
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntisense oligonucleotide modified with serinol nucleic acid (SNA) induces exon skipping in mdx myotubes†
Bao T. Leab,
Keiji Murayamac,
Fazel Shabanpoord,
Hiroyuki Asanumac and
Rakesh N. Veedu *ab
*ab
aCentre for Comparative Genomics, Murdoch University, Perth, Australia-6150. E-mail: R.Veedu@murdoch.edu.au
bPerron Institute for Neurological and Translational Science, Perth, Australia-6009
cDepartment of Biomolecular Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
dFlorey Department of Neuroscience and Mental Health, University of Melbourne, Parkville, VIC 3052, Australia
First published on 6th July 2017
Abstract
Serinol nucleic acid (SNA) is a novel nucleic acid analogue that can form highly stable heteroduplexes with complementary DNA and RNA sequences. Structurally, SNA is a close mimic to peptide nucleic acid (PNA) which is widely used in diagnostic and therapeutic applications. SNA chemistry is relatively new, and so far the scope of SNA has only been explored in improving the efficacy of small interfering RNA and for developing a highly sensitive molecular beacon for diagnostic applications. In this study, we investigated the potential of SNA-modified antisense oligonucleotide (AO) in parallel to PNA-oligo for splice-modulation in an in vitro cellular model of Duchenne muscular dystrophy (DMD). We synthesized a 20-mer SNA and PNA antisense oligonucleotide (AO) designed to induce exon-23 skipping in the mouse dystrophin gene transcript. Our results demonstrated that the SNA AO induced exon-23 skipping at all tested concentrations, whereas the corresponding PNA AO failed to induce any exon-23 skipping upon 24 hours of transfection using Lipofectin transfection reagent. Our results further expands the potential of SNA oligonucleotides in therapeutic applications.
Introduction
Recent clinical translation of novel oligonucleotide therapeutic molecules demonstrates that the use of chemically-modified nucleotide analogues is crucial for the development of successful nucleic acid-based drugs. Although a number of nucleic acid analogues has been reported in recent years, only very small numbers were utilised in the United States Food and Drug Administration (US FDA) approved drugs such as phosphorothioate (PS) DNA, 2′-O-methyl (2′-OMe) RNA, 2′-O-methoxyethyl RNA, 2′-fluoro RNA and phosphorodiamidate morpholino.1 Peptide nucleic acid (PNA, Fig. 1), a prominent class of acyclic nucleic acid analogues, attracted considerable attention in the field of nucleic acid biology for diagnostic and therapeutic applications.2,3 PNA exhibits very high target binding affinity and form extremely stable homo- and hetero-duplex with complementary DNA or RNA.4,5 In addition, PNA also shows very high stability against nucleases.5,6 Recently, the potential of different chemically-modified antisense oligonucleotides (AOs) including twisted intercalating nucleic acids (TINA),7 anhydrohexitol nucleic acid (HNA),8 cyclohexenyl nucleic acid (CeNA),8 altritol nucleic acid (ANA),8 morpholino nucleic acid (MNA)9 and also PNA10,11 has been explored in several studies for exon-skipping in Duchenne muscular dystrophy (DMD), a muscle wasting fatal genetic disease mainly affecting boys caused by the mutations in the dystrophin gene.12,13 However, based on previous reports, the cell delivery of PNA AOs using normal lipid-based transfection reagents is difficult, mainly due to the charge issue.5,6 To improve this limitation, we envisaged the use of serinol nucleic acid-based (SNA) AOs.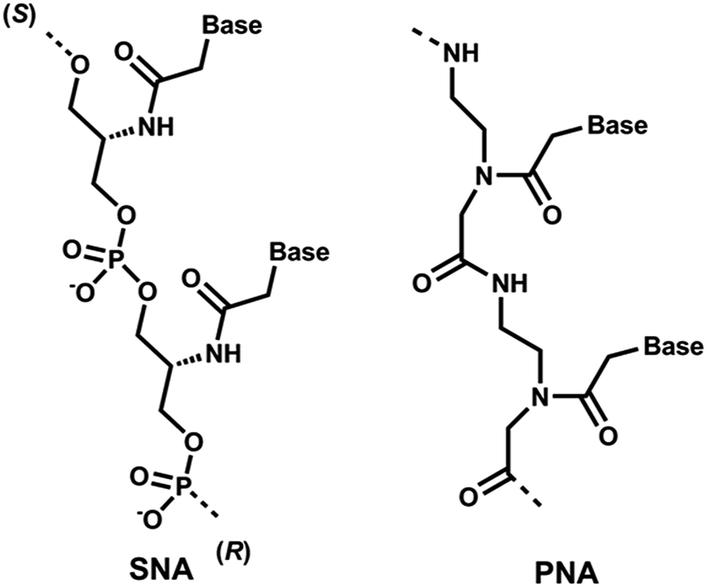 | ||
| Fig. 1 Structural representations of SNA and PNA oligonucleotide building blocks used in this study. | ||
Unlike PNA, SNA building blocks are developed on serinol (2-amino-1,3-propanediol) moiety and each monomer units are linked via normal phosphodiester backbone giving them negative charge (Fig. 1).14 So far, SNA oligonucleotides have been utilised in improving the efficacy of siRNA and for developing molecular beacons for sensitive detection of mRNA molecules.15,16 Generally, SNA oligonucleotides demonstrated high duplex stability with complementary RNA oligonucleotides that highlights the potential applicability of SNA in developing a robust AO capable of effective transfection with normal lipid-based delivery cargos.17 Herein, we investigated the potential of SNA AO to induce exon-23 skipping in H-2Kb-tsA58 mdx mouse myoblasts in vitro and compared its efficacy with PNA AO in parallel to an established 2′-OMePS control AO.
Experimental
Design and synthesis of antisense oligonucleotides used in this study
A 20-mer SNA and PNA AOs and their corresponding 2′-OMePS control AO and a 2′-OMePS-mismatch AO sequence were designed to induce exon-23 skipping in Dmd (Table 1). SNA AO was synthesised by an ABI 3400 DNA/RNA synthesizer using phosphoramidite chemistry as described in previous reports.14,17 The synthesized SNA oligonucleotide was purified using Poly-Pak cartridges and reversed-phase HPLC (Merck LiChrospher 100 RP-18(e) column). After purification, the oligonucleotide was characterized by MALDI-ToF MS analysis. The PNA AO sequence was synthesised by Fmoc solid-phase synthesis. The PNA was assembled on TantaGel XV RAM resin (10 μmol). Fmoc groups were removed by treating the resin with a 20% piperidine solution in DMF for 5 min. The couplings were achieved using 3-fold excess of Fmoc-PNA monomers activated with HATU (3 eq.), DIEA (3 eq.) and 2,5-lutidine (3 eq.). PNA was cleaved from the solid-support with a cocktail of TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIPS:H2O (95:2.5:2.5, v/v/v) for 2 h at room temperature. The crude oligonucleotide was then purified, desalted and verified by MALDI-ToF MS analysis.
:TIPS:H2O (95:2.5:2.5, v/v/v) for 2 h at room temperature. The crude oligonucleotide was then purified, desalted and verified by MALDI-ToF MS analysis.
| Chemistries and AOs names | Sequence, 5′ → 3′ direction | Tm (°C) |
|---|---|---|
| a Complementary synthetic RNA target: 5′-r(AG GUA AGC CGA GGU UUG GCC)-3′. Miss-matched nucleotides are represented in bold underlined letters. See Fig. S5 (ESI) for melting curves. | ||
| PNA | P(GGCCAAACCUCGGCUUACCU) | 85.7 |
| SNA | S(GGCCAAACCUCGGCUUACCU) | 62.3 |
| 2′-OMePS | 2′(GGCCAAACCUCGGCUUACCU) | 60.4 |
| DNA | D(GGCCAAACCUCGGCUUACCU) | 49.8 |
| 2′-OMePS-mismatch | 2′(![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) GCC GCC![[U with combining low line]](https://www.rsc.org/images/entities/b_char_0055_0332.gif) AAC AAC![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif) UCGGU UCGGU![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) ACC) ACC) |
33.4 |
Melting temperature analysis of the antisense oligonucleotides
The SNA, PNA, 2′-OMePS, DNA and the 2′-OMePS-mismatch AOs were prepared at 2 μM concentration in a buffer solution containing 10 mM NaCl, 0.01 mM EDTA and 10 mM sodium phosphate (pH 7.0). Before loading onto a quartz cuvettes of 1 mm path-length, the AOs were hybridized with the complementary RNA sequence (2 μM) in equal volume by denaturing at 95 °C followed by slow cooling to room temperature. Then, the melting temperature measurement was performed using Shimadzu UV-1800 UV spectrophotometer with a temperature range of 20–95 °C (ramp rate = 1.0 °C min−1). Tm values were then calculated by the first derivative.Cell culture and transfection
H-2Kb-tsA58 (H2K) mdx myoblasts were propagated in Dulbecco's Modified Eagle Medium (DMEM) containing 20% fetal bovine serum (FBS), 10% horse serum (HS) supplemented with 0.5% chicken embryo extract.18 Cells were then plated onto a 24 well plate pre-treated with 50 μg ml−1 poly-D-lysine (Sigma) and 100 μg ml−1 Matrigel (Corning) at 2 × 104 cells per well in DMEM 5% HS and incubated for 24 h at 37 °C, 5% CO2 for differentiation into myotubes. SNA and PNA AOs were transfected using Lipofectin in parallel to the corresponding 2′-OMePS control AO and a 2′-OMePS-mismatch AO at a ratio of 2:1 (Lipofectin:AO) with final concentrations of 100, 200 and 400 nM as per the manufacturer's instructions without removing the medium after 3 hours. For naked-AO transfection, the AOs were mixed directly with Optimem reduced serum medium and added to the cells with final concentrations of 100, 200 and 400 nM. All reagents used in this study were supplied from Thermo Fisher Scientific, Australia unless stated.
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR)
Twenty-four hours after transfection, the cells were collected for RNA extraction. Briefly, RNA was extracted using Direct-zol™ RNA MiniPrep Plus with TRI Reagent® (Zymo Research) as per the manufacturer's instructions. The dystrophin transcripts were then amplified by nested RT-PCR across exons 20–26 as described previously.17 PCR products were separated on 2% agarose gels in Tris–acetate–EDTA buffer and the images were captured on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). Densitometry analysis was performed by Image J software.Nuclease stability analysis of the SNA oligonucleotide
Stability of SNA oligonucleotide against 3′ → 5′ exonuclease degradation was investigated using snake venom phosphodiesterase (Sigma), in comparison to an unmodified DNA AO (Table 1) control. Briefly, 10 μM of the oligonucleotides were incubated with 0.08 units per ml snake venom phosphodiesterase in a buffer containing 10 mM Tris–HCl, 100 mM NaCl, and 15 mM MgCl2 in a final volume of 60 μl. Samples were incubated at 37 °C and 10 μl samples were collected at 0, 10, 30, 60 and 120 minutes and quenched with an equal volume of 80% formamide containing bromophenol blue and xylene cyanol gel tracking dyes. The samples were then heated for 5 min at 95 °C and analysed by 20% denaturing polyacrylamide electrophoresis. Quantitation was performed on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France).Results and discussion
In this study, we investigated the potential of SNA modified AO to induce exon-skipping in parallel to PNA modified AO, a closely related analogue, as a control. For this purpose, we synthesised a 20-mer fully-modified SNA and PNA AOs including their corresponding 2′-OMePS AO and 2′-OMePS-mismatch AO controls (Table 1), and evaluated their efficacy to induce Dmd exon-23 skipping in mdx mouse myotubes. First, we performed the melting temperature (Tm) analysis of the AO sequences against a 20-mer synthetic RNA sequence, which is identical to Dmd gene transcript target as described previously18 (Table 1). The results showed that the PNA AO exhibited the highest Tm (85.7 °C, Table 1) compared to all other tested AOs. However, the SNA AO showed higher Tm (62.3 °C) than the 2′-OMePS AO (60.4 °C) and DNA AO (49.8 °C, Table 1). A possible explanation for the lower stability of SNA AO compared to PNA AO could be due to the electrostatic repulsion induced by phosphodiester linkage on SNA AO and flexible conformation of SNA compared to PNA.17 Not surprisingly, the 2′-OMePS-mismatch AO showed very low Tm (33.4 °C) because of the impaired Watson–Crick interactions with the RNA target due to miss-matched nucleotides.Next, we tested the ability of SNA and PNA AOs to induce exon-23 skipping in H2K mdx mouse myoblasts in parallel to their established control 2′-OMePS AO. In short, the H2K mdx myoblasts were cultured as previously and when propagated to 80–90% confluence, the cells were plated onto a 24-well plate for differentiation into myotubes.18 The AOs were then transfected into cells at 100, 200 and 400 nM concentrations using a lipid-based transfection reagent Lipofectin. After 24 hours, the cells were collected and extracted the RNA for analysing exon-skipping efficacy by RT-PCR. The dystrophin transcripts were then amplified across exons 20–26 with a full-length product size of 901 bp, and 688 bp product for exon-23 skipping. The PCR products were then examined on a 2% agarose gel. The results clearly demonstrated that the SNA AO induced exon-23 (full-length band at 901 bp) skipping at all concentrations by yielding the skipped product of 688 bp (Fig. 2A and B), and the skipping efficacy was improved at higher doses. Densitometry analysis to quantify the actual percentage of skipping showed that the percentage of exon-23 skipped product increased from 41% at 100 nM to 52% at 400 nM (Fig. 2B). As previously reported, the established 2′-OMePS control AO induced efficient exon-23 skipping even at 100 nM concentration (Fig. 2A and B).
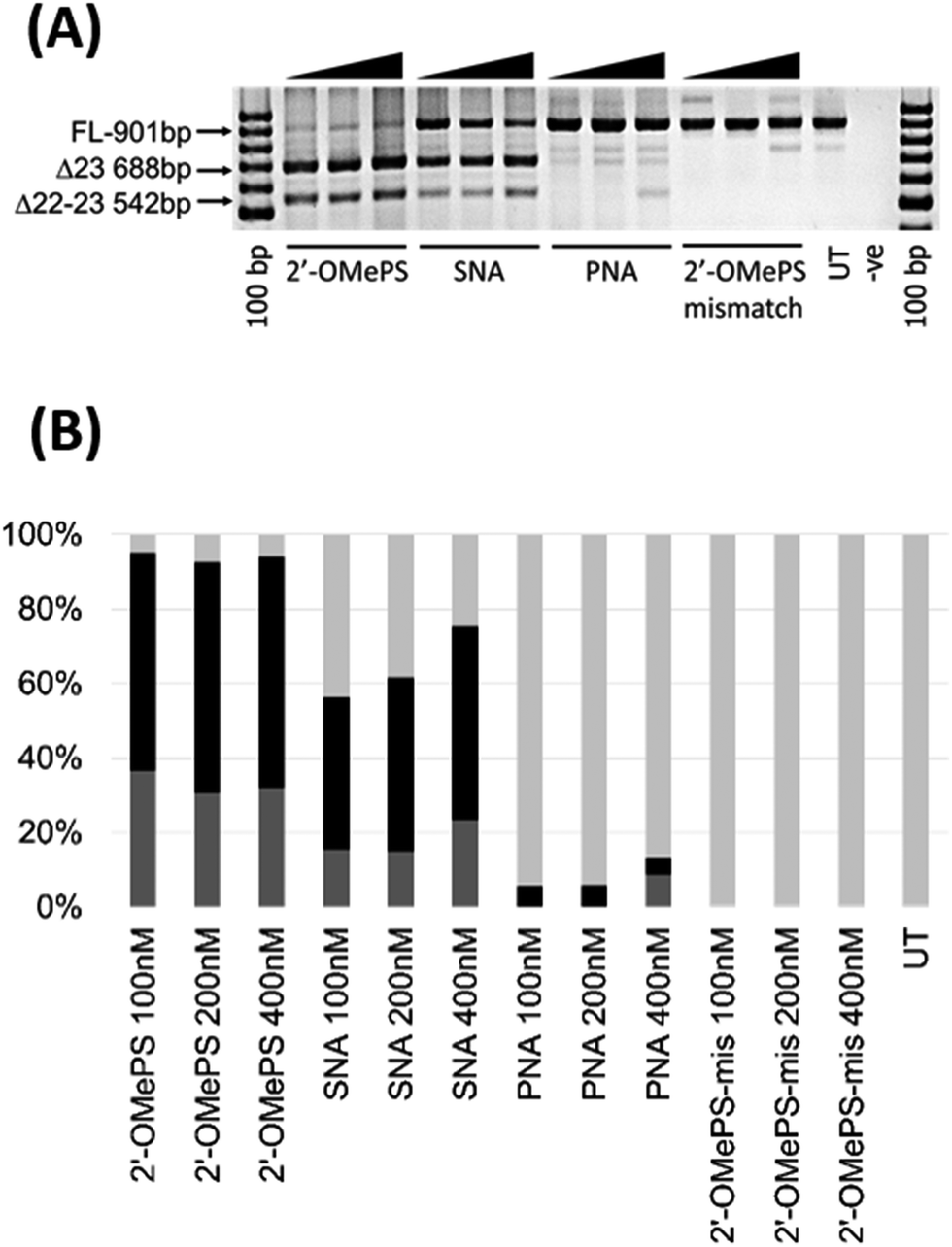 | ||
| Fig. 2 (A) RT-PCR analysis of 2′-OMePS, SNA, PNA and 2′-OMePS-mismatch (2′-OMePS-mis) AOs for exon-23 skipping in mdx mouse myotubes. FL: full length; Δ23: exon-23 skipped product; Δ22–23: dual exon-22/23 skipped product. (B) Densitometry based on RT-PCR analysis of the AOs. Black: percentage of exon-23 skipped product; dark grey: percentage of dual exon-22/23 skipped product and light grey: percentage of full length product; UT: untreated. | ||
But, the PNA AO failed to induce any significant exon-23 skipping at all concentrations. This may be due to poor cellular-uptake of PNA with normal lipid-based transfection reagent Lipofectin as it doesn't complex well. In contrast, SNA AO is negatively charged with an inter-nucleotide phosphodiester linkage and can form complex with Lipofectin. As the SNA and 2′-OMePS AOs showed similar melting temperature, we further designed and synthesised an additional miss-matched 2′-OMePS (2′-OMePS-mismatch, Table 1) sequence to gain more insights on sequence specificity. Not surprisingly, the 2′-OMePS-mismatch AO did not induce any exon-23 skipping at all concentrations. In the case of SNA AO, an additional band at 542 bp was observed, and this is believed to be due to the undesired dual exon-22/23 skipping (Fig. 2A and B). 2′-OMePS AO showed a higher percentage of dual skipping product (32–37%) compared to the SNA AO (16–24%) and PNA AO (8%, showed only at 400 nM).
To further investigate the efficacy of exon-23 skipping, the SNA, PNA and 2′-OMePS AOs were transfected without using the transfection reagent Lipofectin. The AOs were mixed with the media and added directly to the cells and followed the same procedure as discussed above. The results showed that all AOs failed to induce any exon-23 skipping after 24 h of incubation (Fig. 3). This experiment highlights that complexation with lipid-based transfection reagent is important for effective delivery of SNA and 2′-OMePS AOs, and to induce exon-skipping.
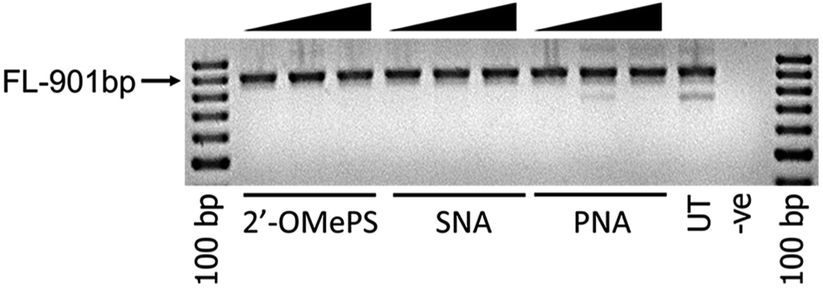 | ||
| Fig. 3 RT-PCR analysis of 2′-OMePS, SNA and PNA AOs for exon-23 skipping in mdx mouse myotubes after naked transfection. FL: full length. | ||
Very high nuclease stability of PNA oligonucleotides has been reported previously, which is not surprising in line with its structure.5,6 Based on this, and to further characterise the properties of SNA oligonucleotides, we tested the stability of SNA AO against exonuclease degradation using snake venom phosphodiesterase, in comparison with a DNA AO. The AOs were incubated in 0.08 units per ml of the enzyme and samples were collected at 0, 10, 30, 60 and 120 minutes intervals. The products were then mixed with formamide loading buffer and analysed on a 20% denaturing polyacrylamide gel. As predicted, the DNA AO was very vulnerable to nuclease attack and completely degraded within 10 minutes of incubation. But, the SNA AO showed very high resistance to enzymatic degradation even after 120 minutes of incubation (Fig. 4) which is indicative of high stability of SNA AOs.
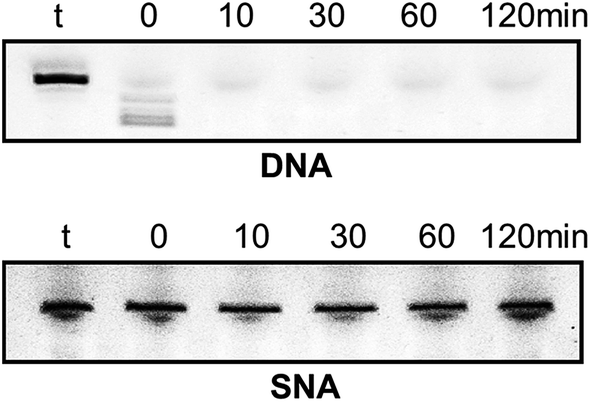 | ||
| Fig. 4 Enzymatic stability analysis of DNA and SNA oligonucleotides; t: AO template without enzyme. | ||
Conclusions
In summary, we have synthesised SNA AO and investigated its potential to induce exon-skipping. We found that SNA-based AO can effectively induce Dmd exon-23 skipping in vitro. Although it's closely related analogue PNA showed very high RNA binding affinity, it failed to induce any significant exon-23 skipping due to poor transfection efficiency with lipid-based transfection reagents. The normal phosphodiester linkages between SNA monomers make them readily complex with lipid-based transfection reagents and render better cellular uptake capability. In addition, SNA-modified AO also showed very high resistance to nuclease degradation. SNA chemistry is relatively new and has not been explored for therapeutic applications compared to PNA. Our study opens up the scope of SNA-modified oligonucleotides in developing novel therapeutic oligonucleotides such as AOs, antimiRs, DNAzymes as fully-modified SNA or in combination with other chemistries (e.g. LNA19,20).Acknowledgements
We thank Prof. Steve Wilton and Prof. Sue Fletcher, and their research group for providing H-2Kb-tsA58 mdx cells and guidance in cell culture protocols. RNV acknowledges funding support from the Department of Health-Western Australia (Merit Award), McCusker Charitable Foundation and Perron Institute for Neurological and Translational Science. BTL thanks the MIPS funding scheme of Murdoch University.References
- K. E. Lundin, O. Gissberg and C. I. E. Smith, Hum. Gene Ther., 2015, 26, 475 CrossRef CAS PubMed.
- J. Matcher, J. Wesoly and H. A. Bluyssen, Mini-Rev. Med. Chem., 2014, 14, 401–410 CrossRef.
- R. Gambari, Expert Opin. Ther. Pat., 2014, 24, 267–294 CrossRef CAS PubMed.
- P. E. Nielsen and M. Egholm, Curr. Issues Mol. Biol., 1999, 1, 89–104 CAS.
- B. Hyrup and P. E. Nielsen, Bioorg. Med. Chem., 1996, 4, 5–23 CrossRef CAS PubMed.
- H. J. Larsen, T. Bentin and P. E. Nielsen, Biochim. Biophys. Acta, 1999, 1489, 159–166 CrossRef CAS.
- B. T. Le, V. V. Filichev and R. N. Veedu, RSC Adv., 2016, 6, 95169–95172 RSC.
- B. T. Le, S. Chen, M. Abramov, P. Herdewijn and R. N. Veedu, Chem. Commun., 2016, 52, 13467–13470 RSC.
- S. Chen, B. T. Le, K. Rahimizadeh, K. Shaikh, N. Mohal and R. N. Veedu, Molecules, 2016, 21, 1582 CrossRef PubMed.
- X. Gao, X. Shen, X. Dong, N. Ran, G. Han, L. Cao, B. Gu and H. Yin, Mol. Ther. Nucleic Acids, 2015, 4, e255 CrossRef CAS PubMed.
- H. Yin, Q. Lu and M. Wood, Mol. Ther., 2008, 16, 38–45 CrossRef CAS PubMed.
- S. D. Wilton, R. N. Veedu and S. Fletcher, Trends Mol. Med., 2015, 21, 417–426 CrossRef CAS PubMed.
- T. L. Bao, R. N. Veedu, S. Fletcher and S. D. Wilton, Expert Opin. Orphan Drugs, 2016, 4, 139–152 CrossRef CAS.
- H. Kashida, K. Murayama, T. Toda and H. Asanuma, Angew. Chem., Int. Ed. Engl., 2011, 50, 1285–1288 CrossRef CAS PubMed.
- K. Murayama, Y. Kamiya, H. Kashida and H. Asanuma, ChemBioChem, 2015, 16, 1298–1301 CrossRef CAS PubMed.
- Y. Kamiya, J. Takai, H. Ito, K. Murayama, H. Kashida and H. Asanuma, ChemBioChem, 2014, 15, 2549–2555 CrossRef CAS PubMed.
- K. Murayama, Y. Tanaka, T. Toda, H. Kashida and H. Asanuma, Chemistry, 2013, 19, 14151–14158 CrossRef CAS PubMed.
- C. J. Mann, K. Honeyman, A. J. Cheng, T. Ly, F. Lloyd, S. Fletcher, J. E. Morgan, T. A. Partridge and S. D. Wilton, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 42–47 CrossRef CAS.
- R. N. Veedu and J. Wengel, Chem. Biodiversity, 2010, 7, 536 CAS.
- R. N. Veedu and J. Wengel, RNA Biol., 2009, 6, 321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06091b |
| This journal is © The Royal Society of Chemistry 2017 |