
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of alkyl chain number and position on 2D self-assemblies†
Yi Hu ,
Kai Miao,
Li Xu*,
Bao Zha,
Xinrui Miao and
Wenli Deng*
,
Kai Miao,
Li Xu*,
Bao Zha,
Xinrui Miao and
Wenli Deng*
College of Materials Science and Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: mslxu@scut.edu.cn; wldeng@scut.edu.cn; Tel: +86-20-22236708
First published on 26th June 2017
Abstract
Utilizing the effects of alkyl chain number and position is an efficient strategy for the fabrication and regulation of diverse self-assembly structures. Based on self-assembling behavior, we explored these two effects with regard to the self-assembly mechanism and thermal, spectral, and crystal analysis. Eight types of anthraquinone derivatives were used, with eight different self-assembly configurations observed at the 1-octanoic acid/HOPG interface, namely six linear structures, one zigzag structure, and one dimer-linear structures. As these anthraquinone derivatives possess different molecular symmetries, which can affect the molecular dipole, the formation mechanism of these self-assembled networks was investigated with regard to dipole–dipole interactions. Furthermore, density functional theory calculations showed that an increasing alkyl chain number led to a rise in intermolecular van der Waals interactions. However, despite the variation in alkyl chain number and position, the strength of hydrogen bonds between the anthraquinone cores was particularly dependent on the assembled structure. This work provides a comprehensive method for fabricating self-assemblies at liquid/solid interfaces in 2D crystal engineering, and we believe it will be of significance toward studying the effects of alkyl chain number and position on structural diversity in supramolecular chemistry.
Introduction
The preparation of nanosized self-assemblies of supramolecules on surfaces or at liquid (gas)/solid interfaces is a powerful method for fabricating complex nanostructures based on a bottom-up approach.1–7 Owing to their precise chemical structures and physical properties, Π-conjugated molecules are usually adopted as targets for constructing well-defined assemblies, which play a crucial role in the development of molecular electronic devices.8–12 Therefore, an efficient strategy for constructing and tuning molecular packing in assembly systems is indispensable for the further development of nanoscience and nanotechnology.Functionalized supramolecules with Π-conjugated cores and alkyl side chains have been explored extensively for the fabrication of two-dimensional (2D) nanopatterns on highly oriented pyrolytic graphite (HOPG) surfaces using scanning tunneling microscopy (STM).13,14 In the last decade, a variety of methods have been reported to induce diverse structures by changing parameters such as alkyl chain length,15 solvent,16 concentration,17 temperature,18 voltage,19 and substrate.14 Although changing the number of substituents (functional groups and alkyl chains) seems to facilitate the fabrication of different configurations, it has rarely been reported due to difficulties in their synthesis.20–22 Wang et al. demonstrated that the number of end-groups had a pronounced effect on the formation of self-assembled monolayers.23 They synthesized a series of oligofluorenes (StOFs) containing one, two, or three carboxyl terminal groups (StOF–COOHn, n = 1, 2, 3), with lamellae (n = 1), zigzag (n = 2), and honeycomb (n = 3) structures observed. As the number of carboxyl group increased, the number of hydrogen bonds increased accordingly, which simultaneously increased the driving force. Therefore, structural diversity can be achieved by changing interactions between the adsorbate molecules. However, adding or removing whole alkyl chains attached to the main skeleton of the molecule remains a synthetic challenge.
Herein, we investigate the effects of alkyl chain number and position on self-assembling behavior, comprising a comprehensive study based on our previous research. We have previously reported self-assemblies of anthraquinone derivatives with one,24,25 two,26 and three side chains.27,28 These studies emphasized the diverse assembled structures induced by changing the alkyl chain lengths. However, self-assemblies of anthraquinone derivatives with different numbers of alkyl chains in different positions have not been explored and compared, and their potential mechanisms have yet to be discussed. Changing the alkyl chain number alters the van der Waals (vdW) interactions. Moreover, different alkyl chain numbers and positions can induce different molecular symmetry, allowing the intermolecular forces to be controlled. In the present study, pentadecyloxy chains are used as substituents that are mono-, bis- or tri-substituted in different positions on anthraquinone derivatives. As a result, eight anthraquinone derivatives have been prepared, as shown in Scheme 1. Nanonetworks were formed under the driving force of hydrogen bonding between the anthraquinone cores and controllable vdW interactions between the molecule and substrate, and between interdigitated alkyl chains. In the assembled structures, the anthraquinone cores were arranged in different modes, and the alkyl chains were interdigitated in different fashions. To compare these forces, density functional theory (DFT) calculations were conducted to determine the binding energies. Furthermore, since these anthraquinone derivatives possessed different molecular symmetries, which can affect molecular dipoles, the formation mechanisms of these self-assembled networks were also investigated with regard to dipole–dipole interactions. Moreover, the thermal and spectral properties changed as the number of alkyl chains was altered. Differential scanning calorimetry (DSC) performance and ultraviolet (UV) absorption spectra were also recorded to further investigate the effects of side chain number and position on the anthraquinone derivatives. Finally, X-ray diffraction (XRD) measurements were performed, with results showing that all the compounds were polycrystalline with diffraction peaks located in different positions, demonstrating large differences in their 3D arrangement, which explains their structural diversity in 2D space. This work not only aids understanding of self-assembly mechanisms for molecules containing different numbers and positions of alkyl chains, but also provides an efficient and comprehensive strategy for fabricating diverse molecular monolayers. We believe that these results will be significant for 2D self-assembly in supramolecular chemistry, and especially in the field of interface science.
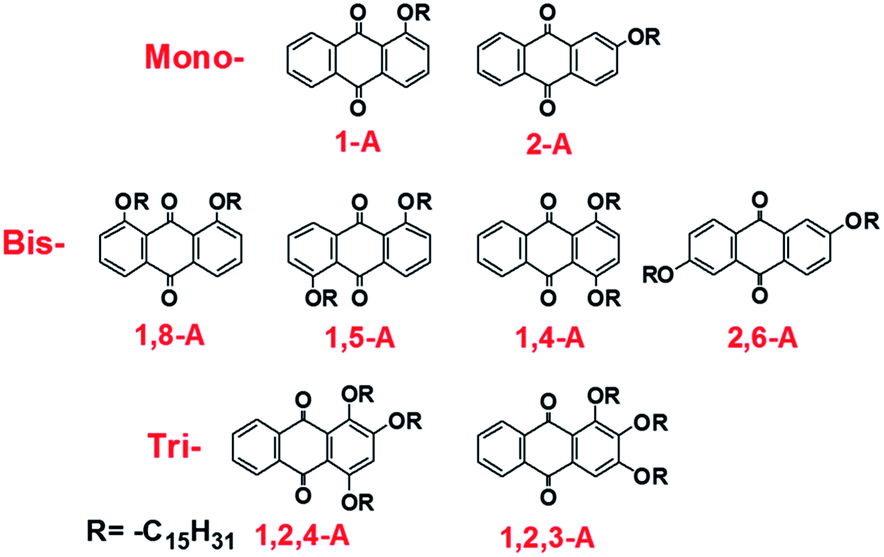 | ||
| Scheme 1 Chemical structures of mono-, bis-, and tri-substituted anthraquinone derivatives. | ||
Experimental section
Anthraquinone derivatives 1-A, 2-A, 1,8-A, 1,5-A, 1,4-A, 2,6-A, 1,2,4-A, and 1,2,3-A were synthesized using previously reported methods.24–27 The solvent, 1-octanoic acid, was purchased from Tokyo Chemical Industry (TCI) and used without purification. Samples were prepared by depositing a droplet (about 1 μL) of solution onto a freshly cleaved atomically flat surface of HOPG (quality ZYB, Bruker, USA). As these molecules showed no concentration effect on the formation of stable self-assembled structures, the concentrations have not been provided in this present work. STM experiments were performed on a Nanoscope IIIa Multimode SPM (Bruker, USA) under ambient conditions (temperature: 15–20 °C, humidity: 45–60%). The tips were mechanically cut from Pt/In wires (80/20). Different tips and samples were used to check the reproducibility of the results. All STM images were recorded in constant current mode and are shown without further processing. The imaging conditions are provided in corresponding figure captions. Materials Studio 4.4 was used to build the network models of the assembled structures. Intermolecular hydrogen bonds and vdW interactions were calculated with DFT, provided by Materials Studio 4.4, using the Forcite package. DSC experiments were conducted with a scan rate of 10 °C min−1 for heating and cooling traces (instrument: NETZSCH DSC 200F3). UV absorption spectra were recorded using an ultraviolet spectrometer (Agilent Cary 60) by dissolving the compounds in CH2Cl2. Powder XRD patterns were recorded using a Bruker D8-ADVANCE diffractometer with Cu Kα radiation. A step-scan mode was adopted with a sampling time of 0.1 s and a scanning step of 0.02°.Results and discussions
When a drop of solution containing an anthraquinone derivative was deposited onto the graphite surface, an ordered monolayer consisting of a large-area domain spontaneously formed (Fig. S1†). As we previously reported,24–28 mono-substituted anthraquinone derivatives 1-A and 2-A can self-assemble into linear structures, denoted as linear I (Fig. 1a) and linear II (Fig. 1b), respectively. In these two linear structures, the molecules are arranged in pairs, with the two partners oriented in opposite directions, as shown by the molecular models overlaid on the STM images. When the alkyl chain number was increased to two, resultant bis-substituted anthraquinone derivatives 1,8-A, 1,5-A, 1,4-A, and 2,6-A arranged to form four more nanostructures, denoted as linear III (Fig. 1c), linear IV (Fig. 1d), zigzag (Fig. 1e), and linear V (Fig. 1f) patterns, respectively. Derivatives 1,8-A and 1,4-A tended to have every two molecules arranged together in a head-to-head fashion. However, 1,5-A and 2,6-A were packed in ordered lines, without oppositely arranged pairs. Moreover, except for the linear structure formed by 1,8-A, the alkyl chains of the other structures were interdigitated. These structural details are indicated by the models in the STM images. Of the tri-substituted anthraquinone derivatives, 1,2,4-A self-assembled into a dimer-linear structure (Fig. 1g). Pairs of molecules packed in an antiparallel fashion were observed in both the dimer and linear ribbons of 1,2,4-A. In this type of self-assembled structure, the chains showed great commensurability with the graphite lattice, meaning that they were extended along a main symmetry axis of the HOPG surface, as indicated by the black arrows at the bottom left of the STM images (Fig. 1). In the linear rows, the three chains belonging to one 1,2,4-A molecule were fully interdigitated with chains from adjacent rows. However, in the dimer rows, we only observe two chains and two anthraquinone cores in the dimer that are too close to each other, leaving no space for the other chain. Therefore, the other chain is believed to be directed toward the liquid phase (Fig. S2†).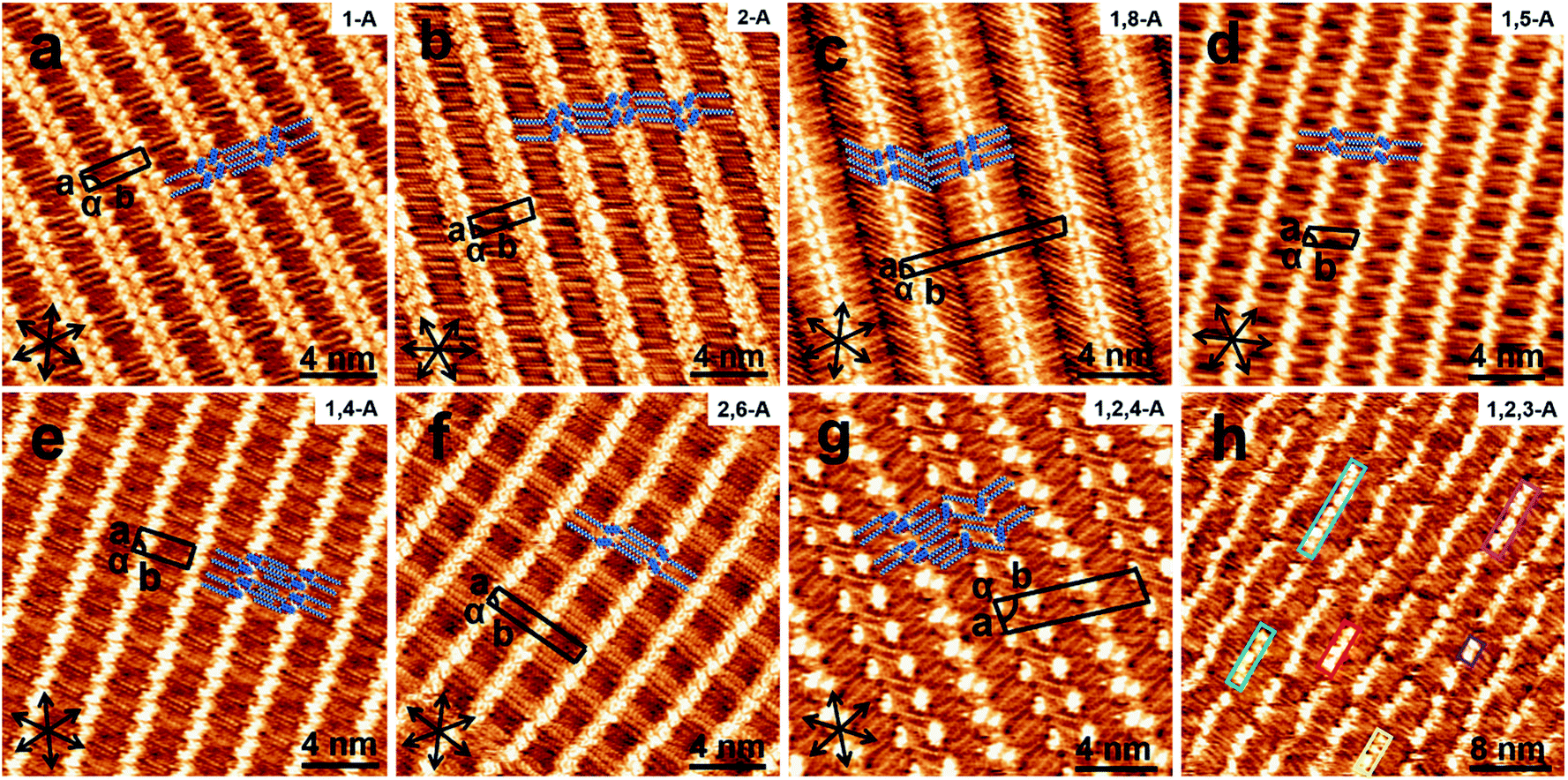 | ||
| Fig. 1 High-resolution STM images of (a) 1-A, (b) 2-A, (c) 1,8-A, (d) 1,5-A, (e) 1,4-A, (f) 2,6-A, (g) 1,2,4-A, and (h) 1,2,3-A at the 1-octanoic acid/HOPG interface, showing linear I, II, III, and IV, zigzag, linear V, dimer-linear, and linear VI structures, respectively. Black arrows in the bottom left of each image represent the three-fold symmetry axis of the graphite surface. Proposed structural models are overlaid. In (h), aggregations consisting of different numbers of molecules are marked with rectangles of different colors. Imaging conditions: It = 450–700 pA, Vbias = 550–900 mV. | ||
Derivative 1,2,3-A is a type of molecule that has not been reported in the field of 2D self-assembly. In the linear ribbons of 1,2,3-A (Fig. 1h, denoted as linear VI), the molecules were not arranged in an orderly fashion, meaning that the ribbons were not continuous. The monolayer was scanned in different positions, but disordered ribbons were found to cover the entire substrate surface (Fig. S3†). Different aggregations constructed from different numbers of molecules were observed, as denoted by rectangles of different colors in Fig. 1h, with purple, yellow, red, green, pink, and blue rectangles representing dimer, trimer, tetramer, pentamer, hexamer, and octamer aggregations, respectively. Notably, the number of molecules in the aggregations was not limited to those illustrated because the appearance of any of the aggregations was random.
Therefore, this kind of configuration was irregular, and the linear ribbons were not straight over a long range. Statistical analysis (from more than twenty images, in an area of 50 × 50 nm2) of the distribution for all types of aggregation was conducted, as shown in Fig. S4.† As the number of molecules in the aggregations changed, the number of each type of aggregation changed, and the surface coverage of the different aggregations was different. This result indicated that 1,2,3-A molecules were, in most cases, arranged as tetramers.
As anthraquinone derivatives self-assemble into diverse structures, we were motivated to explore the effects of alkyl chain number and position on the self-assembly mechanisms. In this present system, dipole–dipole interactions, hydrogen bonds between the anthraquinones, and molecule–molecule and molecule–substrate vdW interactions all significantly affect the stabilization of the nanostructures. For ease of comparison, the geometric characters and calculated packing densities for the observed self-assembled structures are summarized in Table 1.
| Molecule | Structure | Structural model | Symmetry type | Na | Area density (nm2 per molecule) | Hydrogen bonding (kJ mol−1) | vdW interactions (kJ mol−1) |
|---|---|---|---|---|---|---|---|
| a N represents the number of molecules in the unit cell. As 1,2,3-A molecules were arranged in linear ribbons without regularity, the unit cell has not been presented. | |||||||
| 1-A | Linear I |  |
Asymmetric | 2 | 1.64 | −17.0 | −64.7 |
| 2-A | Linear II |  |
Asymmetric | 2 | 1.65 | −25.7 | −62.5 |
| 1,8-A | Linear III | 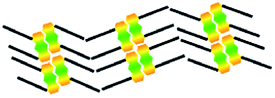 |
Axisymmetric | 4 | 1.76 | −75.4 | −66.0 |
| 1,5-A | Linear IV | 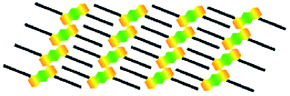 |
Centrosymmetric | 1 | 2.97 | −37.1 | −71.3 |
| 1,4-A | Zigzag |  |
Axisymmetric | 2 | 2.52 | −25.2 | −79.7 |
| 2,6-A | Linear V |  |
Centrosymmetric | 2 | 2.11 | −43.6 | −73.9 |
| 1,2,4-A | Dimer-linear |  |
Asymmetric | 4 | 3.72 | −22.7 | −94.9 |
| 1,2,3-A | Linear VIa | — | Asymmetric | — | — | — | — |
As the number and position of alkyl chains changed, the eight anthraquinone derivatives showed different types of symmetry. Changing the distribution of alkyl chains resulted in different electrostatic potential maps, as shown in Fig. 2. Blue and the red parts in the maps represent positive and negative potentials, respectively. For centrosymmetric molecules 1,5-A and 2,6-A, the potential maps (Fig. 2d and f) clearly show that both the positive and negative centers are located in centrosymmetric positions. Therefore, the dipole moments are offset inside the molecules, resulting in 1,5-A and 2,6-A showing no polarity. Moreover, the 1,5-A and 2,6-A self-assembled into linear IV (Fig. 1d) and linear V (Fig. 1f) structures without molecule pairs, in which molecules were arranged in antiparallel fashion to offset the dipole moments. Molecules other than 1,5-A and 2,6-A were asymmetric or axisymmetric. The calculated electrostatic potential maps in Fig. 2a–c, e, and g show that the positive and/or negative centers were asymmetric. Therefore, it was impossible for these molecules to offset their internal dipole moments. Therefore, these molecules showed polarity and dipole–dipole interactions, which played a significant role in forming the assembled monolayers. As described above, 1-A, 2-A, 1,8-A, 1,4-A, and 1,2,4-A self-assembled into linear I, linear II, linear III, zigzag, and dimer-linear structures, respectively, in which molecule pairs were common and minimized the overall polarity near the self-assembled area to near zero. The dipole offsets in these configurations are illustrated by arrows (Fig. 3a–c, e, and g).
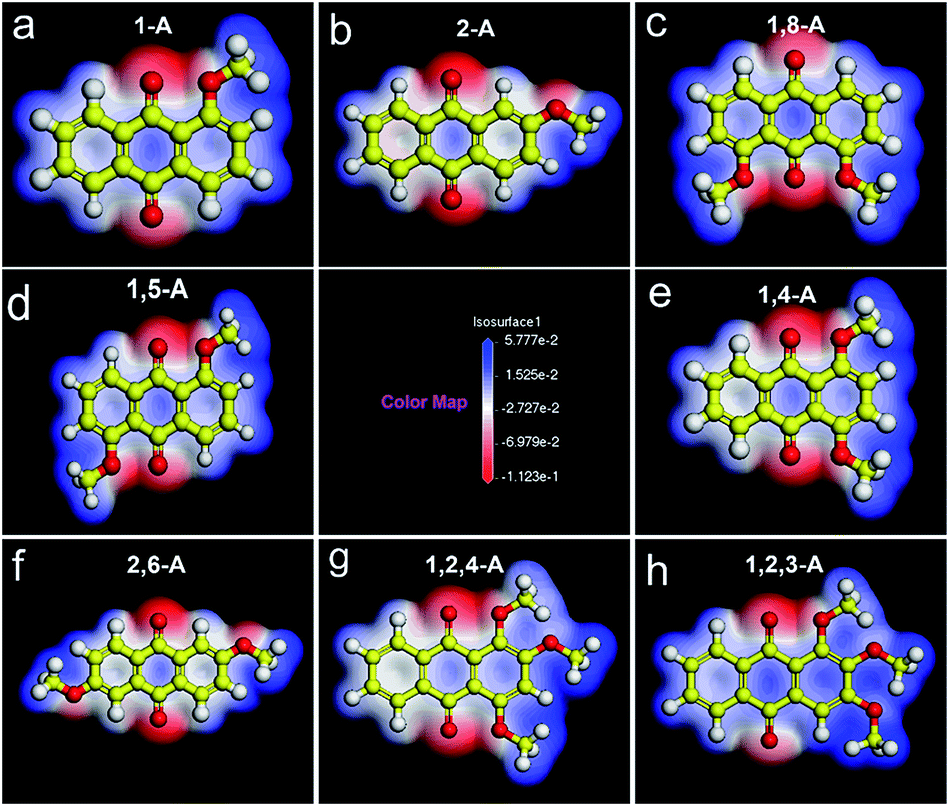 | ||
| Fig. 2 Calculated electrostatic potential maps for (a) 1-A, (b) 2-A, (c) 1,8-A, (d) 1,5-A, (e) 1,4-A, (f) 2,6-A, (g) 1,2,4-A, and (h) 1,2,3-A using Materials Studio 4.4. Alkyl chains were replaced with methyl groups for ease of calculation. Blue and red parts of the color map represent positive and negative potentials, respectively. | ||
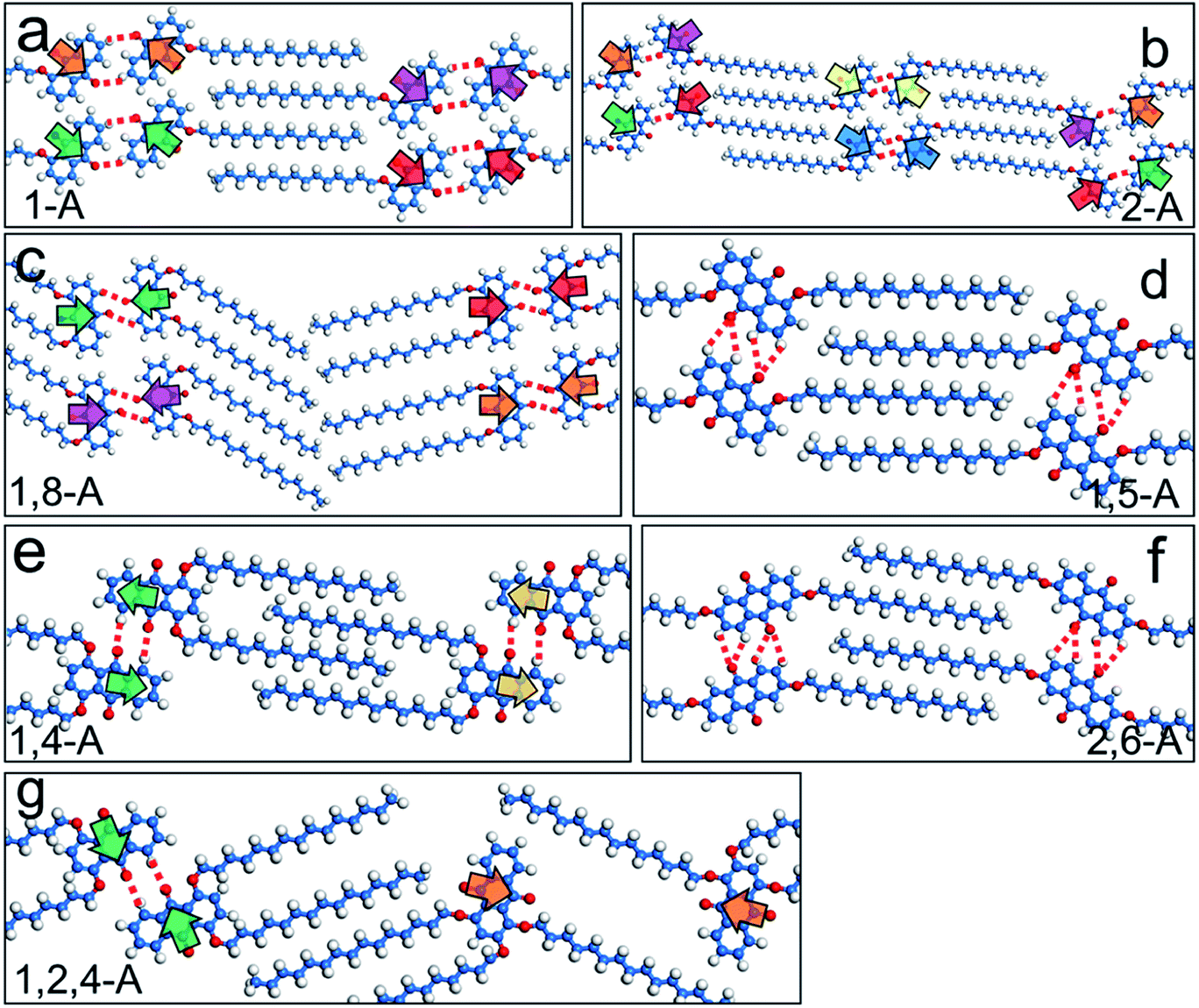 | ||
Fig. 3 Schematic diagrams of self-assembled structures of (a) 1-A, (b) 2-A, (c) 1,8-A, (d) 1,5-A, (e) 1,4-A, (f) 2,6-A, and (g) 1,2,4-A. Offset of dipole moment is illustrated by paired arrows, which are the same color and have opposite orientations. C–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds between the adjacent anthraquinone cores are illustrated with red dotted lines. C hydrogen bonds between the adjacent anthraquinone cores are illustrated with red dotted lines. | ||
Hydrogen bonding interactions have been widely reported as a driving force for self-assembly due to their relative strength, selectivity, and directional nature.29 In the present work, the anthraquinone cores could form C–H⋯OC hydrogen bonds, with the O atom in the carbonyl group acting as the acceptor and the H atom in the adjacent anthraquinone core as the donor, as shown by red dotted lines in Fig. 3. To further confirm the effects of hydrogen bonding on the self-assembled behavior of the anthraquinone derivatives, we performed DFT calculations. The binding energies of the C–H⋯OC hydrogen bonding interactions between the anthraquinone cores of 1-A, 2-A, 1,8-A, 1,5-A, 1,4-A, 2,6-A, and 1,2,4-A were calculated as −17.0, −25.7, −75.4, −37.1, −25.2, −43.6, and −22.7 kJ mol−1 per molecule, respectively. These results showed that changing the alkyl chain number and position changed the self-assembled structures, causing large differences in the binding energies of hydrogen bonds. Furthermore, the strength of these binding energies depended on the self-assembled structures rather than the number and position of the alkyl chains. These calculated results are shown in Table 1.
In the seven types of self-assembled networks (linear VI is not included because of its irregularity) observed, the alkyl chains were densely packed and, in most cases, fully interdigitated with each other (except for the linear III structure of 1,8-A, in which the alkyl chains were packed in a tail-to-tail fashion, and the zigzag structure of 1,4-A, in which the alkyl chains were not fully interdigitated). Moreover, the chains were extended along the main symmetry axis of the HOPG surface. These phenomena indicated that the molecule–molecule and molecule–substrate vdW interactions were the driving forces stabilizing the assembled structures. As the alkyl chains in this work are of the same length, efficient regulation of the vdW interaction was achieved by changing the number of alkyl chains. Furthermore, as the side chain position was changed, the vdW interactions were affected to some extent. To compare the differences in these vdW interactions induced by alkyl chain number and position, we calculated the binding energies of molecule–molecule vdW interactions involved in the nanostructures. For mono-substituted anthraquinone derivatives, the binding energies were −64.7 and −62.5 kJ mol−1 per molecule for 1-A and 2-A, respectively. For the bis-substituted anthraquinone derivatives, the binding energies were −66.0, −71.3, −79.3, and −73.9 kJ mol−1 per molecule for 1,8-A, 1,5-A, 1,4-A, and 2,6-A, respectively. These results showed that vdW interactions between the 1,8-A molecules were weaker than for other bis-substituted anthraquinone derivatives, which was attributed to the non-interdigitated alkyl chains. The alkyl chains of 1,4-A in the zigzag structure were not fully interdigitated, which disfavored vdW interactions. However, the binding energy was stronger than that of the other bis-substituted molecules, which indicated that this kind of configuration was also adequately stable. As the chain was number increased to three, the vdW interactions also increased, as shown by 1,2,4-A, which had a binding energy of −94.9 kJ mol−1 per molecule. These results are shown in Table 1. We concluded that: (i) as the alkyl chain number increased, the binding energy decreased accordingly, meaning that a higher alkyl chain number resulted in stronger vdW interactions; (ii) vdW interactions between molecules with the same number of alkyl chains showed only a tiny difference due to having the same alkyl chains, but different 2D assembled structures.
Self-assembly is a process in which molecules change from a chaotic phase to an ordered phase. Therefore, structural differences induced by changing the alkyl chain number and position must be related to the energy difference between one phase and the other. Accordingly, we conducted a series of DSC experiments to explore how the chain number and position affect the crystallization properties. DSC thermograms are shown in Fig. 4. For the heating trace, the peak temperature in the horizontal ordinate was the melting point (phase-transition temperature). Compounds with the same number of alkyl chains, but in different positions, showed different phase-transition temperatures. Changing the number of alkyl chains also changed the phase-transition temperature drastically. The melting points of 1-A, 2-A, 1,8-A, 1,5-A, 1,4-A, 2,6-A, 1,2,4-A, and 1,2,3-A were 99, 89, 109, 104, 97, 111, 101, and 69 °C, respectively, as summarized in Table S1.† The differences in phase-transition temperature arose from the different amount of energy required to form a uniform phase.30 These DSC results provided strong evidence for the effects of alkyl chain number and position on thermal analysis.
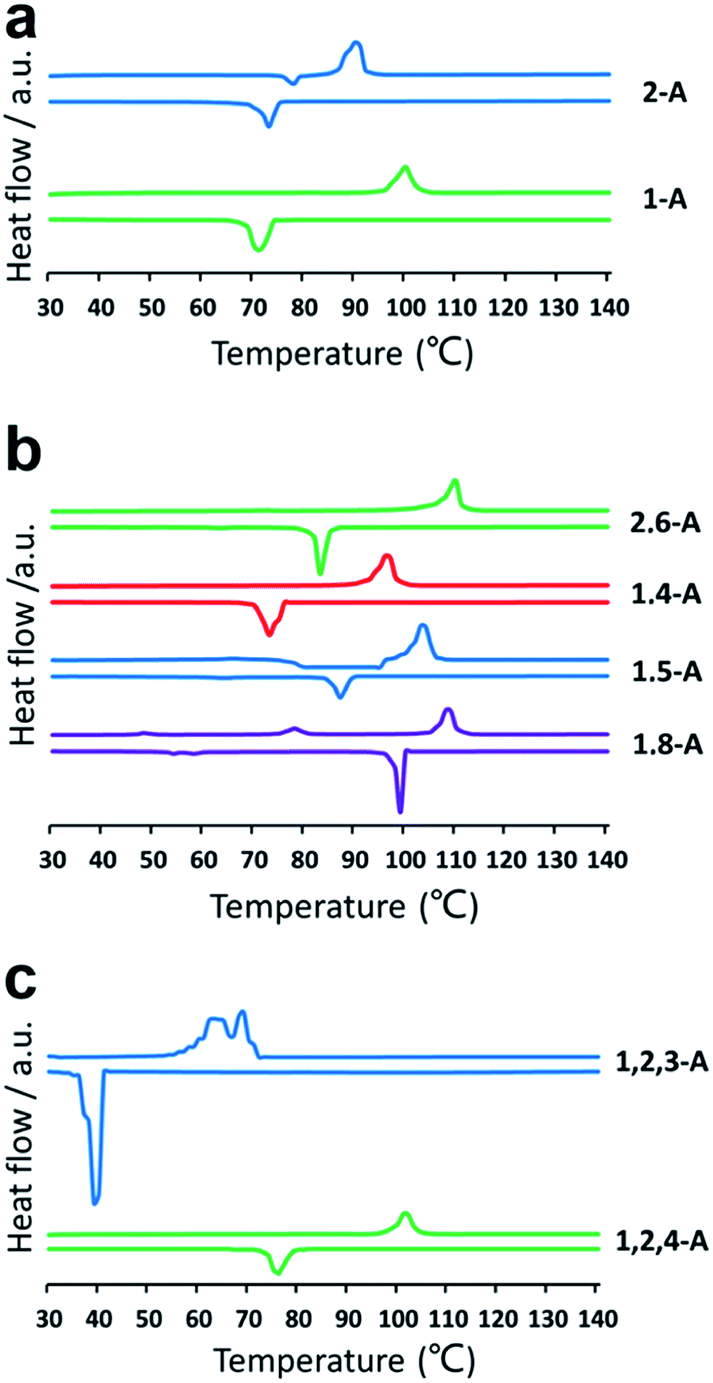 | ||
| Fig. 4 DSC thermograms for (a) mono-, (b) bis-, and (c) tri-substituted anthraquinone derivatives. Heating (above line) and cooling (below line) traces were conducted at a rate of 10 °C min−1. | ||
Substituted alkyl chain can have an immense effect on the physical and chemical properties of organic molecules. To further explore the effects of alkyl chain number and position on this anthraquinone system, careful spectral analysis was essential. Fig. 5 shows the UV absorption spectra of mono-, bis-, and tri-substituted anthraquinone derivatives in CH2Cl2 at the same concentration. The UV absorption peaks for 1-A, 2-A, 1,8-A, 1,5-A, 1,4-A, 2,6-A, 1,2,4-A, and 1,2,3-A were 380, 370, 385, 420, 280, 350, 405, and 365 nm, respectively, as summarized in Table S1.† The characteristic peaks were assigned to Π–Π* electron transitions.31 Although UV absorption does not have a direct relationship with molecular self-assembly, it can reflect even tiny differences caused by different alkyl chain numbers and positions. Therefore, UV characterization can be used to study the effect of alkyl chain number and position and explain structural differences induced by these effects.
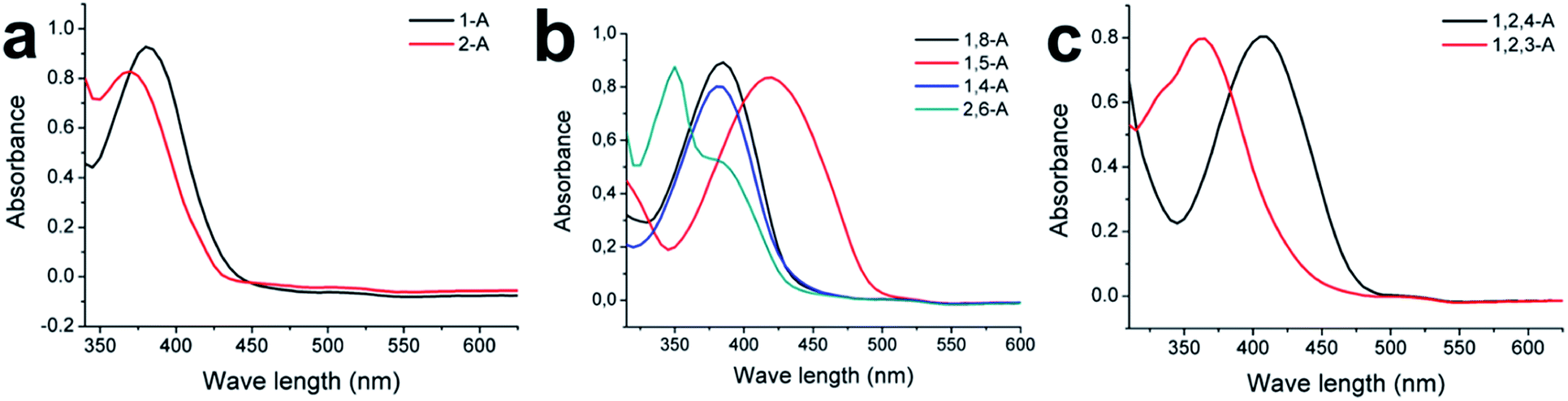 | ||
| Fig. 5 Absorption spectra of (a) mono-, (b) bis-, and (c) tri-substituted anthraquinone derivatives at room temperature. Solvent, CH2Cl2; concentration, 10−5 M. | ||
As a further step, we explored the three dimensional (3D) crystalline structures, because 3D behavior can provide aid understanding of 2D behavior, and vice versa.32 Therefore, these anthraquinone derivatives were detected in the 3D solid state, and XRD was performed. Powder X-ray diffraction patterns for the eight anthraquinone compounds are shown in Fig. 6, and indicate that all are polycrystalline. These results show that, as the position and/or number of alkyl chains changes, the diffraction peaks between these anthraquinone derivatives vary greatly. As a specific peak represents a corresponded lattice plane, these large differences in the peaks represent large differences in their 3D arrangement, and can be regarded as explaining their structural diversity in 2D space.33
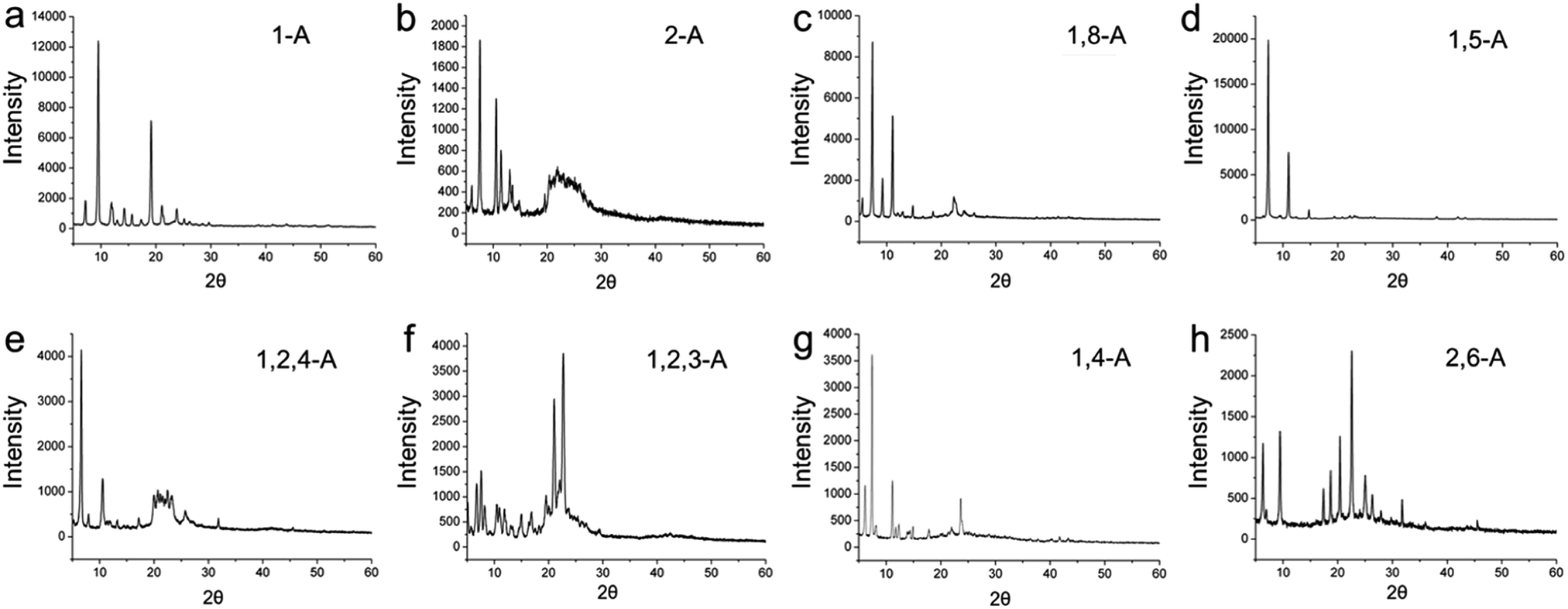 | ||
| Fig. 6 X-ray diffraction patterns for (a, b) mono-, (c, d, g, h) bis-, and (e, f) tri-substituted anthraquinone derivatives. | ||
Conclusions
In summary, we have systematically explored the effects of alkyl chain number and position on the self-assembling behavior of eight anthraquinone derivatives. Asymmetric molecules 1-A, 2-A, and 1,2,4-A, and axisymmetric molecules 1,8-A and 1,4-A, self-assembled into networks in which molecule pairs were common. This indicated that dipole–dipole interactions were favored because molecules were arranged antiparallel to minimize the overall dipole moment. However, centrosymmetric molecules 1,5-A and 2,6-A were arranged in ordered ribbons without antiparallel pairs, which arose from their nonpolarity. Hydrogen bonding interactions were another noncovalent force existing between the anthraquinone cores. Nevertheless, the vdW interactions were controllable, which played an important role in forming the assembled monolayers. As the alkyl chain number was increased from one to two, and then three, the corresponded binding energies showed a sharp decrease, which indicate that the vdW interactions became much stronger. Furthermore, as the number and position of alkyl chains changed, their thermal, spectral and crystal properties were altered. DSC performance, UV absorption spectra, and X-ray diffraction were measured to further investigate the effect of side chain number and position on the anthraquinone derivatives. This study provides an efficient strategy for the fabrication of diverse self-assembled polymorphs with regard to regulating interplay between the molecules. We believe that the results will be of significance in understanding the effects of alkyl chain number and position, and be an important step towards molecular self-assembly in supramolecular chemistry.Acknowledgements
This work is supported by the Natural Science Foundation of China (21573077, 21403072, 51373055), the National Program on Key Basic Research Project (2012CB932900), the China Postdoctoral Science Foundation (2014M552189), the South China University of Technology (SCUT), and Katholieke Universiteit Leuven (University of Leuven). Y. H. acknowledges receipt of a scholarship from China Scholarship Council (CSC).References
- L. Piot, D. Bonifazi and P. Samorì, Adv. Funct. Mater., 2007, 17, 3689–3693 CrossRef CAS.
- E. M. Osteritz and P. Bäuerle, Adv. Mater., 2006, 18, 447–451 CrossRef.
- J. Zhang, B. Li, X. Cui, B. Wang, J. Yang and J. G. Hou, J. Am. Chem. Soc., 2009, 131, 5885–5890 CrossRef CAS PubMed.
- K. E. Plass, A. L. Grzesiak and A. J. Matzger, Acc. Chem. Res., 2007, 40, 287–293 CrossRef CAS PubMed.
- S. Yoshimoto, N. Higa and K. Itaya, J. Am. Chem. Soc., 2004, 126, 8540–8545 CrossRef CAS PubMed.
- D. Zhong, K. Wedeking, T. Blömker, G. Erker, H. Fuchs and L. Chi, ACS Nano, 2010, 4, 1997–2002 CrossRef CAS PubMed.
- A. Miura, P. Jonkheijm, S. De Feyter, A. Schenning, E. W. Meijer and F. C. De Schryver, Small, 2005, 1, 131–137 CrossRef CAS PubMed.
- X. Zhang, T. Chen, Q. Chen, G. J. Deng, Q. H. Fan and L. J. Wan, Chem.–Eur. J., 2009, 15, 9669–9673 CrossRef CAS PubMed.
- J. Lahann, S. Mitragotri, T. N. Tran, H. Kaido, J. Sundaram, I. S. Choi, S. Hoffer, G. A. Somorjai and R. Langer, Science, 2003, 299, 371–374 CrossRef CAS PubMed.
- M. Abe, T. Masuda, T. Kondo, K. Uosaki and Y. Sasaki, Angew. Chem., Int. Ed., 2005, 117, 420–423 CrossRef.
- S. Weigelt, C. Busse, L. Petersen, E. Rauls, B. Hammer, K. V. Gothelf, F. Besenbacher and T. R. Linderoth, Nat. Mater., 2006, 5, 112–117 CrossRef CAS PubMed.
- F. Chiaravalloti, L. Gross, K. H. Rieder, S. M. Stojkovic, A. Gourdon, C. Joachim and F. Moresco, Nat. Mater., 2007, 6, 30–33 CrossRef CAS PubMed.
- J. Adisoejoso, K. Tahara, S. Lei, P. Szabelski, W. Rżysko, K. Inukai, M. O. Blunt, Y. Tobe and S. De Feyter, ACS Nano, 2012, 6, 897–903 CrossRef CAS PubMed.
- T. Balandina, K. Tahara, N. Sändig, M. O. Blunt, J. Adisoejoso, S. Lei, F. Zerbetto, Y. Tobe and S. De Feyter, ACS Nano, 2012, 6, 8381–8389 CrossRef CAS PubMed.
- K. Tahara, S. Furukawa, H. Uji-i, T. Uchino, T. Ichikawa, J. Zhang, W. Mamdouh, M. Sonoda, F. C. De Schryver, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2006, 128, 16613–16625 CrossRef CAS PubMed.
- X. Zhang, Q. Chen, G. J. Deng, Q. H. Fan and L. J. Wan, J. Phys. Chem. C, 2009, 113, 16193–16198 CAS.
- B. Zha, X. R. Miao, P. Liu, Y. Wu and W. L. Deng, Chem. Commun., 2014, 50, 9003–9006 RSC.
- C. Marie, F. Silly, L. Tortech, K. Müllen and D. Fichou, ACS Nano, 2010, 4, 1288–1292 CrossRef CAS PubMed.
- R. Wen, G. B. Pan and L. J. Wan, J. Am. Chem. Soc., 2008, 130, 12123–12127 CrossRef CAS PubMed.
- Y. Kikkawa, E. Koyama, S. Tsuzuki, K. Fujiwara, K. Miyake, H. Tokuhisa and M. Kanesato, Chem. Commun., 2007, 13, 1343–1345 RSC.
- Y. Kikkawa, E. Koyama, S. Tsuzuki, K. Fujiwara and M. Kanesato, Langmuir, 2010, 26, 3376–3381 CrossRef CAS PubMed.
- S. De Feyter, A. Gesquière, M. Klapper, K. Müllen and F. C. De Schryver, Nano Lett., 2003, 3, 1485–1488 CrossRef CAS.
- Z. Ma, Y. Y. Wang, P. Wang, W. Huang, Y. B. Li, S. B. Lei, Y. L. Yang, X. L. Fan and C. Wang, ACS Nano, 2007, 1, 160–167 CrossRef CAS PubMed.
- Y. Hu, K. Miao, B. Zha, X. R. Miao, L. Xu and W. L. Deng, RSC Adv., 2015, 5, 93337–93346 RSC.
- Y. Hu, K. Miao, S. Peng, B. Zha, L. Xu, X. R. Miao and W. L. Deng, CrystEngComm, 2016, 18, 3019–3032 RSC.
- Y. Hu, K. Miao, B. Zha, L. Xu, X. R. Miao and W. L. Deng, Phys. Chem. Chem. Phys., 2016, 18, 624–634 RSC.
- Y. Hu, K. Miao, B. Zha, L. Xu, X. R. Miao and W. L. Deng, Phys. Chem. Chem. Phys., 2016, 18, 13164–13168 RSC.
- Y. Hu, K. Miao, B. Zha, S. Peng, L. Xu, X. R. Miao and W. L. Deng, Adv. Mater. Interfaces, 2016, 3, 1600428 CrossRef.
- K. S. Mali, K. Lava, K. Binnemans and S. Feyter, Chem.–Eur. J., 2010, 16, 14447–14458 CrossRef CAS PubMed.
- J. Y. Wang, J. Yan, L. Ding, Y. Ma and J. Pei, Adv. Funct. Mater., 2009, 19, 1746–1752 CrossRef CAS.
- L. Cui, X. R. Miao, L. Xu, Y. Hu and W. L. Deng, Phys. Chem. Chem. Phys., 2015, 17, 3627–3636 RSC.
- O. Ivasenko, J. M. MacLeod, K. Y. Chernichenko, E. S. Balenkova, R. V. Shpanchenko, V. G. Nenajdenko, F. Rosei and D. F. Perepichka, Chem. Commun., 2009, 10, 1192–1194 RSC.
- P. Liu, X. R. Miao, Z. Li, B. Zha and W. L. Deng, CrystEngComm, 2014, 16, 9690–9696 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of experimental section and additional STM images. See DOI: 10.1039/c7ra05811j |
| This journal is © The Royal Society of Chemistry 2017 |