ar), 134.5 (Ar), 131.1 (Ar), 129.9 (Ar), 127.5 (Ar), 125.8 (Ar), 123.0 (Ar), 121.1 (Ar), 119.2 (Ar), 117.9 (Ar), 114.9 (Ar), 110.0 (NCN), 51.4 (C-7), 29.9 (C-10′), 11.0 (C–CH3), 9.0 (C–CH3).:1, v/v) and subsequent recrystallized from CH2Cl2/diethyl ether to afford an orange solid (927 mg, 82.3% yield, and 1 mmol). Anal. calc. (%) for C58H47BF2IrN9O (1127.08): C, 61.81; H, 4.20; N, 11.18; found (%): C, 61.76; H, 4.16; N, 11.12. IR (KBr): 2088 (vs., NCN), 1538, 1503 (m, CC, CN), 1185 (m, B–F) cm−1. TOF-MS: 1128 [M + H]+. 1H NMR (DMSO-d6): δ 9.04 (d, 1H, H-8b, 3J 7.6), 8.80 (d, 2H, H-4′, 3J 8.0), 8.39 (d, 1H, H-16b, 3J 7.6), 8.05 (d, 1H, H-5b, 3J 7.6), 7.92 (t, 1H, H-7b, 3J 7.6), 7.80 (t, 1H, H-6b, 3J 7.6), 7.60 (d, 2H, H-3b,4b, 3J 7.6), 7.29 (t, 1H, H-15, 3J 7.6), 6.99–7.06 (m, 10H, H-phenyl), 6.79 (s, 2H, H-3,5), 6.61 (d, 2H, H-11a or 12a, 3J 7.6), 6.00 (s, 2H, H-2a,6a), 3.70 (s, 3H, H-7), 3.40 (s, 6H, H-10′), 2.18 (s, 6H, H-9a), 1.49 (s, 6H, H-10a). 13C NMR (DMSO-d6): δ 166.1 (Ar), 162.3 (Ar), 159.9 (Ar), 156.9 (Ar), 154.0 (Ar), 151.2 (Ar), 150.0 (Ar), 148.0 (Ar), 144.9 (Ar), 142.0 (Ar), 139.1 (Ar), 137.1 (Ar), 135.0 (Ar), 132.6 (Ar), 130.9 (Ar), 128.0 (Ar), 125.9 (Ar), 122.6 (Ar), 121.1 (Ar), 118.5 (Ar), 117.1 (Ar), 113.0 (Ar), 109.9 (NCN), 52.5 (C-7), 28.5 (C-10′), 12.1 (C–CH3), 8.0 (C–CH3).
C, CN), 1192 (m, B–F) cm−1. TOF-MS: 365 [M + H]+. 1H NMR (DMSO-d6): δ 8.132 (s, 1H, H–NH), 6.95 (d, 2H, H-11a or 12a, 3J 7.6), 6.77 (d, 2H, H-11a or 12a, 3J 7.6), 6.09 (s, 2H, H-2a and H-6a), 2.28 (s, 6H, H-9a), 1.60 (s, 6H, H-10a). 13C NMR (DMSO-d6): δ 150.0 (Ar), 140.1 (Ar), 138.0 (Ar), 136.0 (Ar), 133.1 (Ar), 131.6 (Ar), 129.9 (Ar), 127.1 (Ar), 116.8 (Ar), 112.0 (NCN), 12.5 (C–CH3), 10.2 (C–CH3).:1, v/v) was refluxed under an inert atmosphere of nitrogen for 12 h. To this solution, PPY (2-phenylpyridine) (155 mg, 1 mmol) and Pcyd-BODIPY (8-(4-phenylcyanamide) BODIPY) (364 mg, 1 mmol) in MeOH/CH2Cl2 (30 mL, 1:2, v/v) was added. The pH of the solution was adjusted to ∼7 using triethylamine. The solution was refluxed again under an inert atmosphere for 24 h. The reaction mixture was then cooled to room temperature and then evaporated under vacuum. The residual orange solid was dissolved in CH2Cl2 and purified by column chromatography on silica gel. The desired product was eluted with CH2Cl2/MeOH (10:1, v/v) and subsequent recrystallized from CH2Cl2/diethyl ether to afford an orange solid (954 mg, 88.6% yield, and 1 mmol). Anal. calc. (%) for C54H45BF2IrN9O (1077.02): C, 60.22; H, 4.21; N, 11.70; found (%): C, 60.18; H, 4.18; N, 11.65. IR (KBr): 2092 (vs., NCN), 1542, 1507 (m, CC, CN), 1187 (m, B–F) cm−1. TOF-MS: 1078 [M + H]+. 1H NMR (DMSO-d6): δ 8.31 (d, 2H, H-4′, 3J 8.0), 8.24 (d, 1H, H-5b, 3J 7.6), 7.96 (t, 1H, H-4b, 3J 7.6), 7.86 (d, 1H, H-12b, 3J 7.6), 7.68 (d, 1H, H-2b, 3J 7.6), 7.25 (t, 2H, H-6′, 3J 7.6), 6.94–7.01 (m, 9H, H-phenyl), 6.75 (s, 2H, H-3,5), 6.61 (d, 2H, H-11a or 12a, 3J 7.6), 6.35 (d, 1H, H-9b, 3J 7.6), 6.00 (s, 2H, H-2a,6a), 3.80 (s, 3H, H-7), 3.49 (s, 6H, H-10′), 2.09 (s, 6H, H-9a), 1.60 (s, 6H, H-10a). 13C NMR (DMSO-d6): δ 162.9 (Ar), 159.9 (Ar), 153.1 (Ar), 149.9 (Ar), 147.9 (Ar), 144.1 (Ar), 141.0 (Ar), 138.5 (Ar), 136.6 (Ar), 134.5 (Ar), 131.1 (Ar), 129.9 (Ar), 127.5 (Ar), 125.8 (Ar), 123.0 (Ar), 121.1 (Ar), 119.2 (Ar), 117.9 (Ar), 114.9 (Ar), 110.0 (NCN), 51.4 (C-7), 29.9 (C-10′), 11.0 (C–CH3), 9.0 (C–CH3).:1, v/v) and subsequent recrystallized from CH2Cl2/diethyl ether to afford an orange solid (927 mg, 82.3% yield, and 1 mmol). Anal. calc. (%) for C58H47BF2IrN9O (1127.08): C, 61.81; H, 4.20; N, 11.18; found (%): C, 61.76; H, 4.16; N, 11.12. IR (KBr): 2088 (vs., NCN), 1538, 1503 (m, CC, CN), 1185 (m, B–F) cm−1. TOF-MS: 1128 [M + H]+. 1H NMR (DMSO-d6): δ 9.04 (d, 1H, H-8b, 3J 7.6), 8.80 (d, 2H, H-4′, 3J 8.0), 8.39 (d, 1H, H-16b, 3J 7.6), 8.05 (d, 1H, H-5b, 3J 7.6), 7.92 (t, 1H, H-7b, 3J 7.6), 7.80 (t, 1H, H-6b, 3J 7.6), 7.60 (d, 2H, H-3b,4b, 3J 7.6), 7.29 (t, 1H, H-15, 3J 7.6), 6.99–7.06 (m, 10H, H-phenyl), 6.79 (s, 2H, H-3,5), 6.61 (d, 2H, H-11a or 12a, 3J 7.6), 6.00 (s, 2H, H-2a,6a), 3.70 (s, 3H, H-7), 3.40 (s, 6H, H-10′), 2.18 (s, 6H, H-9a), 1.49 (s, 6H, H-10a). 13C NMR (DMSO-d6): δ 166.1 (Ar), 162.3 (Ar), 159.9 (Ar), 156.9 (Ar), 154.0 (Ar), 151.2 (Ar), 150.0 (Ar), 148.0 (Ar), 144.9 (Ar), 142.0 (Ar), 139.1 (Ar), 137.1 (Ar), 135.0 (Ar), 132.6 (Ar), 130.9 (Ar), 128.0 (Ar), 125.9 (Ar), 122.6 (Ar), 121.1 (Ar), 118.5 (Ar), 117.1 (Ar), 113.0 (Ar), 109.9 (NCN), 52.5 (C-7), 28.5 (C-10′), 12.1 (C–CH3), 8.0 (C–CH3).4.3. Singlet oxygen quantum yields
The singlet oxygen quantum yields were measured by two different methods, direct and indirect evaluation, as previously reported.374.4. DNA binding studies
The complexes were dissolved in 10 mM Tris–HCl buffer (pH 7.4). The concentration of CT DNA was determined from the UV absorption intensity at 260 nm with ε = 6600 M−1 cm−1.61 The CT DNA solution in Tris–HCl buffer gave an absorbance ratio at 260 and 280 nm (A260/A280) of about 1.8–1.9 indicating that the CT DNA was sufficiently free of protein. The DNA concentration was varied between 0 and 100 μM, while the complex concentration was kept constant at 5.0 μM at room temperature.The intrinsic equilibrium binding constant (Kb) and the binding site size (s) values were obtained from a nonlinear fitting according to the equation:62
| (εa − εf)/(εb − εf) = [b − (b2 − 2Kb2Ct[DNA]/s)1/2]/2KbCt | (1) |
| b = 1 + KbCt + Kb[DNA]/2s | (2) |
The observed linearity in the plot of I0/I vs. the concentration ratio of the complexes to DNA is in good agreement with the linear Stern–Volmer equation:63
| I0/I = 1 + Ksv[Q] |
The apparent binding constant (Kapp) of the complexes could be calculated using the following equation:
| Kapp = KEB[EB]50%/[complex]50% |
The photocleavage of SC pUC19 DNA (0.2 μg, 30 μM) by the complexes 1 and 2 (30 μM, 0.2 μg) in presence of complexes (100 μM) was performed in Tris–HCl/NaCl buffer (50 mM, pH 7.2) by photo-irradiation of the samples with light of 500 nm (10 J cm−2) by agarose gel electrophoresis. Mechanistic studies were performed using different additives as ROS scavengers/quenchers (TEMP, 0.2 mM; DABCO, 0.2 mM; NaN3, 0.2 mM; KI, 0.2 mM; DMSO, 2 μL; catalase, 4 units; SOD, 4 units) prior to the addition of the complexes. After incubation of the sample for 1 h in dark and quenched by gel loading dye, solution was loaded on 1% agarose gel having 1 μg mL−1 ethidium bromide. Electrophoresis was run for 2.0 h at 60 V in Tris–acetate EDTA (TAE) buffer in dark room. The quantification of cleavage products was performed using UVITEC Fire Reader V4 gel documentation system and UVI band software. The error observed in measuring the band intensities was in the range of 4–7%.
4.5. Stability studies
The stability of the complexes in plasma was assessed using a procedure analogous to that recently reported.37 An aliquot (25 mL) of a solution containing the respective complexes 1 and 2 (final concentration 20 μM) and diazepam (final concentration 10 μM) in DMSO was added to human plasma solution (975 mL) to give a total volume of 1000 mL. The resulting mixture was incubated for 72 h at 37 °C with continuous and gentle shaking (ca. 300 rpm). The reaction was stopped by adding acetonitrile (2 mL), and the mixture was centrifuged for 45 min at 1000g at 4 °C. The acetonitrile was evaporated, and the residue was suspended in acetonitrile/H2O (1:1, v/v; 200 mL). The suspension was filtered and the filtrate was analyzed by HPLC-UV. A 0.1 mL aliquot of the solution was injected into an HPLC system (Thermo, USA) connected to a UV/Vis spectrophotometer. A Hypersil Gold Dim (100 × 2.1 mm, Thermo, USA) reversed-phase column was used at a flow rate of 0.5 mL min −1. The runs were performed with a linear gradient of A (acetonitrile (Sigma-Aldrich HPLC-grade)) and B (distilled water containing 0.1% HCOOH): t = 0–3 min, 20% A; t = 7 min, 50% A; t = 20 min, 90% A; t = 23 min, 100% A; t = 25 min, 100% A; t = 28 min, 20% A.
Recoveries of diazepam and complexes 1 and 2 were determined by comparing the peak area ratios of the spiked drug-free plasma to the peak area ratios obtained by direct injection of the solutions of the same concentration of complexes 1 and 2 or diazepam (20 μM for complexes 1 and 2 and 10 μM for diazepam).
4.6. Cell culture
Human cervical cancer (HeLa) and human fibroblast (MRC-5) nontumorigenic cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS, 100 IU mL−1 of penicillin, 100 μg mL−1 of streptomycin and 2 mM Glutamax at 37 °C in a humidified incubator at 5% CO2. The adherent cultures were grown as monolayer and were passaged once in 4–5 days by trypsinizing with 0.25% trypsin–EDTA.4.7. Cytotoxicity
The photocytotoxicity of the complexes was studied using a standard MTT assay.64 Approximately 8000 HeLa cancer cells and MRC-5 nontumorigenic cells were plated separately in two different 96 wells culture plates in DMEM containing 10% FBS. Stock solutions of the complexes were prepared by dissolving the complexes in aqueous solutions with DMSO as the co-solvent and diluting with cell culture medium to the desired concentrations. The final DMSO concentration never exceeded 1% v/v. Cells were then treated with increasing concentrations of compounds for 24 h. Dark controls, or cytotoxicity (CT) assays, refer to assays that include metal complex but were not exposed to light, and light controls refer to light exposed assays that did not contain metal complex. Photocytotoxicity (PCT) assays contained metal complex and were exposed to light. For phototoxicity studies, cells were treated for 4 h only with increasing concentrations of the compounds. After that, the medium was removed and replaced by fresh complete medium prior to 20 min irradiation at 50 nm (10 J cm −2). After 44 h in the incubator, the medium was replaced by 100 mL complete medium containing resazurin (0.2 mg mL−1 final concentration). After 4 h incubation at 37 °C, fluorescence of the highly red fluorescent resorufin product was quantified at 590 nm emission with 540 nm excitation wavelength in a SpectraMax M5 microplate reader. Light doses were evaluated with a Gigahertz Optic X1-1 optometer. Cell counts were carried out immediately after light exposure. All experiments were carried out in triplicate, and the graphed data is the average of three trials.The LED light sources were monitored using an International Light Technologies Powermeter (ILT2400) equipped with SED 623 detector and spectral range 200–2100 nm. Light doses were evaluated with a Gigahertz Optic X1-1 optometer. The light doses employed in this work are comparable to those employed for activation of other metal-based phototoxic compounds (2–11.5 J cm−2 at 400–600 nm).65–69
4.8. ICP-MS
Cells grown in a 60 mm tissue culture dish were incubated with the iridium(III) complexes (100 μM) in glucose-free medium at 37 °C under a 5% CO2 atmosphere for 5 min. The medium was removed, and the cell layer was washed gently with 10 mL PBS. Then, the cells were digested with 65% HNO3 (2 mL) at 50 °C for 2 h and trypsinized and harvested with 20 mL PBS. The solution was analyzed using NexION 2000 B ICP Mass.4.9. DCFDA assays
DCFDA assay was done to quantify ROS on the oxidative production of fluorescent DCF (λem = 525 nm). HeLa cells (2 × 105 cells plated in duplicate) were incubated in the presence or absence of complexes 1 and 2 (concentration = IC50 values in light) for 4 h in the dark with or without photoirradiation in DPBS (500 nm, 1 h). Cells were trypsinized and treated with 1 μM DCFDA for 15 min, and the stained cells were determined by flow cytometry.4.10. Annexin V-FITC/PI assay
HeLa cells (2 × 105) were incubated (concentration = IC50 values in light) for 24 h followed by light treatment in DPBS (500 nm). Cells were further kept for 12 h post-treatment and then trypsinized. Annexin VFITC (2 μL) and propidium iodide (PI; 1 μL) were then added to clear suspensions of cells in 500 μL of a 1× binding buffer and kept in the dark for 10 min. The percent cell population in each quadrant was estimated by FACS analysis. Experiments were performed in triplicate along with untreated controls for reference.4.11. TrxR inhibition
Thioredoxin reductases (Trx-R) sourced from rat liver was obtained from Sigma-Aldrich. It is a buffered aqueous glycerol solution, ≥100 units per mg protein, in 50 mM Tris–HCl, pH 7.5, 300 mM NaCl, 1 mM EDTA, and 10% glycerol. The complexes 1, 2, and auranofin were preincubated for 5 min at room temperature; the reaction started with 2 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), and the increase of the absorbance was monitored at 412 nm over 5 min at 25 °C. The reaction velocity in the presence of inhibitor was normalized relative to the control to generate percent activity and plots of percent activity versus concentration were constructed to obtain IC50 values (that is, the concentration that inhibited 50% of enzyme activity).Acknowledgements
We are grateful for general support from the Department of Chemistry, Isfahan University of Technology (IUT).References
- R. Bonnett, Chemical aspects of photodynamic therapy, CRC Press, 2000 Search PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- M. R. Hamblin and T. Hasan, Photochem. Photobiol. Sci., 2004, 3, 436–450 CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- http://www.theralase.com/pressrelease/health-canada-approves-clinical-trialapplication-anti-cancer-drug/.
- P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2016, 15, 481 CAS.
- J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2015, 14, 2014 CAS.
- T. Sainuddin, J. McCain, M. Pinto, H. Yin, J. Gibson, M. Hetu and S. A. McFarland, Inorg. Chem., 2016, 55, 83–95 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, J. Heier, S. Ferrari and G. Gasser, Dalton Trans., 2014, 43, 4287–4294 RSC.
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- L. He, Y. Li, C.-P. Tan, R.-R. Ye, M.-H. Chen, J.-J. Cao, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- L. K. McKenzie, I. V. Sazanovich, E. Baggaley, M. Bonneau, V. Guerchais, J. A. Williams, J. A. Weinstein and H. E. Bryant, Chemistry, 2017, 23, 234–238 CrossRef CAS PubMed.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466–3479 CrossRef CAS.
- R. E. Doherty, I. V. Sazanovich, L. K. McKenzie, A. S. Stasheuski, R. Coyle, E. Baggaley, S. Bottomley, J. A. Weinstein and H. E. Bryant, Sci. Rep., 2016, 6, 22668, DOI:10.1038/srep22668.
- A. Kando, Y. Hisamatsu, H. Ohwada, T. Itoh, S. Moromizato, M. Kohno and S. Aoki, Inorg. Chem., 2015, 54, 5342–5357 CrossRef CAS PubMed.
- R. Cao, J. L. Jia, X. C. Ma, M. Zhou and H. Fei, J. Med. Chem., 2013, 56, 3636–3644 CrossRef CAS PubMed.
- C. Yang, F. Mehmood, T. L. Lam, S. L. F. Chan, Y. Wu, C. S. Yeung, X. G. Guan, K. Li, C. Y. S. Chung, C. Y. Zhou, T. T. Zou and C. M. Che, Chem. Sci., 2016, 7, 3123–3136 RSC.
- W. H. Wang, Z. F. Mao, M. D. Wang, L. J. Liu, D. W. J. Kwong, C. H. Leung and D. L. Ma, Chem. Commun., 2016, 52, 3611–3614 RSC.
- D. Maggioni, M. Galli, L. DAlfonso, D. Inverso, M. V. Dozzi, L. Sironi, M. Iannacone, M. Collini, P. Ferruti, E. Ranucci and G. D'Alfonso, Inorg. Chem., 2015, 54, 544–553 CrossRef CAS PubMed.
- L. He, Y. Li, C. P. Tan, R. R. Ye, M. H. Chen, J. J. Cao, L. N. Ji and Z. W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- F. Xue, Y. Lu, Z. Zhou, M. Shi, Y. Yan, H. Yang and S. Yang, Organometallics, 2015, 34, 73–77 CrossRef CAS.
- C. Wang, L. Lystrom, H. Yin, M. Hetu, S. Kilina, S. A. McFarland and W. Sun, Dalton Trans., 2016, 45, 16366–16378 RSC.
- J. S. Nam, M. G. Kang, J. Kang, S. Y. Park, S. J. C. Lee, H. T. Kim, J. K. Seo, O. H. Kwon, M. H. Lim, H. W. Rhee and T. H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed.
- X. Jiang, J. Peng, J. Wang, X. Guo, D. Zhao and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3591–3600 CAS.
- L. K. McKenzie, I. V. Sazanovich, E. Baggaley, M. Bonneau, V. Guerchais, J. A. G. Williams, J. A. Weinstein and H. E. Bryant, Chem.–Eur. J., 2017, 23, 234–238 CrossRef CAS PubMed.
- W. Lv, Z. Zhang, K. Y. Zhang, H. Yang, S. Liu, A. Xu, S. Guo, Q. Zhao and W. Huang, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 (Angew. Chem., 2016, 128, 10101–10105) CrossRef CAS PubMed.
- Q.-X. Zhou, W.-H. Lei, Y.-J. Hou, Y.-J. Chen, C. Li, B.-W. Zhang and X.-S. Wang, Dalton Trans., 2013, 42, 2786–2791 RSC.
- T. Wang, Y. Hou, Y. Chen, K. Li, X. Cheng, Q. Zhou and X. Wang, Dalton Trans., 2015, 44, 12726–12734 RSC.
- P. Majumdar, X. Yuan, S. Li, B. L. Guennic, J. Ma, C. Zhang, D. Jacqueminde and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC.
- R. Lincoln, A. M. Durantini, L. E. Greene, S. R. Martínez, R. Knox, M. C. Becerra and G. Cosa, Photochem. Photobiol. Sci., 2017, 16, 178–184 CAS.
- K. Krumova and G. Cosa, J. Am. Chem. Soc., 2010, 132, 17560–17569 CrossRef CAS PubMed.
- M. Jazestani, H. Chiniforoshan, L. Tabrizi and P. McArdle, J. Biomol. Struct. Dyn., 2017, 35, 2055–2065 CAS.
- L. Tabrizi and H. Chiniforoshan, Dalton Trans., 2017, 46, 2339–2349 RSC.
- L. Tabrizi, P. McArdle, A. Erxleben and H. Chiniforoshan, Inorg. Chim. Acta, 2015, 438, 94–104 CrossRef CAS.
- L. Tabrizi and H. Chiniforoshan, Invest. New Drugs, 2016, 34, 723–732 CrossRef CAS PubMed.
- L. Tabrizi and H. Chiniforoshan, Dalton Trans., 2016, 45, 18333–18345 RSC.
- M. Jazestani, H. Chiniforoshan, L. Tabrizi, P. McArdle and B. Notash, Inorg. Chim. Acta, 2016, 450, 402–410 CrossRef CAS.
- L. Tabrizi and H. Chiniforoshan, BioMetals, 2017, 30, 59–70 CrossRef CAS PubMed.
- R. J. Crutchley, Coord. Chem. Rev., 2001, 219–221, 125–155 CrossRef CAS.
- L. Tabrizi and H. Chiniforoshan, RSC Adv., 2016, 6, 93349–93355 RSC.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- (a) J. Karolin, L. B. A. Johansson, L. Strandberg and T. Ny, J. Am. Chem. Soc., 1994, 116, 7801–7806 CrossRef CAS; (b) G. Meng, S. Velayudham, A. Smith, R. Luck and H. Liu, Macromolecules, 2009, 42, 1995–2001 CrossRef CAS; (c) J. M. Zimbron, K. Passador, B. Gatin-Fraudet, C.-M. Bachelet, D. Plażuk, L.-M. Chamoreau, C. Botuha, S. Thorimbert and M. Salmain, Organometallics, 2017 DOI:10.1021/acs.organomet.7b00250; (d) T. Rohand, W. Qin, N. Boens and W. Dehaen, Eur. J. Org. Chem., 2006, 4658–4663 CrossRef CAS; (e) X. Yin, Y. Li, Y. Zhu, X. Jing, Y. Li and D. Zhu, Dalton Trans., 2010, 39, 9929–9935 RSC.
- S. P.-Y. Li, C. T.-S. Lau, M.-W. Louie, Y.-W. Lam, S. H. Cheng and K. K.-W. Lo, Biomaterials, 2013, 34, 7519–7532 CrossRef CAS PubMed.
- J. X. Zhang, J. W. Zhou, C. F. Chan, T. C. K. Lau, D. W. J. Kwong, H. L. Tam, N. K. Mak, K. L. Wong and W. K. Wong, Bioconjugate Chem., 2012, 23, 1623–1638 CrossRef CAS PubMed.
- S. P. Foxon, M. A. H. Alamiry, M. G. Walker, A. J. H. M. Meijer, I. V. Sazanovich, J. A. Weinstein and J. A. Thomas, J. Phys. Chem. A, 2009, 113, 12754–12762 CrossRef CAS PubMed.
- J. Yang, Q. Cao, W.-L. Hu, R.-R. Ye, L. He, L.-N. Ji, P. Z. Qin and Z.-W. Mao, Dalton Trans., 2017, 46, 445–454 RSC.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed.
- G. Zuber, J. C. Quada and S. M. Hecht Jr, J. Am. Chem. Soc., 1998, 120, 9368–9369 CrossRef CAS.
- S. M. Hecht, J. Nat. Prod., 2000, 63, 158–168 CrossRef CAS.
- S. Saha, R. Majumdar, M. Roy, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2009, 48, 2652–2663 CrossRef CAS PubMed.
- F. J. Meyer-Almes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS PubMed.
- R. F. Pasternack, M. Caccam, B. Keogh, T. A. Stephenson, A. P. Williams and A. E. J. Gibbst, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS.
- B. Maity, M. Roy, B. Banik, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Organometallics, 2010, 29, 3632–3641 CrossRef CAS.
- E. Delaey, F. van Laar, D. De Vos, A. Kamuhabwa, P. Jacobs and P. de Witte, J. Photochem. Photobiol., B, 2000, 55, 27–35 CrossRef CAS.
- P. Majumdar, X. Yuan, S. Li, B. L. Guennic, J. Ma, C. Zhang, D. Jacqueminde and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC.
- K. K.-W. Lo, W. H.-T. Law, J. C.-Y. Chan, H.-W. Liu and K. Y. Zhang, Metallomics, 2013, 5, 808–812 RSC.
- W. H.-T. Law, L. C.-C. Lee, M.-W. Louie, H.-W. Liu, T. W.-H. Ang and K. K.-W. Lo, Inorg. Chem., 2013, 52, 13029–13041 CrossRef CAS PubMed.
- C. Liu, Z. Liu, M. Li, X. Li, Y. S. Wong, S. M. Ngai, W. Zheng, Y. Zhang and T. Chen, PLoS One, 2013, 8, e53945 CAS.
- A. Cui, X. Peng, J. Fan, X. Chen, Y. Wu and B. Guo, J. Photochem. Photobiol., A, 2007, 186, 85–92 CrossRef CAS.
- Y. Gultneh, A. R. Khan, D. Blaise, S. Chaudhry, B. Ahvani, B. B. Marvey and R. J. Butcher, J. Inorg. Biochem., 1999, 75, 7–18 CrossRef CAS.
- L. Tabrizi, M. Fooladivanda and H. Chiniforoshan, BioMetals, 2016, 29, 981–993 CrossRef CAS PubMed.
- J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421 CrossRef CAS PubMed.
- J. X. Zhang, J. W. Zhou, C. F. Chan, T. C. K. Lau, D. W. J. Kwong, H. L. Tam, N. K. Mak, K. L. Wong and W. K. Wong, Bioconjugate Chem., 2012, 23, 1623 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, V. P. Munk, F. S. Mackay and P. J. Sadler, Chem. Res. Toxicol., 2010, 23, 413 CrossRef CAS PubMed.
- C. Mari, H. Huang, R. Rubbiani, M. Schulze, F. Würthner, H. Chao and G. Gasser, Eur. J. Inorg. Chem., 2017, 2017, 1745–1752 CrossRef CAS.
- A. K. Renfrew, N. S. Bryce and T. Hambley, Chem.–Eur. J., 2015, 21, 15224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05579j |
| This journal is © The Royal Society of Chemistry 2017 |