
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved performance of immobilized lipase by interfacial activation on Fe3O4@PVBC nanoparticles
Zhiming Chen *,
Leilei Liu and
Renchun Yang
*,
Leilei Liu and
Renchun Yang
School of Biological and Chemical Engineering, Anhui Polytechnic University, Wuhu 241000, PR China. E-mail: zmchen@ahpu.edu.cn
First published on 13th July 2017
Abstract
An effective strategy for enhancement of catalytic activity and stability of immobilized lipase by interfacial activation on Fe3O4@PVBC (Fe3O4@polyvinylbenzyl chloride) nanoparticles was developed, which involved the fabrication of core–shelled Fe3O4@PVBC nanoparticles via an emulsion polymerization process and the subsequent immobilization of lipase in phosphate buffer. Due to the magnetic nature of Fe3O4 cores and the presence of chloromethyl-functionalized polymer shells, the Fe3O4@PVBC nanoparticles were employed as valid magnetic carriers for lipase immobilization. Bradford assays indicated that the loading amount of lipase on the Fe3O4@PVBC nanoparticles was calculated to be 162.5 mg protein per g particles. The catalytic activity of the immobilized lipase retained about 99.6 ± 3.3% of the free enzyme activity, which was attributed to interfacial activation of lipase by Fe3O4@PVBC nanoparticles. Thermal and urea tolerance tests revealed that the immobilized lipase exhibited much better stabilities. Additionally, the immobilized lipase retained more than 69.8% of its initial activities after 10 times of reuse. It is believed that the results of the present investigation may provide a versatile approach for designing and fabricating biocatalysts with high activation and stability.
Introduction
Enzymes have attracted much research interest due to their high effectiveness, specificity and mild reaction conditions.1–9 Lipases are ubiquitous enzymes with various biological activities, including high activities, chemo-, regio-, and stereo-selectivities and are widely used in enantioselective hydrolysis and esterification, chiral resolution, synthesis of enantioenriched monomers and macromolecules for polymerization, and other enzymatic reactions.10,11 However, the major drawbacks for their industrial applications are their poor chemical and thermal durability as well as difficulty in recovery and reuse of the enzyme.12–15 To lengthen performance of enzyme for practical applications, immobilization of lipase on various supports is considered to be an effective strategy to improve the handling, stability and recycling.16,17 Previous studies show that the immobilization will produce slight distortions in the enzymes structure and may alter the properties of the enzyme.18,19 Due to the uncontrollable of the changes of proteins structure, the immobilization of lipase was usually associated with a decrease in enzyme activity or a worsening of other catalytic features.19–21 Therefore, the development of strategy for the fabrication of immobilized lipase with high activity and stability is of vital importance for the immobilization of lipase.The increasing knowledge of enzyme structures and mechanism should enable more controlled immobilizations. As we all know that a “lid” composed of either one or two amphiphilic α-helices covers the active site of lipase, blocking substrate access in the closed conformation.22–24 The side of the “lid” facing towards the active site is hydrophobic and the side facing away is hydrophilic.22 In general, increasing the hydrophobicity of the surface in the immediate area surrounding the active site, exposing a large hydrophobic area which can interact with the hydrophobic interface and the catalytic triad becomes accessible to the hydrophobic substrate.25–30 It is expected that hydrophobic carriers would provide more access of the substrate to the active site of the immobilized lipase.
Nowadays, Fe3O4 nanoparticles have been showing great potential applications in lipase immobilization due to their unique magnetic properties, ready availability, and environmentally friendly merits.31,32 Correspondingly, considerable attention has been paid to the fabrication of Fe3O4 nanocomposites to enhance the performance of immobilized lipase.33–40 But due to the complexity of the immobilization process and enzyme structure, most improvements in catalytic performance of the reported immobilization are not always really related to the production of a more active or selective enzyme molecule.18 Bearing this in mind, we fabricated chloromethyl-functionalized Fe3O4@PVBC nanoparticles with a core–shell structure as magnetic carriers for lipase immobilization. The immobilization of lipase on the core–shell Fe3O4@PVBC nanoparticles was conducted and the catalytic performance of the immobilized lipase was then investigated by the hydrolysis of p-nitrophenyl palmitate (pNPP) in phosphate buffer. Hydrolytic activity tests reveal that the immobilized lipase retains about 99.6 ± 3.3% of the free enzyme activity. It was found that thermal stability and urea tolerance of the immobilized lipase were strengthened. Additionally, the immobilized lipase retained more than 69.8% of its initial activities after 10 times reuse.
Experimental
Materials
Candida rugosa lipase (type VII), pNPP, p-nitrophenol (pNP), Coomassie Brilliant Blue G-250 and bovine serum albumin (BSA) protein were purchased from Sigma-Aldrich. Vinylbenzyl chloride (VBC) was purchased from J&K Scientific Ltd. (China) and was purified by vacuum distillation. All other chemicals and reagents were purchased from Aladdin Chemical Co. Ltd. (Shanghai, China), and used without further purification. Deionized water was used throughout.Preparation of Fe3O4@PVBC nanoparticles
The Fe3O4 nanoparticles were synthesized by a solvothermal treatment of Fe(acac)3 in the presence of oleic acid and oleylamine, as described previously.41 After that, the as-obtained Fe3O4 nanoparticles was mixed in 36 mL water with 0.5 mL dodecanol and 200 mg sodium dodecyl sulfate (SDS), and was treated by ultrasound for 10 min with 650 W protected by ice-water bath to obtain Fe3O4 emulsion. 0.5 mL VBC was dispersed in 24 mL solution contained 50 mg SDS and 0.3 mL dodecanol by 30 min of ultrasonic bath to obtain the monomer emulsion. Fe3O4 emulsion, monomer emulsion and 20 mg potassium persulfate were added to three-neck flask and stirred for 30 min at 400 rpm in nitrogen atmosphere. Then the reactor was placed in 80 °C water bath for 24 h to obtain Fe3O4@PVBC nanoparticles suspension. Subsequently, the as-obtained Fe3O4@PVBC nanoparticles were collected, washed with ethanol (25%, v/v) and water several times, and dried in a vacuum oven at 30 °C for 8 h.Immobilization of lipase
10 mg Fe3O4@PVBC nanoparticles were dispersed in 2 mL of 1 M ammonium solution by sonication for 2 min. Subsequently, the mixture was incubated at 30 °C in a shaker operating at 120 times min−1 for 4 h. After the reaction, the Fe3O4@PVBC nanoparticles were washed 2 times with water, dispersed in 5 mL glutaraldehyde (2% v/v) by sonication for 2 min, and incubated at 30 °C in a shaker operating at 120 times min−1 for 9 h. Then the Fe3O4@PVBC nanoparticles were washed 2 times with phosphate buffer (0.1 M, pH 7.5), and dispersed in 2 mL phosphate buffer (0.1 M, pH 7.5) containing 0.2 mg lipase for the bond with lipase covalently. The reaction was conducted at 30 °C for 2 h. Ultimately, the immobilized lipase was recovered by magnetic separation, washed with 2 mL phosphate buffer (0.1 M, pH 7.5) to remove excess lipase, and stored at 4 °C until they were used for activity related tests. Using the same procedures, the loading amount tests were conducted in 4 mL phosphate buffer (0.1 M, pH 7.5) containing 2 mg lipase. The loading of lipase on Fe3O4@PVBC nanoparticles was analysed by the Bradford method using BSA as a standard. The amount of immobilized lipase was calculated by subtracting the amount of free lipase from the total amount of the lipase used for the immobilization.Lipase activity assay
The enzymatic activities of free and immobilized lipase were assayed by the hydrolysis of pNPP in phosphate buffer (0.1 M) following the method described previously.41 Thermal stabilities of free and immobilized lipase were studied by measuring the residual activities of the enzyme after incubation in phosphate buffer (0.1 M, pH 7.5) at 50 °C for 0–10 h. Urea tolerance of free and immobilized lipase were investigated by measuring the residual activities of the enzyme after incubation in 1 mL phosphate buffer (0.1 M, pH 7.5) contained 0–6 mmol urea at 40 °C for 1 h. The reusability of the immobilized lipase was determined by hydrolysis of pNPP by the recovered immobilized lipase with magnetic separation and compared with the first running (activity defined as 100%).Characterization
The powder X-ray diffraction (XRD) pattern of the Fe3O4@PVBC nanoparticles was recorded on a Shimadzu XRD-6000 X-ray diffractometer. The morphologies of the samples were investigated by using a scanning electron microscopy (SEM) of Model S-4800 (Hitachi) and a transmission electron microscopy (TEM) of Model JEM-2010 (JEOL). X-ray photoelectron spectroscopy (XPS) analysis of the surface was studied by an ESCALAB 250Xi spectrometer (Thermo Scientific) equipped with an Al Kα X-ray radiation source. Water contact angles measurement was measured by contact angle measurement instrument (JCY-4, Shanghai Fangrui Instrument Co. Ltd.). Before measurements, a small amount of Fe3O4@PVBC nanoparticles was pressed at 5 MPa into a disc. Fourier transform infrared spectroscopy (FT-IR) spectra were obtained on a Vectortm 22. The magnetic properties of the products were detected at 300 K using a superconducting quantum interference device (SQUID, Quantum Design MPMS3).Results and discussion
As described in Fig. 1, Fe3O4@PVBC nanoparticles were fabricated for lipase immobilization, which involves the preparation of magnetic Fe3O4 nanoparticles in a solvothermal process and the subsequent polymerization of VBC and Fe3O4 nanoparticles in the solution containing dodecanol and sodium dodecyl sulphate. Fig. 2 shows typical XRD patterns of Fe3O4@PVBC nanoparticles and reveals that the product is assigned to Fe3O4 with a face-centered cubic (fcc) structure. All the positions of the peaks are in good agreement with values in the Joint Committee on Powder Diffraction Standards (JCPDS) card file no. 77-1545. Calculations with the Debye–Scherrer equation (d = 0.89λ/B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) for the five strongest peaks (220, 311, 400, 511, and 440) gave mean grain sizes of 14.9 nm for the Fe3O4@PVBC nanoparticles.
cosθ) for the five strongest peaks (220, 311, 400, 511, and 440) gave mean grain sizes of 14.9 nm for the Fe3O4@PVBC nanoparticles.
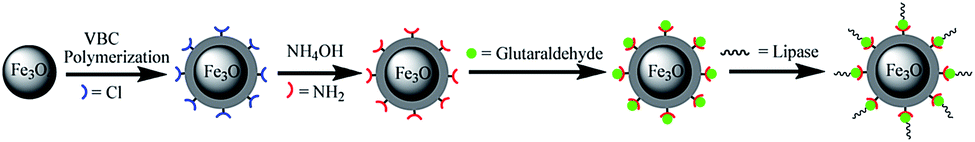 | ||
| Fig. 1 Schematic representation the fabrication procedure of Fe3O4@PVBC nanoparticles for lipase immobilization. | ||
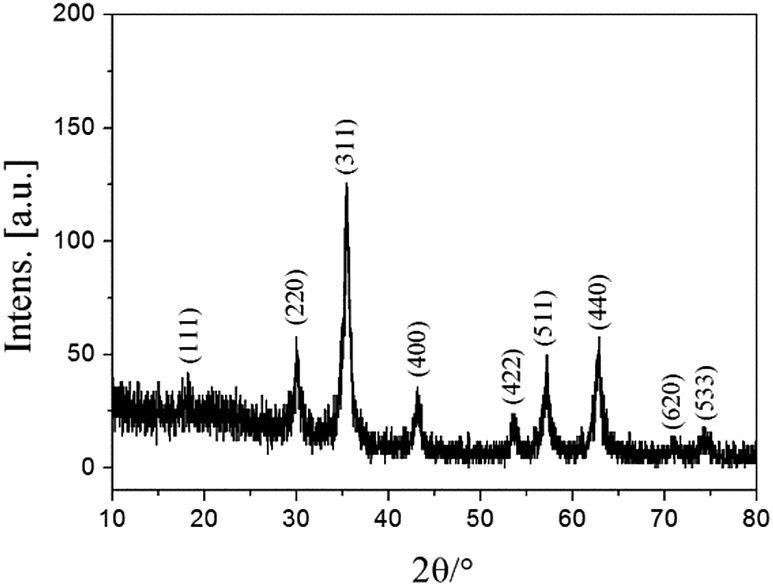 | ||
| Fig. 2 XRD patterns of Fe3O4@PVBC nanoparticles. | ||
The typical SEM image of the as-synthesized products in Fig. 3a indicates that these magnetic nanocomposites exhibit an obvious spherical shape, and the diameters of the Fe3O4@PVBC nanoparticles are 25–45 nm. In Fig. 3b the dark/light different contrast was clearly observed, suggesting a different phase composition, which indicated the formation of Fe3O4@PVBC core–shell nanostructures. It is clearly seen that the Fe3O4 nanoparticles are completely encapsulated in a polymer shell and that there are no obvious voids between the core and the shell. The size of polymer shell is approximately 2–6 nm. Selected-area electron diffraction (SAED) in Fig. 3c of the prepared Fe3O4@PVBC nanoparticles further confirmed that they are fcc magnetite. More detailed structural information about the Fe3O4@PVBC nanoparticles was obtained using a high resolution TEM (HRTEM). Fig. 3d shows that an amorphous polymer layers coating covered the Fe3O4 nanoparticles. The lattice fringe calculated from the HRTEM image was 0.30 nm, fitting well with the (220) planes of fcc Fe3O4 structure.
 | ||
| Fig. 3 (a) SEM and (b) TEM, (c) SAED and (d) HRTEM images of Fe3O4@PVBC nanoparticles. | ||
The typical C 1s, Cl 2p, and Fe 2p X-ray photoelectron spectroscopy (XPS) survey spectra of the Fe3O4@PVBC nanoparticles are depicted in Fig. 4a–c. The asymmetric C 1s peak was decomposed into two peaks at 284.6 and 285.9 eV, which were ascribed to C atoms in aromatic and aliphatic part of the PVBC.42,43 The Cl 2p peak consists of a spin–orbit split (Cl 2p3/2 and Cl 2p1/2) doublet at the BE of 200.1 and 201.6 eV, and is attributed to the chlorine atom signals associated with R–Cl groups.44,45 The Fe 2p1/2 and Fe 2p3/2 peaks are found to be located at 723.9 and 710.7 eV. No Fe 2p signals of γ-Fe2O3 can be clearly identified on the magnetic samples, indicating that the product is Fe3O4 rather than γ-Fe2O3.46 Fig. 4d shows the wettability of a water droplet on the surface of a Fe3O4@PVBC disc that was prepared by pressing Fe3O4@PVBC particles. The static contact angle of water is 135.5 ± 2.0°, indicating that Fe3O4@PVBC nanocomposites are hydrophobic in nature. The hydrophobic character has been further confirmed by observed the system of water and Fe3O4@PVBC nanoparticles, which the magnetic nanocomposites are non-disperse in water. If Fe3O4@PVBC nanoparticles were mixed with ammonia, the products were transformed to amino-functionalized polymer via an amination process and became dispersible in aqueous environments. It is observed that the Fe3O4@PVBC nanoparticles are well dispersed in ammonia solution.
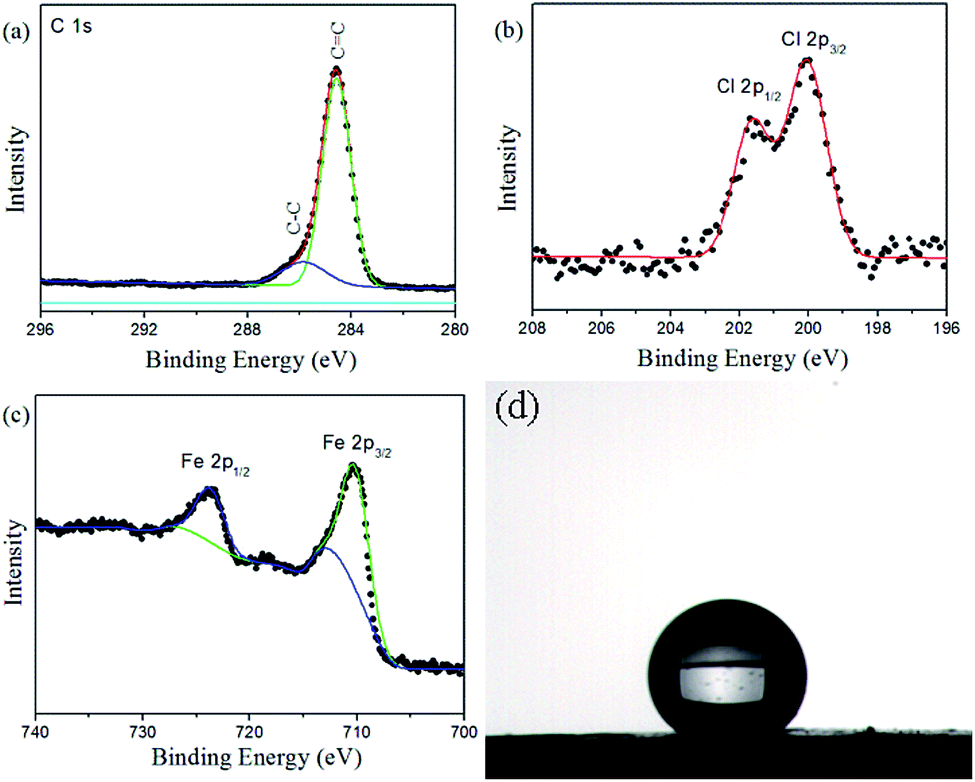 | ||
| Fig. 4 (a) C 1s, (b) Cl 2p and (c) Fe 2p XPS spectra of Fe3O4@PVBC nanoparticles, and (d) image of a water droplet sitting on Fe3O4@PVBC nanoparticles. | ||
The –CH2Cl functional groups of the Fe3O4@PVBC nanoparticles was further determined by FT-IR analysis. The strong peak at 572 cm−1 corresponds to the characteristic band of Fe–O vibration.47 In Fig. 5a, apart from the peaks (2920, 2851, 1636, 1511, 1456, 1420, 1062 and 823 cm−1) corresponding to the benzene ring and methylene which result from the polymer shell, the vibration band at 1263 and 653 cm−1 were assigned to stretching vibrations of CH2–Cl.48 All these results reveal that a large amount of –CH2Cl functional groups are bonded to the polymer shell in the Fe3O4@PVBC nanoparticles, which made possible the amination of the nanoparticles. To confirm this hypothesis, the Fe3O4@PVBC nanoparticles were incubated with ammonia solution. It is observed that the signals at 3410–3450 and 1627 cm−1 for –NH2 stretching vibration, and 1384 cm−1 for C–N stretching vibration appeared (Fig. 5b), indicating that the Fe3O4@PVBC nanoparticles were transformed into amino-functionalized polymer.49
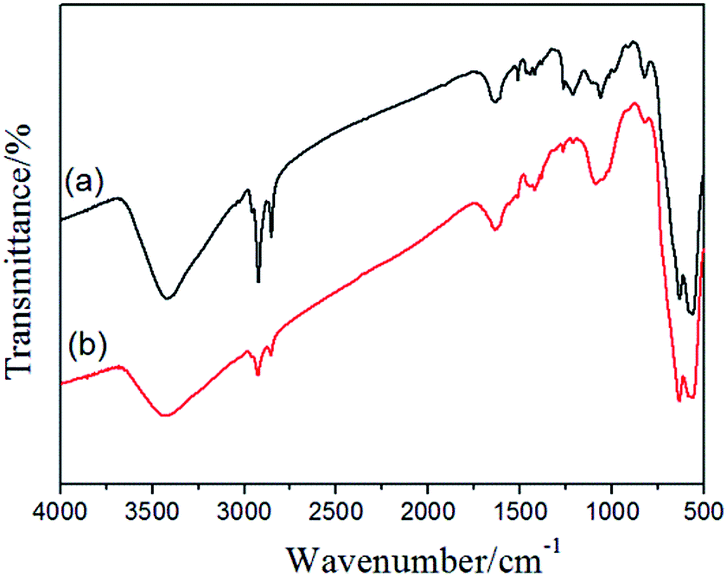 | ||
| Fig. 5 FT-IR spectra of (a) Fe3O4@PVBC and (b) the amino-functionalized Fe3O4@PVBC nanoparticles. | ||
Magnetic measurements of the Fe3O4@PVBC nanoparticles were investigated by a SQUID magnetometer at 300 K with an applied magnetic field ranging from −40 to 40 kOe. As illustrated in Fig. 6, the isothermal magnetization of Fe3O4@PVBC samples showed a rapid increase with increasing applied magnetic field. The saturation magnetization, remanent magnetization and coercivity are 46.8 emu g−1, 2.6 emu g−1, and 31.2 Oe, respectively. The partially enlarged curves demonstrate that the Fe3O4@PVBC sample has the ferromagnetic behaviour (the down inset of Fig. 6). The magnetic separability of the Fe3O4@PVBC nanoparticles was tested in 1 M ammonia solution by placing a magnet near the glass bottle (the up inset of Fig. 6). The dark brown particles were attracted toward the magnet within 20 s, demonstrating directly that the Fe3O4@PVBC nanoparticles possess magnetic properties. This will provide an easy and efficient way to separate Fe3O4@PVBC nanoparticles from a suspension system.
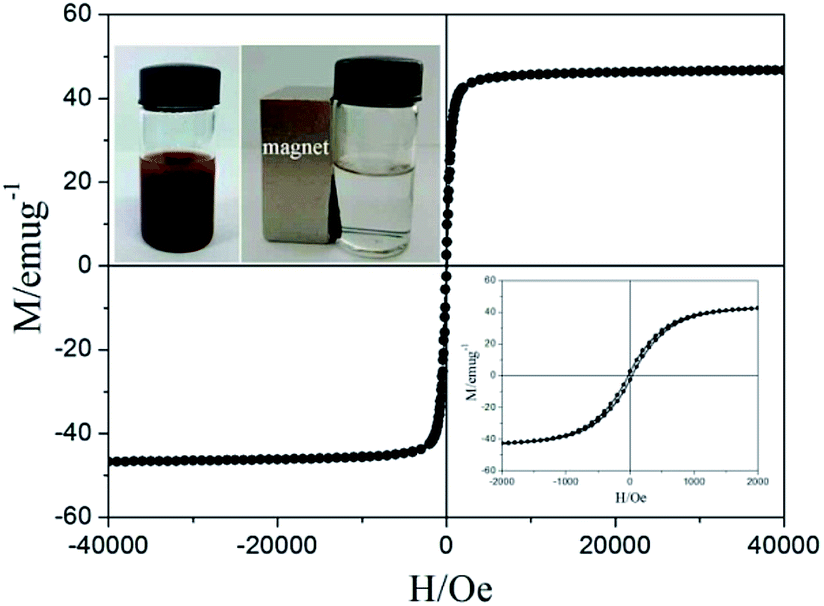 | ||
| Fig. 6 Magnetic hysteresis loops and enlarged view (the down inset) of Fe3O4@PVBC nanoparticles at 300 K, and the up insets: Fe3O4@PVBC nanoparticles suspended in 1 M ammonia solution (left) and separated from the solution under an external magnetic field (right). | ||
To prove the application of Fe3O4@PVBC nanoparticles in biotechnology, the products were used as supports for lipase immobilization. As described in Fig. 1, the magnetic polymer functionalized with aldehyde group were achieved and used to bond with lipase covalently. According the Bradford method, the loading amount of lipase on the Fe3O4@PVBC nanoparticles was calculated to be 162.5 mg protein per g particles. The high lipase loading amount might be attributed to the existence of abundant –CH2Cl on Fe3O4@PVBC supports for the attachment of enzymes. The catalytic activities of free and immobilized lipase were determined by measuring the initial hydrolysis rate of pNPP. By taking the maximum activity value of the immobilized and free lipase under optimal conditions as 100%, the activities obtained from other conditions were expressed as relative activities. Hydrolytic activity tests reveal that the maximum activities of the immobilized and free lipase were both at pH 7.5 and 40 °C. However, hydrolytic activity tests reveal that the immobilized lipase retains about 99.6 ± 3.3% of the free enzyme activity. The high catalytic activity remaining after immobilization may be attributed to interfacial activation of lipase by Fe3O4@PVBC nanoparticles, which is similar to the hyperactivation of lipase adsorbed on hydrophobic materials in previous reported literatures.25–27 The hydrophobic interface could provide a better micro-aqueous environment for lipase molecules, a better dispersion support to avoid agglomeration, and flexibility in opening of the α-helix lid to achieve active catalytic sites which tends to offer a better improvement in enzyme activity.50
For practical applications, it is usually required that biocatalysts possess high stabilities and keep catalytic activity in relatively tough environmental conditions. Therefore, the thermal stabilities and denaturation of the free and immobilized lipase were studied. Fig. 7a shows the comparison of thermal stabilities of free and immobilized lipase at 50 °C in phosphate buffer (0.1 M, pH 7.5). The residual activity of both free and immobilized lipase exhibited a similar trend, whereas the immobilized lipase decreased less and more slowly than the free one. The free lipase lost about 69% of its initial activity after 10 h of incubation, but the immobilized lipase could retain about 60% of its activity after this period of time. These results demonstrated that the thermal stability of immobilized lipase was much better than the free one, which could be explained by the formation of covalent bonds between the lipase molecules and the magnetic carriers enhancing the enzyme rigidity, protected it from unfolding and prevented the conformation transition of the enzyme at high temperature.38,39 Fig. 7b illustrates the comparison of urea tolerance of free and immobilized lipase. It is observed that the lipase remained protected against the urea denaturation after immobilization. The immobilized lipase exhibited strong urea tolerance to retain 69.3% of the residual activity in the presence of 6 M urea, while the free one only retained approximately 22.2% of the residual activity in 6 M urea. The possible explanation of the urea tolerance of lipase could be that the active sites of the enzyme molecules become less accessible for the urea molecules after covalent attached onto the magnetic carriers.51 The free and immobilized lipase were stored at 4 °C in phosphate buffer (0.1 M, pH 7.5) and measured for a period of 14 days. The free lipase lost about 19% of its initial activity within 14 days, while the residual activity of immobilized lipase could retain about 88% of its activity after this period of time. These results demonstrated that storage stability of the immobilized lipase was enhanced.
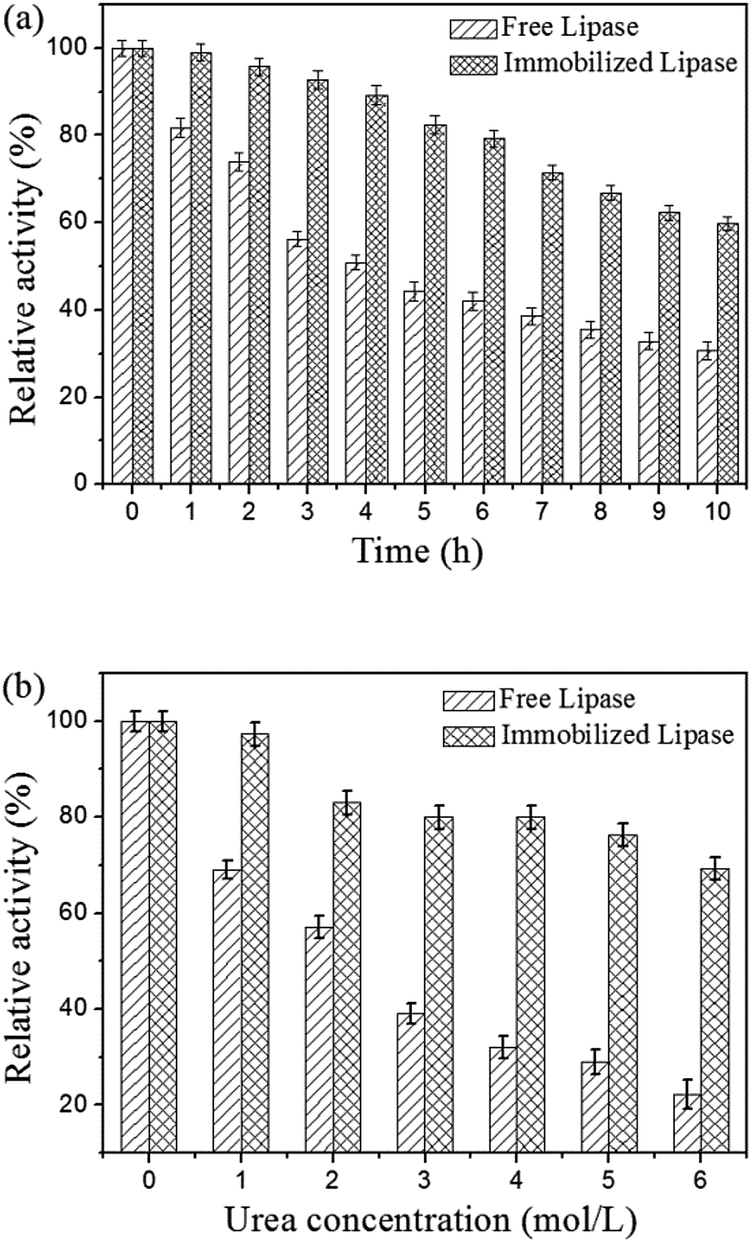 | ||
| Fig. 7 (a) Thermal stabilities of free and immobilized lipase at 50 °C and (b) effect of urea concentration on the activity of free and immobilized lipase. | ||
Reusability is the most important advantage of the immobilized lipase compared to the free lipase. To evaluate the reusability, the immobilized lipase was washed with phosphate buffer (0.1 M, pH 7.5) after one catalysis run and reintroduced into a fresh pNPP solution for another hydrolysis at 40 °C. As shown in Fig. 8, the immobilized lipase shows good stability and retains more than 69.8% of its initial activity after 10 consecutive reuses. This result confirmed that the immobilized lipase on Fe3O4@PVBC nanoparticles has a good durability and magnetic recovery. The decrease of activity is ascribed to the denaturation of the immobilized lipase molecules and the leakage of immobilized enzyme upon the recovery process.
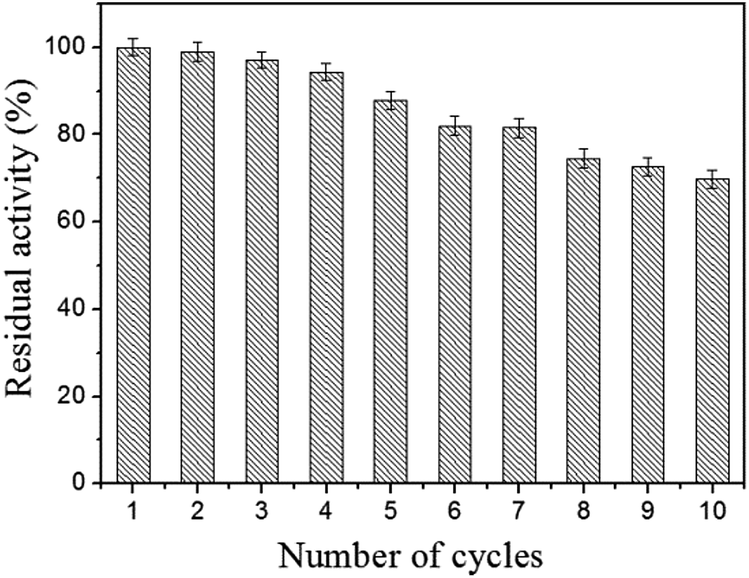 | ||
| Fig. 8 Reusability of the immobilized lipase. | ||
Conclusions
In summary, we reported an effective strategy for enhancement of catalytic activity and stability of immobilized lipase by interfacial activation on Fe3O4@PVBC nanoparticles. The Fe3O4@PVBC nanoparticles were functionalized with –CH2Cl groups, which provides the potential application in lipase immobilization. Bradford assays indicated that the lipase loading amount on Fe3O4@PVBC nanoparticles was 162.5 mg protein per g. The relative activity of the immobilized lipase reaches up to 99.6 ± 3.3% compared with the free one, which is attributed to the enhanced interfacial activity of lipase due to the interaction between lipase and the surface of Fe3O4@PVBC nanoparticles. Hydrolytic activity tests indicate that the immobilized lipase exhibited much better thermal stability and urea tolerance. Additionally, the immobilized lipase retained more than 69.8% of its initial activities after 10 times reuse. The combination of interfacial activation, improved stability and easy recovery of magnetic Fe3O4@PVBC nanoparticles presents an attractive process for lipase immobilization and provides a promising catalyst for practical applications.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21471002 and 51572004), the Outstanding Youth Science Foundation of Anhui Polytechnic University (2015JQ01) and the Young and Middle-Aged Talent Program of Anhui Polytechnic University (2016BJRC001).Notes and references
- M. Held, A. Schmid, J. B. van Beilen and B. Witholt, Pure Appl. Chem., 2000, 72, 1337–1343 CrossRef CAS.
- J. Y. Song, P. Su, Y. Yang, T. Wang and Y. Yang, J. Mater. Chem. B, 2016, 4, 5873–5882 RSC.
- J. J. Sun, R. Yendluri, K. Liu, Y. Guo, Y. Lvov and X. H. Yan, Phys. Chem. Chem. Phys., 2017, 19, 562–567 RSC.
- C. Zhang, X. R. Wang, M. Hou, X. Y. Li, X. L. Wu and J. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 13831–13836 CAS.
- Z. X. Li, Y. Ding, S. M. Li, Y. B. Jiang, Z. Liu and J. Ge, Nanoscale, 2016, 8, 17440–17445 RSC.
- X. L. Wu, C. Yang, J. Ge and Z. Liu, Nanoscale, 2015, 7, 18883–18886 RSC.
- X. L. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077–5085 CAS.
- X. L. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- J. Ge, J. D. Lei and R. N. Zare, Nano Lett., 2011, 11, 2551–2554 CrossRef CAS PubMed.
- M. Kalantari, M. Kazemeini, F. Tabandeh and A. Arpanaei, J. Mater. Chem., 2012, 22, 8385–8393 RSC.
- C. H. Lee, T. S. Lin and C. Y. Mou, Nano Today, 2009, 4, 165–179 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49–61 CrossRef CAS.
- E. Yilmaz, M. Sezgin and M. Yilmaz, J. Mol. Catal. B: Enzym., 2011, 69, 35–41 CrossRef CAS.
- X. H. Li, H. Zhu, J. Feng, J. W. Zhang, X. Deng, B. F. Zhou, H. L. Zhang, D. S. Xue, F. S. Li, N. J. Mellors, Y. F. Li and Y. Peng, Carbon, 2013, 60, 488–497 CrossRef CAS.
- Y. Kuwahara, T. Yamanishi, T. Kamegawa, K. Mori and H. Yamashita, ChemCatChem, 2013, 5, 2527–2536 CrossRef CAS.
- Y. Kuwahara, T. Yamanishi, T. Kamegawa, K. Mori, M. Che and H. Yamashita, Chem. Commun., 2012, 48, 2882–2884 RSC.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- G. Fernández-Lafuente, J. M. Palomo, C. Mateo, R. Munilla, C. Ortiz, Z. Cabrera, J. M. Guisán and R. Fernández-Lafuente, Biomacromolecules, 2006, 7, 2610–2615 CrossRef PubMed.
- J. C. S. dos Santos, O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, ChemCatChem, 2015, 7, 2413–2432 CrossRef CAS.
- A. R. M. Yahya, W. A. Anderson and M. Moo-Young, Enzyme Microb. Technol., 1998, 23, 438–450 CrossRef CAS.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisán, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS PubMed.
- M. T. Reetz, Adv. Mater., 1997, 9, 943–954 CrossRef CAS.
- I. Mahmood, I. Ahmad, G. Chen and L. Huizhou, Biochem. Eng. J., 2013, 73, 72–79 CrossRef CAS.
- K. Solanki and M. N. Gupta, New J. Chem., 2011, 35, 2551–2556 RSC.
- K. C. Badgujar and B. M. Bhanage, Ind. Eng. Chem. Res., 2014, 53, 18806–18815 CrossRef CAS.
- J. Mukherjee and M. N. Gupta, Bioresour. Technol., 2016, 209, 166–171 CrossRef CAS PubMed.
- W. W. Zhang, X. L. Yang, J. Q. Jia, N. Wang, C. L. Hu and X. Q. Yu, J. Mol. Catal. B: Enzym., 2015, 115, 83–89 CrossRef CAS.
- V. Patel, C. Shah, M. Deshpande and D. Madamwar, Appl. Biochem. Biotechnol., 2016, 178, 1630–1651 CrossRef CAS PubMed.
- Y. J. Cui, Y. F. Li, Y. Yang, X. Liu, L. Lei, L. C. Zhou and F. Pan, J. Biotechnol., 2010, 150, 171–174 CrossRef CAS PubMed.
- X. Wang, P. P. Dou, P. Zhao, C. M. Zhao, Y. Ding and P. Xu, ChemSusChem, 2009, 2, 947–950 CrossRef CAS PubMed.
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844–857 RSC.
- M. C. R. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. M. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- Y. J. Li, G. W. Zhou, W. T. Qiao and Y. Y. Wang, Mater. Sci. Eng., B, 2009, 162, 120–126 CrossRef CAS.
- W. Z. Shen, L. W. Ren, H. Zhou, S. C. Zhang and W. B. Fan, J. Mater. Chem., 2011, 21, 3890–3894 RSC.
- M. Y. Chang and R. S. Juang, Enzyme Microb. Technol., 2005, 36, 75–82 CrossRef CAS.
- Y. Yong, Y. X. Bai, Y. F. Li, L. Lin, Y. J. Cui and C. G. Xia, Process Biochem., 2008, 43, 1179–1185 CrossRef CAS.
- H. Zhu, C. Hou, Y. J. Li, G. H. Zhao, X. Liu, K. Hou and Y. F. Li, Chem.–Asian J., 2013, 8, 1447–1454 CrossRef CAS PubMed.
- X. Liu, X. Chen, Y. F. Li, X. Y. Wang, X. M. Peng and W. W. Zhu, ACS Appl. Mater. Interfaces, 2012, 4, 5169–5178 CAS.
- Z. M. Chen, L. L. Liu, X. D. Wu and R. C. Yang, RSC Adv., 2016, 6, 108583–108589 RSC.
- H. H. Jiang, X. Y. Han, Z. L. Li, X. C. Chen, Y. H. Hou, L. G. Gai, D. C. Li, X. R. Lu and T. L. Fu, Colloids Surf., A, 2012, 401, 74–80 CrossRef CAS.
- D. E. Weibel, F. Kessler and G. V. d. S. Mota, Polym. Chem., 2010, 1, 645–649 RSC.
- G. L. Li, D. L. Zeng, L. Wang, B. Y. Zong, K. G. Neoh and E. T. Kang, Macromolecules, 2009, 42, 8561–8565 CrossRef CAS.
- D. Tastet, M. Save, F. Charrier, B. Charrier, J. B. Ledeuil, J. C. Dupin and L. Billon, Polymer, 2011, 52, 606–616 CrossRef CAS.
- Y. Tian, B. B. Yu, X. Li and K. Li, J. Mater. Chem., 2011, 21, 2476–2481 RSC.
- J. Hong, D. M. Xu, P. J. Gong, H. J. Ma, L. Dong and S. D. Yao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 850, 499–506 CrossRef CAS PubMed.
- W. T. Lu, Z. G. Shao, G. Zhang, Y. Zhao and B. L. Yi, J. Power Sources, 2014, 248, 905–914 CrossRef CAS.
- Z. M. Chen, W. H. Xu, L. Jin, J. J. Zha, T. X. Tao, Y. Lin and Z. Wang, J. Mater. Chem. A, 2014, 2, 18339–18344 CAS.
- K. C. Badgujar and B. M. Bhanage, Bioprocess Biosyst. Eng., 2017, 40, 741–757 CrossRef CAS PubMed.
- S. Rauf, A. Ihsan, K. Akhtar, M. A. Ghauri, M. Rahman, M. A. Anwar and A. M. Khalid, J. Biotechnol., 2006, 121, 351–360 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |