
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heat shock protein 90 and serine/threonine kinase B-Raf inhibitors have overlapping chemical space†
A. Anighoro a,
L. Pinzib,
G. Marvertic,
J. Bajorath*a and
G. Rastelli*b
a,
L. Pinzib,
G. Marvertic,
J. Bajorath*a and
G. Rastelli*b
aDepartment of Life Science Informatics, B-IT, LIMES Program Unit Chemical Biology and Medicinal Chemistry, Rheinische Friedrich-Wilhelms-Universität, Dahlmannstr. 2, D-53113 Bonn, Germany. E-mail: bajorath@bit.uni-bonn.de
bDepartment of Life Sciences, University of Modena and Reggio Emilia, Via Campi 103, 41125, Modena, Italy. E-mail: giulio.rastelli@unimore.it
cDepartment of Biomedical, Metabolic and Neurosciences, University of Modena and Reggio Emilia, Via Campi 287, 41125, Modena, Italy
First published on 15th June 2017
Abstract
Heat shock protein 90 (Hsp90) and B-Raf are validated targets for anticancer drug discovery. Although there is strong evidence that concomitant inhibition of Hsp90 and B-Raf may provide significant therapeutic benefits, molecules endowed with dual activity against the two targets have not been reported. For the first time, we show that Hsp90 and B-Raf inhibitors have overlapping chemical space and we disclose the first-in-class dual inhibitors. The compounds were identified through a computational strategy especially devised for detecting ligands with dual-target activity. Although the two targets had only remote binding site similarity, we were able to identify dual inhibitors with well-balanced in vitro potencies and relatively low molecular weight. Remarkably, they also inhibited the V600E mutant form of B-Raf with similar potency. This study provides the first direct proof that designing dual ligands of Hsp90 and a kinase is possible, thus opening the way to new interesting possibilities in drug discovery.
Introduction
Hsp90 and B-Raf are two established anticancer drug targets. Hsp90 is an ubiquitous molecular chaperone responsible for the assembly and regulation of many signal transduction and regulatory client proteins.1 Hsp90 refolds, stabilizes and regulates the trafficking of many proteins involved in uncontrolled proliferation and apoptotic resistance, including multiple protein kinases, steroid hormone receptors, mutated p53, survivin and others.1,2 Mapping of the interaction network of Hsp90 clearly demonstrated that this chaperone is a key node in multiple pathways and cellular and disease processes.3 Interestingly, many mutant and overexpressed oncoproteins are client proteins of Hsp90.1,2 Unfortunately, despite the availability of a large arsenal of Hsp90 inhibitors belonging to different chemical classes, none of them has yet been approved as a drug, due to non-optimal safety profiles or lack of efficacy.4,5 B-Raf is a member of the Ser/Thr protein kinase family involved in the MAPK signaling cascade, which plays a pivotal role in the regulation of many essential cell processes such as signal transduction, cell proliferation and cell survival.6 Activating mutations in B-Raf such as V600E bypass the regulation of MAPK signaling pathway and stimulate uncontrolled cell proliferation.7 This poses a great challenge for therapeutic intervention because B-Raf mutations are associated with resistance to conventional chemotherapeutic drugs. Although a few B-Raf inhibitors have received approval for the treatment of certain types of cancer, responses are often temporary, rarely complete, and restricted by the onset of drug resistance.8 Therefore, there is a pressing need to develop new drugs with improved clinical profiles.Notably, both B-Raf and B-RafV600E are client proteins of Hsp90.1,2 Therefore, inhibition of this chaperone would contribute to degradation of B-Raf and mutant B-Raf.9 Importantly, several recent studies demonstrated that combinations of various Hsp90 and B-Raf inhibitors provided significant synergistic effects.10–15 Indeed, drug combinations based on Hsp90 and B-Raf inhibitors (such as AT13387 and dabrafenib, or XL888 and vemurafenib) are currently under evaluation in clinical trials against mutated melanoma (http://ClinicalTrials.gov identifiers: NCT02721459, NCT02097225, NCT01657591). In addition, inhibition of Hsp90 may be effective in patients with intrinsic or acquired resistance to RAF inhibitors.10–15 Finally, B-Raf inhibitors may paradoxically activate the MAP kinase pathway, and inhibition of Hsp90 may help overcome such resistance mechanisms.16,17 Therefore, although drug development has been focused on targeting Hsp90 and B-Raf independently, considerable evidence has accumulated indicating that a combined inhibition of the two targets may be clinically relevant.
Despite the high therapeutic relevance of combination therapies, a single molecule with dual activity may show potential advantages, including but not limited to superior efficacy, a more predictable pharmacokinetic profile, and lower toxicity.18 In light of all these considerations, we reasoned that a multitarget drug, i.e. a small molecule that is able to hit multiple targets, should be of particular interest. This concept is in line with polypharmacology, which potentially limits drawbacks arising from the use of a single-target drug or a combination of multiple drugs.18–20 To the best of our knowledge, this possibility has not been explored in Hsp90 and B-Raf, and molecules endowed with dual activity are not available. Although the design of dual inhibitors of Hsp90 and B-Raf is hampered by the fact that the binding sites of the two targets significantly differ, we recently presented evidence from structural and computational analysis that inhibitors of Hsp90 and various kinases display at least remotely similar protein–ligand interaction patterns.21 These insights also motivated the search for dual inhibitors of Hsp90 and B-Raf. To these ends, a combined ligand- and structure-based virtual screening protocol was devised, which enabled the identification of the first-in-class compounds with dual activity. Remarkably, the compounds were also active against mutant B-RafV600E. This study provides the first evidence that Hsp90 and B-Raf inhibitors share a common chemical space, a finding that may open new avenues for future drug discovery.
Results and discussion
The analysis of available X-ray structures of Hsp90 and B-Raf in complex with various inhibitors (see Experimental and ESI sections†) identified two active compounds containing a similar scaffold (Table 1, Fig. 1A and S1†) including 3RR (Hsp90 inhibitor) and L1E (B-Raf inhibitor). This scaffold was involved in well-defined interactions with both targets. Therefore, on the basis of visual inspection of the co-crystallized complexes of these inhibitors, a simple pharmacophore model was generated comprising a hydrogen bond donor, a hydrogen bond acceptor, and an aromatic center (Fig. 1B). The donor and acceptor formed hydrogen bonds with the hinge region of B-Raf and residues Asp93 and Thr184 of Hsp90.22,23 In addition, the aromatic feature centered on the phenyl ring was involved in hydrophobic contacts with both targets. Visual inspection of the two X-ray complexes suggested that the inclusion of pharmacophoric features derived from the larger ligand L1E that are not shared by 3RR would have probably resulted in large solvent-exposed moieties in Hsp90 structures. Therefore, such features were not included.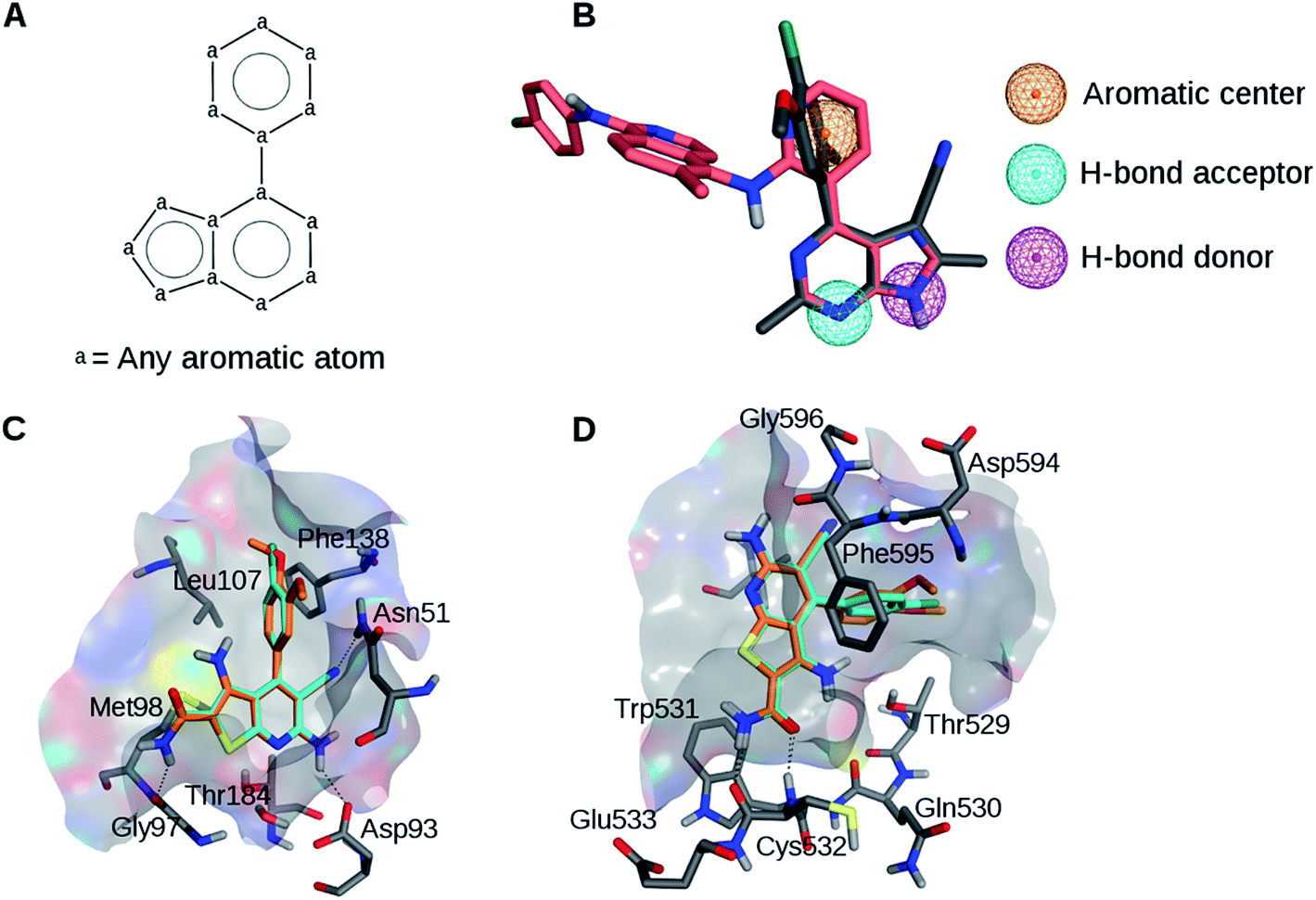 | ||
| Fig. 1 Ligand alignments and pharmacophores. (A) Common scaffold identified for B-Raf and Hsp90 inhibitors in co-crystal structures 3IDP and 3RLR. (B) Pharmacophore model based on the aligned B-Raf (pink) and the Hsp90 (gray) inhibitors. (C) Compounds 1 (cyan) and 2 (orange) docked to Hsp90. (D) Compounds 1 and 2 docked to B-Raf. | ||
A substructure search was carried out to pre-select a subset of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 167 database compounds based on the Hsp90/B-Raf inhibitor scaffold shown in Fig. 1A. The obtained library of compounds was subjected to pharmacophore-guided docking to both targets, in order to prioritize compounds establishing the key interactions discussed above, leading to the selection of 20 candidates for dual inhibition, which were prioritized by comparing target-based docking ranks and visual inspection.
167 database compounds based on the Hsp90/B-Raf inhibitor scaffold shown in Fig. 1A. The obtained library of compounds was subjected to pharmacophore-guided docking to both targets, in order to prioritize compounds establishing the key interactions discussed above, leading to the selection of 20 candidates for dual inhibition, which were prioritized by comparing target-based docking ranks and visual inspection.
Evaluation of the candidate compounds in Hsp90 and B-Raf enzyme assays identified two dual inhibitors with micromolar activity against both targets. Fig. 2 shows the structures of the newly identified dual inhibitors, and their activities are reported in Table 2. Dose–response curves for these compounds are shown in Fig. S2 and S3 of the ESI.† Both compounds contain a thieno[2,3-d]pyrimidine scaffold and are distinguished by substitutions on the attached phenyl ring.24,25 The compounds were also tested in an orthogonal assay to demonstrate that they did not elicit false-positive signals (Fig. S4†).
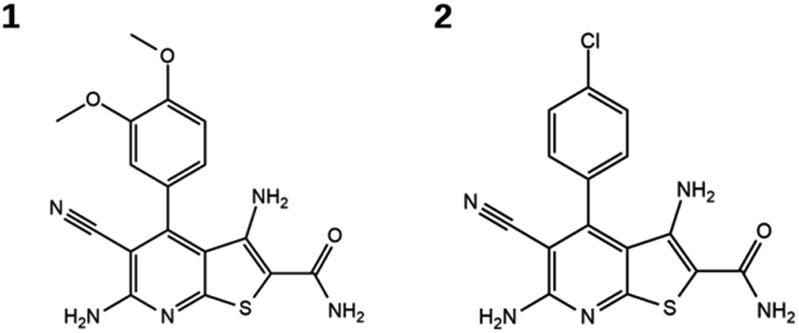 | ||
| Fig. 2 Dual inhibitors of Hsp90 and B-Raf. | ||
| Compound | IC50 B-Raf (μM) | IC50 B-RafV600E (μM) | IC50 Hsp90 (μM) |
|---|---|---|---|
| 1 | 28.9 ± 1.7 | 9.1 ± 0.4 | 1.2 ± 0.1 |
| 2 | 1.5 ± 0.1 | 2.5 ± 0.2 | 7.6 ± 0.6 |
Putative binding modes of the dual inhibitors in Hsp90 and B-Raf are shown in Fig. 1C and D, respectively. In Hsp90, both compounds are likely to form a hydrogen bond network with Asn251, Asp93, and Thr184. The dimethoxyphenyl moiety of 1 and the corresponding chlorophenyl moiety of 2 are likely to form hydrophobic contacts with several residues including Leu107 and Phe138. This binding mode was consistent with the crystallographic structure of a Hsp90 inhibitor containing this scaffold (Fig. S5†).25
In B-Raf, the amide group of the inhibitors is thought to hydrogen bond to the hinge residue Cys532. In addition, the cyano group and the differently substituted phenyl ring are proximal to residues of the DFG motif.
The two inhibitors were also tested on the mutant B-RafV600E, and were both found to be active with IC50 values comparable to, or better than, those obtained for wild type B-Raf (Table 2, Fig. S2†). These findings were considered particularly significant because B-RafV600E is a known escape mutant for therapeutic intervention.
Another advantage of the newly identified dual inhibitors is that they are small in size. We note that chimeras with dual activity are generally sought by merging two ligands with a suitable linker. This inevitably increases molecular weight, which may not only increase the likelihood of promiscuity, but also lead to poorer ADME/PK properties.20 By contrast, the newly identified inhibitors leave considerable room for further chemical optimization. For example, analysis of the predicted binding modes suggests that modifications at the phenyl ring of 1 and 2 may provide valuable opportunities for dual activity optimization. Larger groups are sterically tolerated and could provide additional van der Waals contacts with both targets. Moreover, polar substituents in meta- or para-position may provide additional hydrogen bonds with the backbone of Phe138 in Hsp90 and with Lys483 and Glu501 in B-Raf. These interactions have been observed in several Hsp90 and B-Raf crystal structures (e.g. PDB codes 1OSF and 2QG0 for Hsp90, and 3D4Q, 3PPK, and 3OG7 for B-Raf). Moreover, the cyano group is solvent-exposed in B-Raf and is not required for Hsp90 activity,25 and could be replaced with other small hydrogen bonding groups. While chemical optimization is beyond the scope of the present work, the identified dual ligands constitute valuable starting points for further drug development.
Conclusions
In conclusion, we have reported on the identification of the first Hsp90 and B-Raf dual inhibitors and provided proof of concept that Hsp90 and B-Raf kinase inhibitors have overlapping chemical space. The identified compounds share a thieno[2,3-d]pyrimidine scaffold that is also found in other Hsp90 and B-Raf inhibitors but has never been reported to represent compounds with dual activity. Interestingly, both targets belong to different families and adopt different folds. Binding site similarities estimated with a method particularly efficient in detecting binding site similarity in the absence of sequence or fold conservation confirmed that Hsp90 and B-Raf binding sites significantly differ (see Experimental section for details). Thus, demonstration of dual inhibition properties was far from being obvious.Remarkably, both compounds displayed similar activity against mutant B-RafV600E, which is of considerable potential for cancer treatment.
If synergy exists between two targets, as is the case for Hsp90 and B-Raf,9–15 it is conceivable that dual inhibitors with only moderate activity might still produce superior pharmacological effects in terms of efficacy and safety as compared with highly potent compounds that only inhibit a single target.26,27 Remarkably, the discovered compounds displayed a balanced level of activity among Hsp90, B-Raf, and mutant B-Raf, which makes them especially promising in a polypharmacology approach context18,26,27 More in general, this study provides the first direct proof that designing dual ligands of Hsp90 and a kinase is possible, thus opening the way to new interesting possibilities in drug discovery based on rational polypharmacology approaches.
Experimental section
X-ray structures and pharmacophore model
For B-Raf, eight crystal structures in complex with potent inhibitors were selected for which high confidence activity data were available (Table S1†).28 In addition, for Hsp90, 19 structures of inhibitor complexes with Ki values of less than 10 μM were selected (Table S1†). Analysis of these structures identified two inhibitors with a structurally related scaffold for B-Raf with Protein Data Bank (PDB)29 code 3IDP22 and Hsp90 with PDB code 3RLR23 (Fig. 1A). The bound conformations of these inhibitors (termed L1E and 3RR) were superposed and a basic pharmacophore model was derived using the Molecular Operating Environment 2014.09 (MOE)30 that included a hydrogen bond donor, a hydrogen bond acceptor, and an aromatic center (Fig. 1B). These features were likely to engage in key interactions with both targets.22,23Compound database and pre-filtering
A database of 4805970 unique compounds was obtained by merging the catalogs of 10 vendors (including AMRI, Aronis, Asinex, ChemBridge, Enamine, InterBioScreen, Life Chemicals, Maybridge, Otava and Vitas-MLab). A SMARTS string representing the shared scaffold (Fig. 1A) was used to filter the database with OpenBabel 2.3.2 (ref. 31) that permitted possible heteroatoms and substitutions preserving the aromatic character of the rings (Fig. 1A). On the basis of this search, 15167 compounds were pre-selected and prepared for docking with MOE by computing protonation states and partial charges and optimizing structures by energy minimization.
Protein structure preparation
In X-ray structure 3IDP, 15 residues (residues from 598 to 613) of the activation loop were missing.22 An initial conformation of this loop was obtained by adding the missing residues with MOE followed by relaxation of the loop structure and refinement with ModLoop.32 This tool generates 300 loop conformations and returns the one with the most favorable energy score. The 3IDP and 3RLR template structures were prepared for docking with MOE by adding hydrogen atoms and calculating atomic partial charges according to the MMFF94x force field. All water molecules were removed except for one water molecule in Hsp90 (number 2 according to 3RLR residues numbering) that was known to be involved in a conserved network of hydrogen bonds between Hsp90 and several inhibitors.23,33,34Docking
Docking calculations on Hsp90 and B-Raf binding were performed with the Dock module of MOE.30 The binding sites were defined by the coordinates of the co-crystallized ligands. The pharmacophore model (Fig. 1B) was used to guide docking of the 15167 pre-selected compounds. In each case, 1000 docking poses were generated with the pharmacophore placement function of MOE. Docking poses were ranked on the basis of force field scoring and for each ligand, the top 10 poses were selected and subjected to energy refinement and re-scoring. Ultimately, the best-scoring poses were ranked and retained if they matched the pharmacophore model in a post-filtering step. For both targets, highly ranked compounds were visually inspected to aid in the final selection of 20 candidate compounds for experimental evaluation. Test compounds were also computationally screened for pan assay interference compounds (PAINS)35 using publicly available filters36 and aggregating compounds using the ZINC/UCSF aggregator advisor.37
Enzyme assays
Candidate compounds were assayed for in vitro inhibitory activity against both targets.Orthogonal assay
To test for potential false-positive Hsp90 and/or B-Raf assay readouts as a consequence of compound aggregation, an orthogonal enzyme assay was carried out at NMS focusing on inhibition of Poly(ADP-Ribose)-Polymerase 1 (PARP-1). Recombinant PARP-1 preparations were >80% pure on the basis of SDS-PAGE, N-terminal sequence analysis and electrospray mass spectrometry. A fluorescence polarization assay was conducted to evaluate the displacement of a PARP-1 Binding Probe (PBP) synthesized as described previously.44 The NAD+ competitive PARP-1 inhibitor olaparib was used as a control.45 The compounds were solubilized, serially diluted and dispensed in 384-well black plates by following the same procedure described for the Hsp90 assays. 19 μl of the reaction mixture (final concentration of 250 nM PARP-1 protein and 50 nM PBP) were dispensed in 384-well black plate. Protein and probe were diluted in 50 mM Tris/HCl pH 8, 150 mM NaCl, 10 mM MgCl2, 0.001% Triton. Plate reading (G factor calculated with negative control was 1.160) and IC50 determinations were then performed as described for Hsp90. Results are shown in Fig. S4.†Binding site similarity calculations
Hsp90 and B-Raf binding sites were compared using VolSite and Shaper.46 Crystal structures of human B-Raf and Hsp90 were collected from the PDB (accessed on February 2017)29 and chains were split, leading to two final sets of 119 and 244 structures for B-Raf and Hsp90, respectively. For each set, structures were aligned with the Align Binding Sites tool available in Maestro (version 10.3)47 and a representative structure for each significantly different conformation was selected, obtaining a final set of 9 representative conformations for B-Raf and 11 for Hsp90. The proteins were prepared with the Protein Preparation Wizard utility of Maestro 10.3. Binding sites were then described with VolSite and compared with Shaper, using default settings.46 All ligand and water molecules were removed from the representative structures, except for water molecules involved in a conserved network of hydrogen bonds between Hsp90 and the co-crystallized inhibitors.34 An average RefTversky similarity score of 0.33 was obtained, which was below the statistical threshold of 0.35 that distinguishes similar from dissimilar binding sites.46Conflicts of interest
The authors declare no competing financial interest.Notes and references
- J. Li and J. Buchner, J. Biomed. Sci., 2013, 36, 106–117 Search PubMed.
- P. C. Echeverría and D. Picard, in The Molecular Chaperones Interaction Networks in Protein Folding and Degradation, ed. W. A. Houry, Springer-Verlag, New York, 2014, ch. 5, pp. 133–150 Search PubMed.
- P. C. Echeverría, A. Bernthaler, P. Dupuis, B. Mayer and D. Picard, PLoS One, 2011, 6, e26044 Search PubMed.
- L. Neckers and P. Workman, Clin. Cancer Res., 2012, 18, 64–76 CrossRef CAS PubMed.
- R. Bhat, S. R. Tummalapalli and D. P. Rotella, J. Med. Chem., 2014, 57, 8718–8728 CrossRef CAS PubMed.
- W. Zhang and H. T. Liu, Cell Res., 2002, 12, 9–18 CrossRef PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. C. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Nature, 2002, 417, 949–954 CrossRef CAS PubMed.
- H. E. Haarberg and K. S. M. Smalley, Drug Discovery Today: Technol., 2014, 11, 27–32 CrossRef PubMed.
- O. M. Grbovic, A. D. Basso, A. Sawai, Q. Ye, P. Friedlander, D. Solit and N. Rosen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 57–62 CrossRef CAS PubMed.
- K. H. T. Paraiso, H. E. Haarberg, E. Wood, V. W. Rebecca, Y. A. Chen, Y. Xiang, A. Ribas, R. S. Lo, J. S. Weber, V. K. Sondak, J. K. John, A. A. Sarnaik, J. M. Koomen and K. S. M. Smalley, Clin. Cancer Res., 2012, 18, 2502–2514 CrossRef CAS PubMed.
- Z. Solárová, J. Mojžiš and P. Solár, Int. J. Oncol., 2015, 46, 907–926 Search PubMed.
- J. Acquaviva, D. L. Smith, J.-P. Jimenez, C. Zhang, M. Sequeira, S. He, J. Sang, R. C. Bates and D. A. Proia, Mol. Cancer Ther., 2014, 13, 353–363 CrossRef CAS PubMed.
- G. Zhang, D. T. Frederick, L. Wu, Z. Wei, C. Krepler, S. Srinivasan, Y. C. Chae, X. Xu, H. Choi, E. Dimwamwa, O. Ope, B. Shannan, D. Basu, D. Zhang, M. Guha, M. Xiao, S. Randell, K. Sproesser, W. Xu, J. Liu, G. C. Karakousis, L. M. Schuchter, T. C. Gangadhar, R. K. Amaravadi, M. Gu, C. Xu, A. Ghosh, W. Xu, T. Tian, J. Zhang, S. Zha, Q. Liu, P. Brafford, A. Weeraratna, M. A. Davies, J. A. Wargo, N. G. Avadhani, Y. Lu, G. B. Mills, D. C. Altieri, K. T. Flaherty and M. Herlyn, J. Clin. Invest., 2016, 126, 1834–1856 Search PubMed.
- R. Suzuki, S. Kikuchi, T. Harada, N. Mimura, J. Minami, H. Ohguchi, Y. Yoshida, M. Sagawa, G. Gorgun, D. Cirstea, F. Cottini, J. Jakubikova, Y.-T. Tai, D. Chauhan, P. G. Richardson, N. Munshi, K. Ando, T. Utsugi, T. Hideshima and K. C. Anderson, PLoS One, 2015, 10, e0143847 Search PubMed.
- T. Smyth, K. H. T. Paraiso, K. Hearn, A. M. Rodriguez-Lopez, J. M. Munck, H. E. Haarberg, V. K. Sondak, N. T. Thompson, M. Azab, J. F. Lyons, K. S. M. Smalley and N. G. Wallis, Mol. Cancer Ther., 2014, 13, 2793–2804 CrossRef CAS PubMed.
- P. I. Poulikakos, C. Zhang, G. Bollag, K. M. Shokat and N. Rosen, Nature, 2010, 464, 427–430 CrossRef CAS PubMed.
- M. Phadke, G. T. Gibney, C. J. Rich, I. V. Fedorenko, Y. A. Chen, R. R. Kudchadkar, V. K. Sondak, J. Weber, J. L. Messina and K. S. M. Smalley, J. Invest. Dermatol., 2015, 135, 2542–2544 CrossRef CAS PubMed.
- A. Anighoro, J. Bajorath and G. Rastelli, J. Med. Chem., 2014, 57, 7874–7887 CrossRef CAS PubMed.
- G. Rastelli and L. Pinzi, Front. Pharmacol., 2015, 6, 157 Search PubMed.
- J.-U. Peters, J. Med. Chem., 2013, 56, 8955–8971 CrossRef CAS PubMed.
- A. Anighoro, D. Stumpfe, K. Heikamp, K. Beebe, L. M. Neckers, J. Bajorath and G. Rastelli, J. Chem. Inf. Model., 2015, 55, 676–686 CrossRef CAS PubMed.
- A. L. Smith, F. F. DeMorin, N. A. Paras, Q. Huang, J. K. Petkus, E. M. Doherty, T. Nixey, J. L. Kim, D. A. Whittington, L. F. Epstein, M. R. Lee, M. J. Rose, C. Babij, M. Fernando, K. Hess, Q. Le, P. Beltran and J. Carnahan, J. Med. Chem., 2009, 52, 6189–6192 CrossRef CAS PubMed.
- P.-P. Kung, P.-J. Sinnema, P. Richardson, M. J. Hickey, K. S. Gajiwala, F. Wang, B. Huang, G. McClellan, J. Wang, K. Maegley, S. Bergqvist, P. P. Mehta and R. Kania, Bioorg. Med. Chem. Lett., 2011, 21, 3557–3562 CrossRef CAS PubMed.
- G. K. Packard, P. Papa, J. R. Riggs, P. Erdman, L. Tehrani, D. Robinson, R. Harris, G. Shevlin, S. Perrin-Ninkovic, R. Hilgraf, M. A. McCarrick, T. Tran, Y. Fleming, A. Bai, S. Richardson, J. Katz, Y. Tang, J. Leisten, M. Moghaddam, B. Cathers, D. Zhu and S. Sakata, Bioorg. Med. Chem. Lett., 2012, 22, 747–752 CrossRef CAS PubMed.
- P. A. Brough, X. Barril, J. Borgognoni, P. Chene, N. G. M. Davies, B. Davis, M. J. Drysdale, B. Dymock, S. A. Eccles, C. Garcia-Echeverria, C. Fromont, A. Hayes, R. E. Hubbard, A. M. Jordan, M. R. Jensen, A. Massey, A. Merrett, A. Padfield, R. Parsons, T. Radimerski, F. I. Raynaud, A. Robertson, S. D. Roughley, J. Schoepfer, H. Simmonite, S. Y. Sharp, A. Surgenor, M. Valenti, S. Walls, P. Webb, M. Wood, P. Workman and L. Wright, J. Med. Chem., 2009, 52, 4794–4809 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, Drug Discovery Today, 2007, 12, 156–160 CrossRef CAS PubMed.
- G. Bottegoni, A. D. Favia, M. Recanatini and A. Cavalli, Drug Discovery Today, 2012, 17, 23–34 CrossRef CAS PubMed.
- N. Furtmann, Y. Hu and J. Bajorath, J. Med. Chem., 2015, 58, 252–264 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- Molecular Operating Environment, version 2014.09, Chemical Computing Group, Inc Search PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- A. Fiser and A. Sali, Bioinformatics, 2003, 19, 2500–2501 CrossRef CAS PubMed.
- N. G. M. Davies, H. Browne, B. Davis, M. J. Drysdale, N. Foloppe, S. Geoffrey, B. Gibbons, T. Hart, R. Hubbard, M. R. Jensen, H. Mansell, A. Massey, N. Matassova, J. D. Moore, J. Murray, R. Pratt, S. Ray, A. Robertson, S. D. Roughley, J. Schoepfer, K. Scriven, H. Simmonite, S. Stokes, A. Surgenor, P. Webb, M. Wood, L. Wright and P. Brough, Bioorg. Med. Chem., 2012, 20, 6770–6789 CrossRef CAS PubMed.
- M. Sgobba and G. Rastelli, ChemMedChem, 2009, 4, 1399–1409 CrossRef CAS PubMed.
- J. Baell and M. A. Walters, Nature, 2014, 513, 481–483 CrossRef CAS PubMed.
- ZINC PAINS patterns search http://zinc15.docking.org/patterns/home/(accessed June 17, 2016).
- J. J. Irwin, D. Duan, H. Torosyan, A. K. Doak, K. T. Ziebart, T. Sterling, G. Tumanian and B. K. Shoichet, J. Med. Chem., 2015, 58, 7076–7087 CrossRef CAS PubMed.
- K. E. Mercer and C. A. Pritchard, Biochim. Biophys. Acta, 2003, 1653, 25–40 CAS.
- C. K. Weber, J. R. Slupsky, H. A. Kalmes and U. R. Rapp, Cancer Res., 2001, 61, 3595–3598 CAS.
- Y. Heakal, M. Kester and S. Savage, Ann. Pharmacother., 2011, 45, 1399–1405 CrossRef CAS PubMed.
- X. Huang, J. Biomol. Screening, 2003, 8, 34–38 CrossRef CAS PubMed.
- K. Lundgren, H. Zhang, J. Brekken, N. Huser, R. E. Powell, N. Timple, D. J. Busch, L. Neely, J. L. Sensintaffar, Y. Yang, A. McKenzie, J. Friedman, R. Scannevin, A. Kamal, K. Hong, S. R. Kasibhatla, M. F. Boehm and F. J. Burrows, Mol. Cancer Ther., 2009, 8, 921–929 CrossRef CAS PubMed.
- R. Howes, X. Barril, B. W. Dymock, K. Grant, C. J. Northfield, A. G. S. Robertson, A. Surgenor, J. Wayne, L. Wright, K. James, T. Matthews, K.-M. Cheung, E. McDonald, P. Workman and M. J. Drysdale, Anal. Biochem., 2006, 350, 202–213 CrossRef CAS PubMed.
- G. Papeo, N. Avanzi, S. Bettoni, A. Leone, M. Paolucci, R. Perego, F. Quartieri, F. Riccardi-Sirtori, S. Thieffine, A. Montagnoli and R. Lupi, J. Biomol. Screening, 2014, 19, 1212–1219 CrossRef PubMed.
- K. A. Menear, C. Adcock, R. Boulter, X. Cockcroft, L. Copsey, A. Cranston, K. J. Dillon, J. Drzewiecki, S. Garman, S. Gomez, H. Javaid, F. Kerrigan, C. Knights, A. Lau, V. M. Loh, I. T. W. Matthews, S. Moore, M. J. O'Connor, G. C. M. Smith and N. M. B. Martin, J. Med. Chem., 2008, 51, 6581–6591 CrossRef CAS PubMed.
- J. Desaphy, K. Azdimousa, E. Kellenberger and D. Rognan, J. Chem. Inf. Model., 2012, 52, 2287–2299 CrossRef CAS PubMed.
- Maestro, version 10.3 2015, Schrödinger, LLC, New York, NY Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05889f |
| This journal is © The Royal Society of Chemistry 2017 |