DOI:
10.1039/C7RA06270B
(Paper)
RSC Adv., 2017,
7, 39228-39236
2,6-Di(thiophenyl)-1,5-dihydrodipyrrolopyrazine (DT-DPP) structural isomers as donor–acceptor–donor molecules and their optoelectronic investigation†
Received
5th June 2017
, Accepted 4th July 2017
First published on 10th August 2017
Abstract
Herein we report the synthesis and characterization of two new D–A–D molecules 1,5-dimethyl-2,6-di(thiophen-2-yl)-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (2DT-DPP) and 1,5-dimethyl-2,6-di(thiophen-3-yl)-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (3DT-DPP) via Pd catalyzed C–C and C–N coupling reactions. The dipyrrolopyrazine scaffold comprises an electron-accepting core flanked by two thiophene moieties. To reveal the molecular geometry and molecular packing of 3DT-DPP, X-ray single crystal analysis was carried out. The surface morphological analysis shows the formation of microrods. The photophysical properties of these systems were characterized by UV-vis, fluorescence spectroscopy, and cyclic voltammetry. The thermal properties reveal that both the isomers are thermally stable up to 300 °C. This study clearly demonstrates that DT-DPP can be used to build D–A molecules to produce promising conjugated materials for various optoelectronic applications.
1. Introduction
In recent years, pyrazine and pyrrole fused rings with extended π-conjugation have attracted considerable interest owing to their potential applications in organic field-effect transistors (OFETs),1,2 light-emitting diodes (LEDs),3–9 and organic photovoltaics (OPVs).10,11 Compared to the hole-transporting donor units, the development of an electron transporting acceptor counterparts is still lagging, and new organic semiconductors are highly desirable to realize their practical applications in flexible electronic devices. Therefore, one of the main challenges is to design and synthesize new electron accepting units to develop small organic semiconductors. Consequently, the development of new routes to nitrogen-containing acenes remains important for the next generation of semiconductor devices and technologies. Donor–acceptor alternation of π-conjugated semiconductors is commonly used to tune the optoelectronic properties, thereby improving the OFET performance.12
Thiophene bearing scaffold has been intensively studied for OFET13–18 and in OPVs.19 Takimiya's group reported p-type OFETs with a relatively high mobility. Heteroacenes are interesting owing to the opportunities for tuning the electronic structure, stability, solubility, and molecular packing.20–25 The introduction of an electro-negative nitrogen atom lowers the LUMO level. N-Rich heteroarene are predicted as n-type organic semiconductors.26,27 Miao et al. for the first time reported N-heteroarene and applied to n-type OFETs and investigated their electronic structures, molecular packing, and semiconductor properties.28,29 Their research results indicated that these molecules exhibited the electron mobility up to 3.3 cm2 V−1 s−1.
Conjugated low band gap small molecules containing alternating electron rich donor (D) and electron-poor acceptor (A) units have emerged as a popular approach to improve the device efficiency.30–32 Therefore, the most appealing strategy is the incorporation of electron-deficient group with N-rich heteroarene and electron-donating groups with thiophene. Recently, we reported regioselective synthesis of dipyrrolopyrazine derivative33 and further studied self assembly of these molecules. Inspired by our previous work, herein, we designed and synthesized two structural isomers 2DT-DPP and 3DT-DPP and studied the influence of thiophene on the photophysical properties compared to corresponding DP-DPP. Dihyrodipyrrolopyrazine (DPP) is an electron acceptor unit flanked by electron-donating thiophene moieties has been used to construct D–A–D molecules for new organic semiconductors. It has been shown that the introduction of a thiophene unit creates material that is planar and provide tight packing of the compound.34 The optical, thermal, and electrochemical properties together with single crystal X-ray analysis were described. The self-assembling properties of these synthesized scaffolds were also investigated. DT-DPP structural isomers have demonstrated the capacity to form microrods. The thermal properties of compounds were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). DPP scaffold holds two thiophene units coplanar which definitely have a substantial influence on the optical and electronic properties. The photophysical studies of DT-DPP isomers suggest that these materials are promising candidates for optoelectronics (Fig. 1).
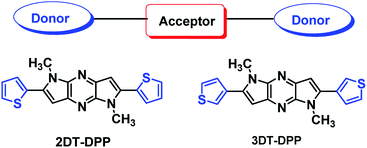 |
| | Fig. 1 Chemical structure of DT-DPP isomers. | |
2. Results and discussion
2.1. Synthesis
The synthesis of structural isomers 2DT-DPP and 3DT-DPP begins from commercially available 3,5-dibromo-6-chloropyrazin-2-amine. The synthetic strategy is depicted in Scheme 1. Compounds 1a, b were prepared by Pd catalysed Sonogashira cross-coupling reaction35 between 3,5-dibromo-6-chloropyridin-2-amine and ethynylthiophene affording compound 5-bromo-6-chloro-3-(thiophenylethynyl)pyrazin-2-amine derivatives 1a, b in excellent yield (90–93%). t-BuOK induced intramolecular cyclization of acetylene 1a, b resulted in bicyclic compound 2a, b, and subsequent methylation of bicyclic scaffold with MeI led to intermediate 3a, b. Methyl group in the pyrrole moiety was introduced to provide good solubility and to enhance the molecular self-organization.
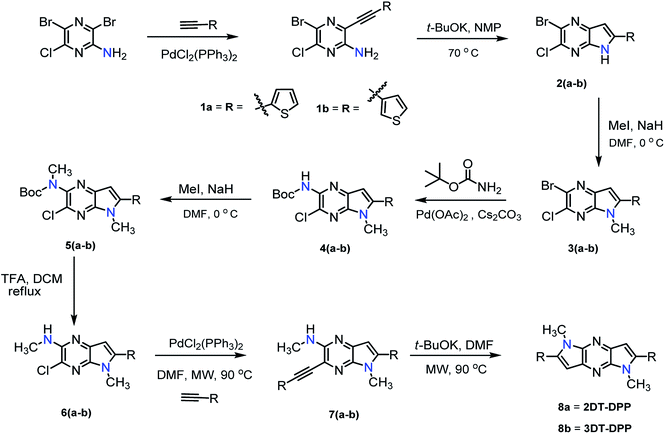 |
| | Scheme 1 Synthetic route of 2DT-DPP and 3DT-DPP isomers. | |
The Pd(OAc)2 catalyzed Buchwald cross-coupling reaction between 3a, b and tert-butyl carbamate afforded compound 4a, b in 70–75% yield. Subsequently amine 4a, b was prepared by the N-methylation, followed by Boc deprotection using TFA to furnish the desired compound 3-chloro-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-2-amine derivative 6a, b. To this end, compounds 6a, b were coupled to appropriate ethynylthiophene under the Sonogashira cross-coupling conditions, employing microwave irradiation (MW) to afford compound 7a, b in good yield. The structural isomers 2DT-DPP and 3DT-DPP were obtained via base induced intramolecular cyclization of 7a, b under MW in good yield.
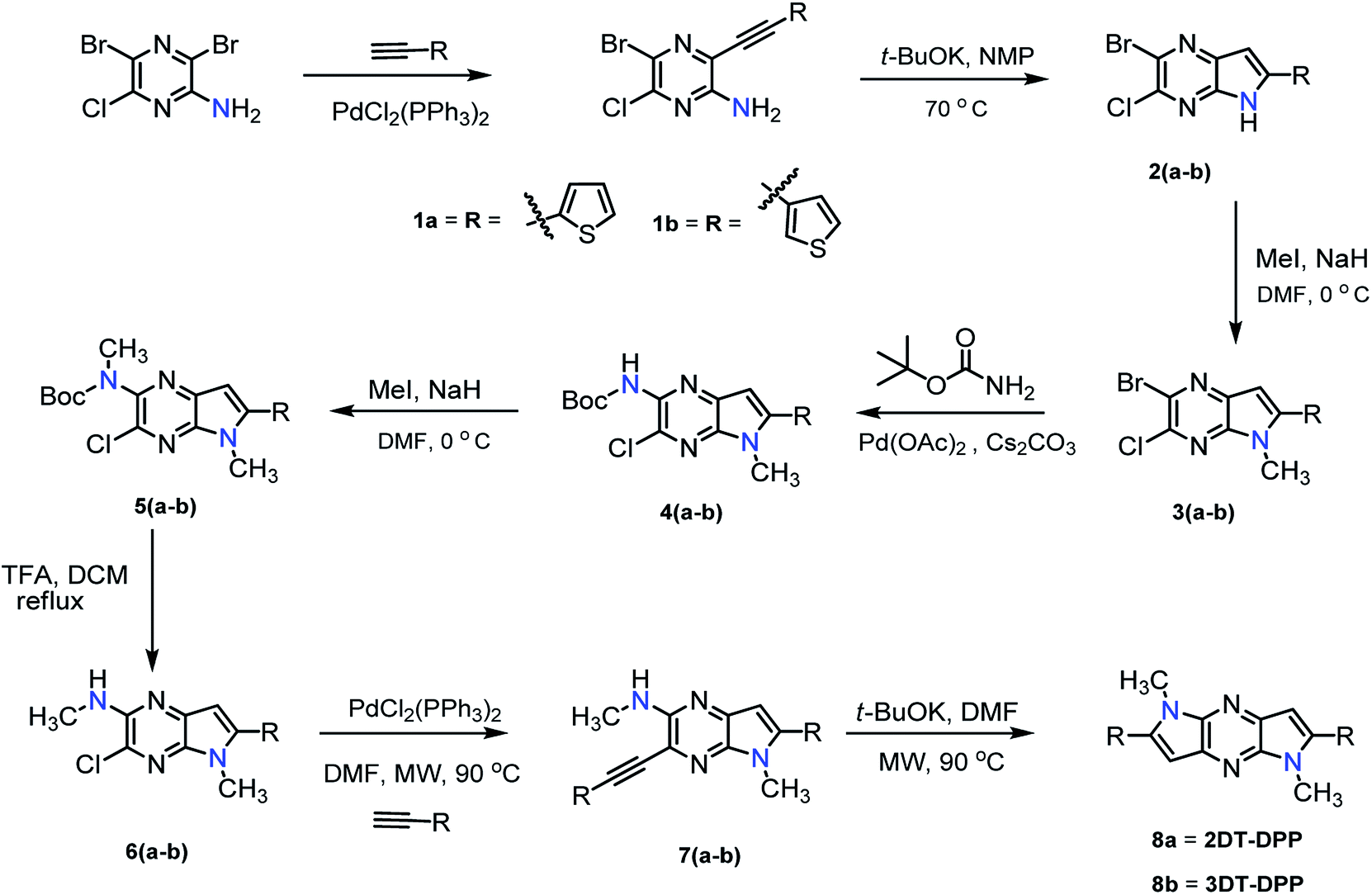
The newly synthesised DT-DPP derivatives were characterized by 1H NMR, 13C NMR, HRMS and X-ray crystallography. The structure of 3DT-DPP was further confirmed by the COSY and 2D-NOE as shown in Fig. 2 (spectra, SI data). As expected, both the isomers can be readily soluble in common organic solvents such as CHCl3, CH2Cl2, MeOH, and THF. The thermogravimetric analysis and differential scanning calorimetry techniques reveals that 2DT-DPP and 3DT-DPP are thermally stable and can be stored at ambient conditions for longer time without any noticeable decomposition.
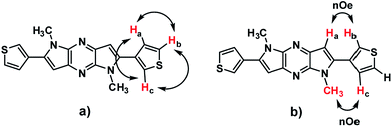 |
| | Fig. 2 (a) COSY and (b) NOE studies of 3DT-DPP. | |
2.2. Crystallographic analysis
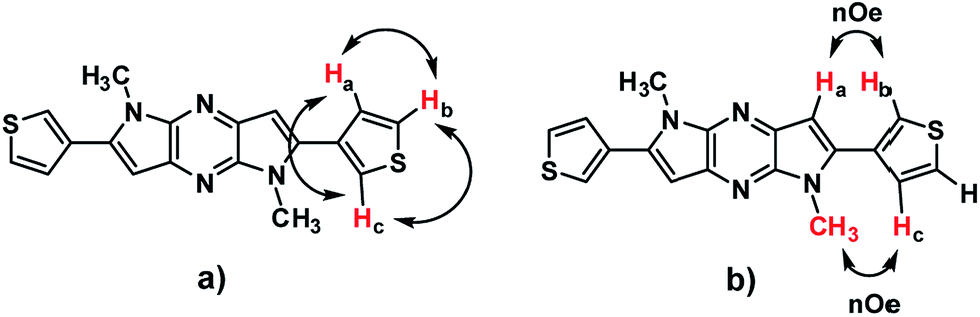
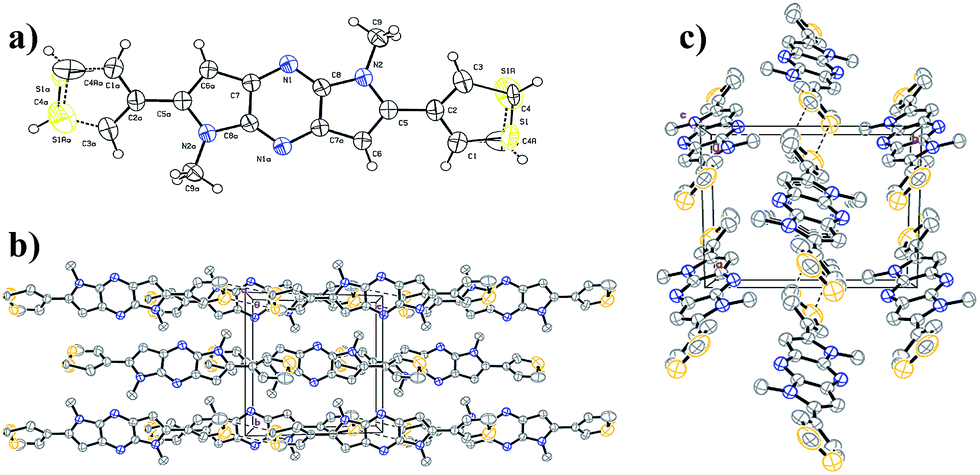
The single crystal X-ray analysis provides useful information about packing in the crystals for the design of organic electronic materials. Single crystal of 3DT-DPP was grown by recrystallization from DCM, and the crystallographic data are provided in the ESI.† We note that the packing mode of 3DT-DPP is similar to DP-DPP. The X-ray crystallographic analysis of 3DT-DPP shows strong π–π stacking, forming face-to-face interactions, and adopts a planar backbone, which is better for charge carrier transport between molecules as shown in Fig. 3. The molecules of 3DT-DPP crystallize in the monoclinic system with space group P2(1)/n and the unit cell dimensions of (a) 9.101(2) Å, (b) 9.9832(2) Å, (c) 9.6509(2) Å, α = 90°, β = 115.463°(10), and γ = 90°. The terminal flanking thiophene moieties were treated as a rotational disorder with S1 and C4 atoms, as similar results reported in literature.36,37 Molecules are assembled to form layers through intermolecular interactions, an intermolecular short S⋯S contact is observed between the columns to give a sheet-like network forming additional interaction between the neighboring 3DT-DPP scaffold. This efficient packing could allow the charges to circulate in 2-dimentional arrangement.
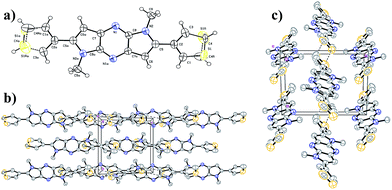 |
| | Fig. 3 (a) ORTEP drawing of single crystal of 3DT-DPP. (b) Crystal packing viewed along a axis. (c) Top view of molecular packing of 3DT-DPP showing S⋯S interactions. Hydrogen atoms and a number of molecules are omitted for clarity. | |
3. Photophysical properties
3.1. Optical properties
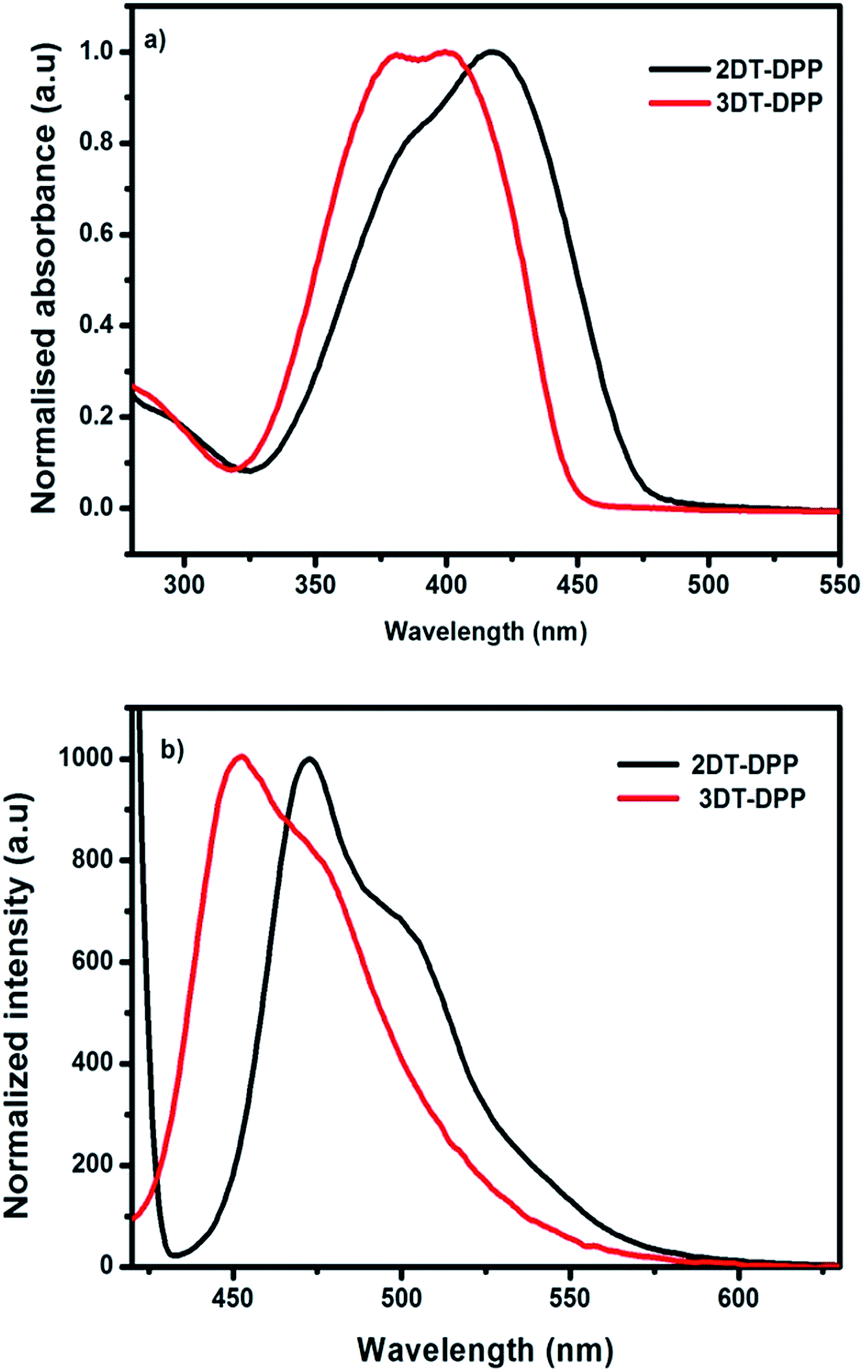
The optical properties of the prepared compounds were obtained in solution-state by UV-vis spectrum using dichloromethane. The photophysical data were summarized in Table 1. The incorporation of an electron-rich unit, such as thiophene, decreases the band gap. The absorption maximum of 2DT-DPP is at 418 nm, whereas 3DT-DPP exhibits two absorption bands at 380 and 400 nm. Absorption spectra of solid film are added in ESI data.† The optical absorption of 3DT-DPP core is slightly hypsochromically shifted with respect to that of 2DT-DPP as observed in Fig. 4a. The higher energy absorbance were attributed to localized π–π* transitions while the lower energy bands were associated with an intramolecular charge transfer (ICT). The DT-DPP isomers exhibit an optical band gap of 2.59 and 2.74 eV, respectively. The optical band gap values were estimated from the onset of the absorption spectra. Both DT-DPP isomers display a broad absorption over much of the visible region, suggesting the possible application as an active layer in organic photovoltaic cells. The emission properties were also characterized in dichloromethane. The fluorescence maxima of the compounds followed the trend of UV-vis absorption spectroscopy (Fig. 4b). The λem values for 2DT-DPP and 3DT-DPP were found to be 473 and 454 nm, respectively.
Table 1 Optical and electrochemical properties of DT-DPP isomers
| Molecules |
λabsa [nm] |
λemb [nm] |
Eoptgc [eV] |
Eoxd [eV] |
HOMOe [eV] |
LUMOf [eV] |
| Absorption spectra. Emission spectra. Optical band gap calculated from the UV-vis absorption onset. Onset voltage of the first oxidation potential. Determined from onset voltage of the first oxidation potential with reference to ferrocene at −4.8 eV. Calculated from Eg and HOMO values. |
| 2DT-DPP |
418 |
473 |
2.59 |
0.74 |
−5.54 |
−2.95 |
| 3DT-DPP |
380![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
454 |
2.74 |
0.77 |
−5.57 |
−2.83 |
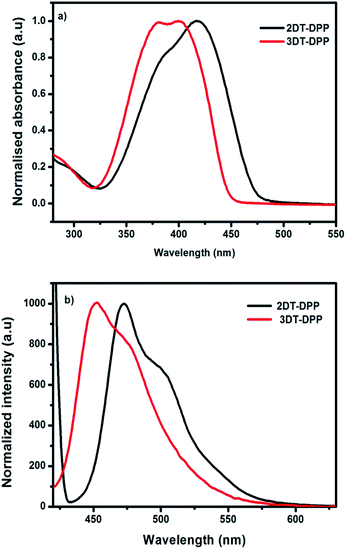 |
| | Fig. 4 (a) Normalised UV-vis absorption spectra of 2DT-DPP and 3DT-DPP in dichloromethane. (b) Emission spectra of compounds in dichloromethane when excited at 380 nm. | |
3.2. Electrochemical properties
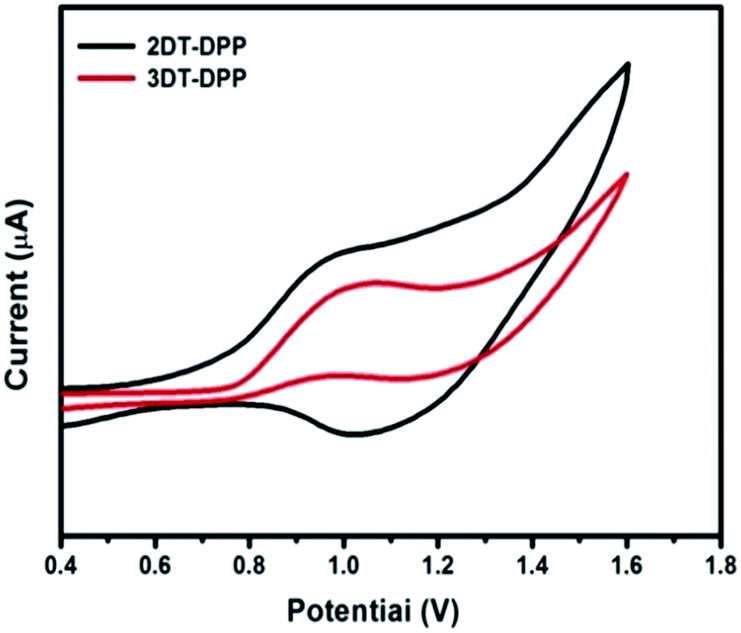
Cyclic voltammetry (CV) was used to investigate the electrochemical behaviour of the compounds and estimate their HOMO and LUMO energy levels. CV was carried out in three-electrode cell with a Pt, Pt wire and Ag/AgCl as the working electrode, auxiliary electrode and reference electrode, respectively. 0.1 M nBu4NPF6 prepared in anhydrous DCM was used as an electrolyte, at the scan rate of 50 mV s−1. Ferrocene was used as the internal standard. The onset oxidation potentials of 2DT-DPP and 3DT-DPP were found to be at 0.74 and 0.77 eV respectively. The HOMO energy levels, estimated from the oxidation onset, vary from −5.54 to −5.57 eV. The LUMO levels were estimated from the HOMO value and the optical bandgap (Fig. 5).
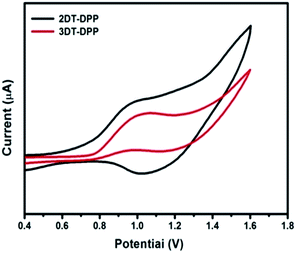 |
| | Fig. 5 Cyclic voltammograms of DT-DPP recorded in dichloromethane, nBu4NPF6 as a supporting electrolyte at a scan rate of 50 mV s−1. | |
3.3. Thermal properties
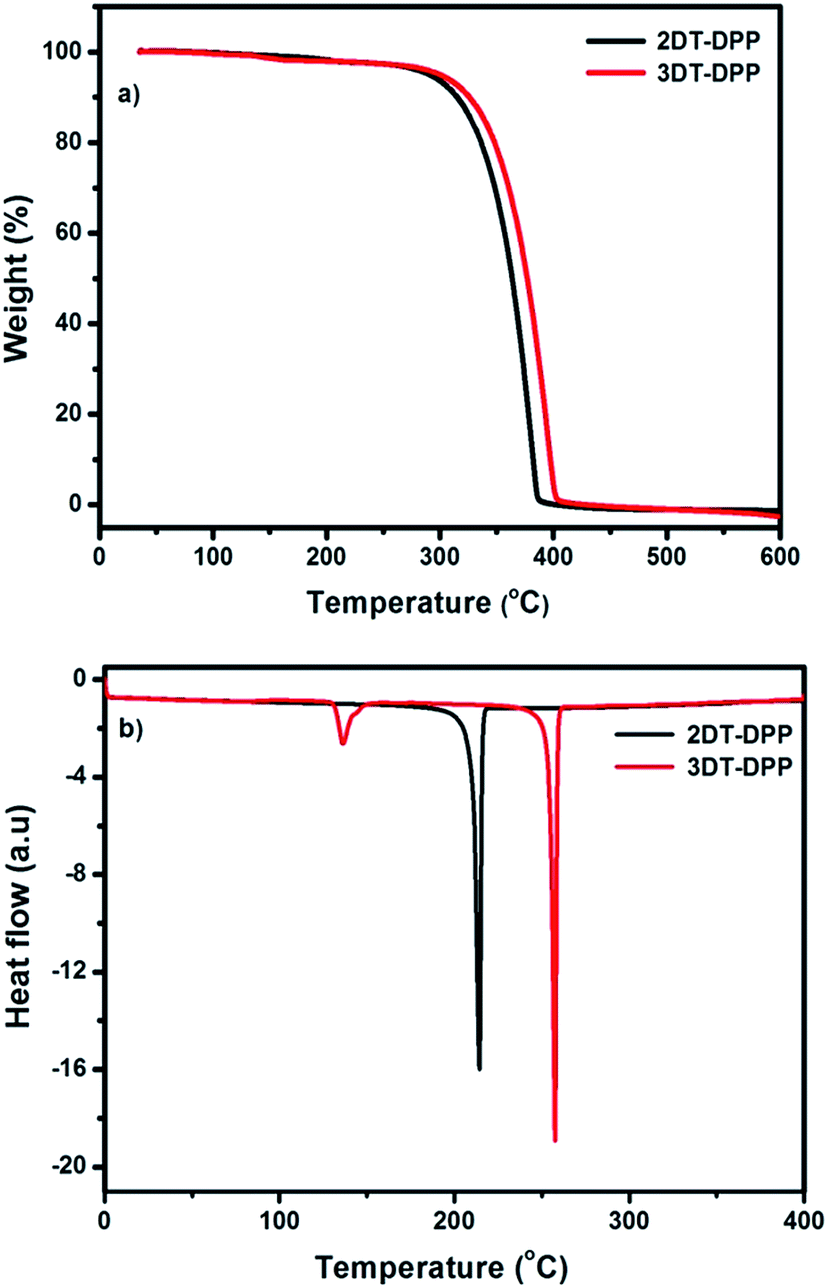
The thermal properties of DT-DPP isomers were determined by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere at a heating rate of 10 °C min−1 as shown in Fig. 6. The phase transition properties of these compounds were analyzed by DSC. Both the compounds 2DT-DPP and 3DT-DPP exhibited a sharp endothermic melting peak at 215 and 257 °C, respectively. This indicates that both isomers were highly crystalline in nature. The TGA results suggested that both compounds exhibited good thermal stability owing to their rigid structure and highly extended conjugation, showing a weight loss of less than 5% in the temperature range 302–308 °C, and indicating sufficient thermal stability for optoelectronic applications. The differences in the thermal degradation of compounds might be because of the structural variation. The physical properties of the isomers are summarized in Table 2.
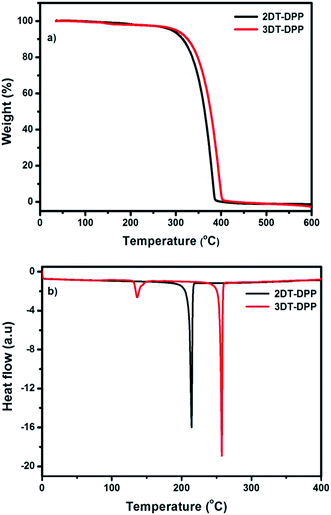 |
| | Fig. 6 (a) TGA and (b) DSC plots of 2DT-DPP and 3DT-DPP obtained at a heating rate of 10 °C min−1 under nitrogen atmosphere. | |
Table 2 Thermal properties of DT-DPP isomers
| Compound |
TGAa [°C] |
DSCb [°C] |
Tgb [°C] |
| Degradation temperature (Td) observed from the TGA corresponding to 5% weight loss at 10 °C min−1 under nitrogen. Melting temperature (Tm) and glass transition temperature (Tg) observed from DSC at a heating rate of 10 °C min−1 under nitrogen. |
| 2DT-DPP |
302 |
215 |
— |
| 3DT-DPP |
308 |
257 |
142 |
4. Thin film morphology and XRD studies
The film morphology is an important parameter for device performance. The morphological studies of the DT-DPP derivatives were carried out by X-ray diffraction (XRD) and scanning electron microscopy (SEM). Thin films of isomers were prepared on a silicon wafer by drop-casting from their hot solutions of dichloromethane and THF, then annealed at 50 °C for 2 h in vacuum before recording their XRD patterns.
4.1. Thin film XRD
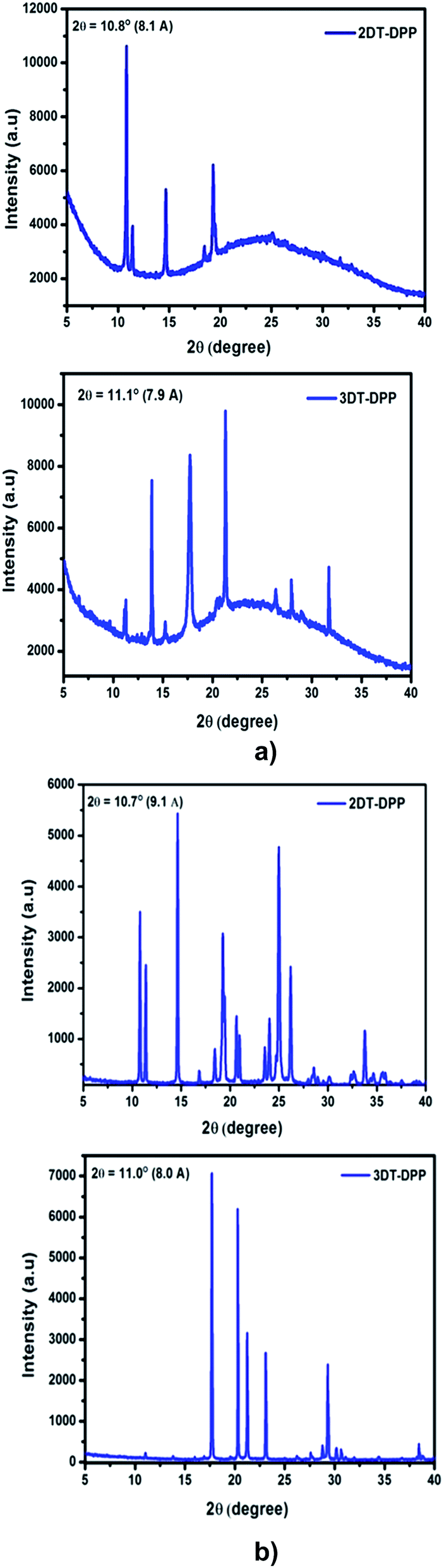
The thin film XRD patterns of 2DT-DPP and 3DT-DPP displayed sharp and intense primary diffraction peaks at 2θ = 10.8° and 11.1°, and the corresponding d-spacing values are 8.1 Å and 7.9 Å, respectively. This indicates both the structural isomers easily form well-ordered and layered molecular structure in thin films.38 The XRD analysis revealed a large number of well defined diffraction peaks, indicating crystalline nature(Fig. 7a).
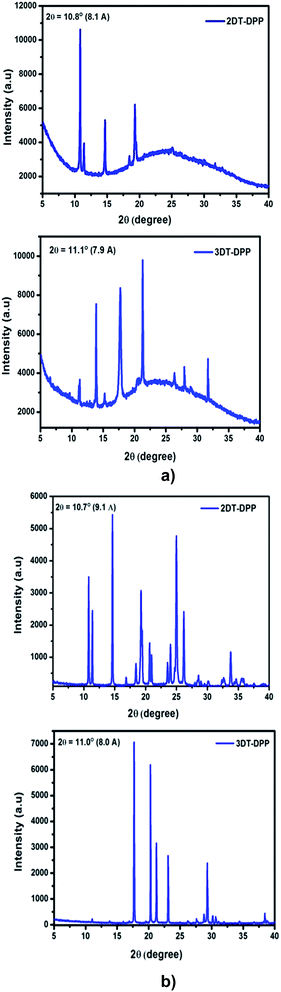 |
| | Fig. 7 (a) X-ray diffraction pattern of DT-DPP thin film vacuum deposited at low substrate temperature on bare silica substrate. (b) Powder X-ray diffractograms of DT-DPP structural isomers obtained from DCM. | |
4.2. Powder XRD
The powder XRD patterns of 2DT-DPP and 3DT-DPP isomers reveal highly ordered structures in both the cases as shown in Fig. 7b and exhibits similar diffraction peaks and d-spacing values as seen in the corresponding thin film XRD spectra along with some additional diffraction peaks. The intense diffraction peak in both isomer is likely attributed to the π–π stacking order.
4.3. Surface morphology
The surface morphological studies of DT-DPP structural isomers were analysed by SEM. Thin films were prepared from drop casting DCM and THF solution of 2DT-DPP and 3DT-DPP depositing on bare silica substrate followed by drying in the air. Self-assembled structures of DT-DPP isomers were fabricated using different solvents. The SEM measurements confirmed the significant morphological difference depending on the solvents used,22,39 with DCM as a solvent, formation of microrods with 2.6 μm in length were observed, and in THF segregated to form flower like microstructures (Fig. 8A). The SEM image of 3DT-DPP also reveals interesting surface morphology. 3DT-DPP forms microrods with 1–4 μm in length in DCM, whereas in THF, it aggregates to form coral like microstructure (Fig. 8B). The optical images of these structural isomers are provided in the ESI (Fig. 1 and 2†).
 |
| | Fig. 8 (A) SEM images of 2DT-DPP thin film deposited on silica substrate using hot DCM solution. (a) Aggregated needle shape microstructures. (b) Microrods and (c) SEM images of self assembled flower-like supramolecular architectures of 2DT-DPP precipitated from its hot solutions in THF. (B) SEM images of 3DT-DPP thin film deposited on silica substrate using hot THF and DCM solution (a) aggregated microrods (b) flower like microstructure. | |
5. Experimental section
5.1. General methods
All the chemicals and reagents were purchased from Sigma Aldrich and used without further purification. Air sensitive reactions were carried out under a nitrogen atmosphere. 1H NMR and 13C NMR spectra were recorded in δ (ppm) units relative to a deuterated solvent using a Bruker 500 MHz NMR instrument. Liquid chromatography/mass spectrometry (6400 series Triple Quadrupole LC/MS/Agilent) analysis was performed using an ESI mass spectrometer. High-resolution mass spectrometry was performed using a 6550 iFunnel Q-TOF LC/MS system (Agilent). UV-vis absorption spectra were acquired using a Varian cary-50 Spectrophotometer. CV was carried out using ZIVE-SP2 LAB with three electrode configuration. TGA and DSC were performed with TGA 3 plus and DSC 2 STAR system (mettler toledo) respectively, under nitrogen at a heating rate of 10 °C min−1. XRD was analysed with Smartlab (Rigaku) using Cu Kα radiation (λ = 1.5406) and single crystal XRD was analysed using a Bruker D8 Discover X-ray Diffractometer. SEM analysis was carried out using an FEI Quanta 3D FEG.
5.2. 5-Bromo-6-chloro-3-(thiophenylethynyl)pyrazin-2-amine derivatives (1)
To a stirred solution of 3,5-dibromo-6-chloro-pyrazin-2-amine (1.5 g, 5.2 mmol) in anhydrous THF (30 mL) TEA (1.6 g, 15 mmol), Pd(PPh3)2Cl2 (0.37 g, 0.52 mmol), CuI (0.1 g, 0.52 mmol) then 2-ethynylthiophene (0.57 mL, 5.2 mmol) were added under nitrogen atmosphere and stirred for 3 h. The reaction mixture was diluted with water and extracted with EA. The combined organic extract was concentrated in vacuum and then purified by column chromatography (5% EA: hexane) to give the desired compound.
5.2.1. 5-Bromo-6-chloro-3-(thiophen-2-ylethynyl)pyrazin-2-amine (1a). Pale yellow crystals, yield: 84%, mp: 180–182 °C. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 5.1 Hz, 1H), 7.42–7.37 (m, 1H), 7.06 (dd, J = 5.0, 3.8 Hz, 1H), 5.24 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 153.0, 145.9, 134.2, 129.6, 127.5, 123.9, 122.1, 120.7, 91.4, 86.1. LC-MS (ESI): m/z = 316.00 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C10H5BrClN3S, 313.9154; found 313.9167.
5.2.2. 5-Bromo-6-chloro-3-(thiophen-3-ylethynyl)pyrazin-2-amine (1b). Pale yellow solid, yield: 89%, mp: 162–163.5 °C. 1H NMR (500 MHz, CDCl3) δ 7.67 (s, 1H), 7.35 (s, 1H), 7.24 (t, J = 11.9 Hz, 1H), 5.30 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 153.2, 145.7, 131.2, 129.6, 126.1, 123.7, 122.4, 120.0, 93.2, 82.1. LC-MS (ESI): m/z = 316.00 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C10H5BrClN3S, 313.9154; found 313.9157.
5.3. 2-Bromo-3-chloro-6-(thiophenyl)-5H-pyrrolo[2,3-b]pyrazine derivatives (2)
5-bromo-6-chloro-3-(thiophen-2-ylethynyl)pyrazin-2-amine (1.0 g, 3.1 mmol) was dissolved in anhydrous NMP (15 mL) and a solution of t-BuOK (0.78 g, 6.2 mmol) in NMP (10 mL) was added dropwise under nitrogen atmosphere. The reaction mixture was refluxed at 80 °C for 1 h and then cooled to ambient temperature. The suspension was diluted with EA filtered through a celite bed and diluted with water. The organic layer was washed with excess water dried over anhydrous MgSO4, filtered and concentrate to afford yellow solid. The crude material thus obtained is used directly without further purification.
5.3.1. 2-Bromo-3-chloro-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazine (2a). Yellow solid, yield: 90%, mp: >240 °C. 1H NMR (500 MHz, DMSO) δ 13.04 (s, 1H), 7.80 (s, 1H), 7.79 (s, 1H), 7.25 (s, 1H), 6.94 (s, 1H). 13C NMR (126 MHz, DMSO) δ 140.7, 140.5, 139.6, 138.9, 133.6, 130.4, 129.5, 129.1, 127.5, 97.0. LC-MS (ESI): m/z = 316.00 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C10H5BrClN3S, 313.9154; found 313.9170.
5.3.2. 2-Bromo-3-chloro-6-(thiophen-3-yl)-5H-pyrrolo[2,3-b]pyrazine (2b). Yellow solid, yield: 91%, mp: >240 °C. 1H NMR (500 MHz, DMSO) δ 12.90 (s, 1H), 8.33–8.16 (m, 1H), 7.77 (d, J = 3.8 Hz, 1H), 7.76 (d, J = 5.1 Hz, 1H), 7.07 (d, J = 1.8 Hz, 1H). 13C NMR (126 MHz, DMSO) δ 140.9, 139.5, 138.7, 137.5, 131.6, 129.1, 127.4, 125.4, 123.6, 96.2. LC-MS (ESI): m/z = 316.00 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C10H5BrClN3S, 313.9154; found 313.9156.
5.4. 2-Bromo-3-chloro-5-methyl-6-(thiophenyl)-5H-pyrrolo[2,3-b]pyrazine derivatives (3)
2-bromo-3-chloro-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazine (0.9 g, 2.8 mmol) was dissolved in DMF (25 mL), followed by adding NaH (60% dispersion in mineral oil, 0.15 g, 3.4 mmol) under nitrogen at 0 °C, and the resulting reaction mixture was stirred for 30 min. MeI (0.9 mL, 13.6 mmol) was then added and stirring continued for 2 h. The reaction mixture was quenched in ice-cold water and the precipitate was collected by vacuum filtration. The crude solid was dissolved in EA and purified by column chromatography (20% EA: hexane).
5.4.1. 2-Bromo-3-chloro-5-methyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazine (3a). Pale yellow solid, yield: 88%, mp: 174.8–176 °C. 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 4.7 Hz, 1H), 7.41 (s, 1H), 7.21 (s, 1H), 6.77 (s, 1H), 3.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 140.7, 140.2, 140.2, 137.8, 131.7, 131.5, 128.4, 128.3, 128.2, 100.1, 30.3. LC-MS (ESI): m/z = 330.00 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C11H7BrClN3S, 327.9311; found 327.9323.
5.4.2. 2-Bromo-3-chloro-5-methyl-6-(thiophen-3-yl)-5H-pyrrolo[2,3-b]pyrazine (3b). Pale yellow crystals, yield: 92%, mp: 202–204 °C. 1H NMR (500 MHz, CDCl3) δ 7.60 (dd, J = 2.9, 1.2 Hz, 1H), 7.52 (dd, J = 5.0, 2.9 Hz, 1H), 7.36 (dd, J = 5.0, 1.2 Hz, 1H), 6.71 (s, 1H), 3.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 142.7, 140.1, 139.9, 138.0, 131.2, 131.2, 127.6, 127.0, 125.4, 99.6, 30.3. LC-MS (ESI): m/z = 330.00 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C11H7BrClN3S, 327.9311; found 327.9312.
5.5. tert-Butyl-3-chloro-5-methyl-6-(thiophenyl)-5H-pyrrolo[2,3-b]pyrazin-2-yl carbamate derivative (4)
An air-dried glass reaction vessel equipped with a magnetic stir bar was charged with, in order: 1,4-dioxane (15 mL), t-amyl alcohol (3 mL),2-bromo-3-chloro-5-methyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazine (0.84 g, 2.5 mmol), Pd(OAc)2 (0.057 g, 0.25 mmol), xantphos (0.25 g, 0.43 mmol), Cs2CO3 (1.66 g, 5 mmol) and tert-butyl carbamate (0.3 g, 2.5 mmol). The suspension was refluxed at 90 °C for 3 h. Once the reaction was complete by TLC, it was diluted with EA and filtered through a bed of celite. The filtrate was concentrated in vacuum. The residue was purified by silica chromatography (25% EA: hexane) to afford the desired product as a crystalline solid.
5.5.1. tert-Butyl-3-chloro-5-methyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (4a). Pale yellow solid, yield: 72%, mp: 136.2–137 °C. 1H NMR (500 MHz, CDCl3) δ 7.51–7.47 (m, 1H), 7.38–7.34 (m, 1H), 7.19 (d, J = 3.2 Hz, 1H), 7.18 (d, J = 1.2 Hz, 1H), 6.83 (s, 1H), 3.94 (s, 3H), 1.56 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 151.4, 139.3, 139.2, 138.4, 135.8, 132.4, 131.7, 128.0, 127.8, 127.7, 100.7, 81.4, 30.1, 28.2. LC-MS (ESI): m/z = 365.20 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H17ClN4O2S, 365.0839; found 365.0856.
5.5.2. tert-Butyl-3-chloro-5-methyl-6-(thiophen-3-yl)-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (4b). Pale yellow solid, yield: 73%, mp: 155.7–157 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 (dd, J = 2.9, 1.2 Hz, 1H), 7.49 (dd, J = 5.0, 2.9 Hz, 1H), 7.34 (dd, J = 5.0, 1.1 Hz, 1H), 7.17 (s, 1H), 6.76 (s, 1H), 3.89 (s, 3H), 1.56 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 151.4, 141.4, 139.0, 138.3, 136.0, 131.8, 131.5, 127.8, 126.7, 124.7, 100.1, 81.4, 30.1, 28.2. LC-MS (ESI): m/z = 365.10 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H17ClN4O2S, 365.0839; found 365.0843.
5.6. tert-Butyl-3-chloro-5-methyl-6-(thiophenyl)-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl)carbamate (5)
To a solution of tert-butyl 3-chloro-5-methyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (2.1 g, 5.7 mmol) in DMF (30 mL) NaH (60% dispersion in mineral oil, 0.28 g, 6.9 mmol) is added under nitrogen atmosphere and stirred at 0 °C. After 30 min, MeI (0.35 mL, 5.7 mmol) was added and stirring continued for 2 h. After the completion of the reaction by TLC, the reaction mixture was poured in ice-cold water and the precipitate was collected by vacuum filtration. The crude solid was dissolved in EA and purified by column chromatography (25% EA: hexane).
5.6.1. tert-Butyl-3-chloro-5-methyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl)carbamate (5a). Light brown solid, yield: 85.5%, mp: 196–197.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.51–7.47 (m, 1H), 7.38–7.34 (m, 1H), 7.19 (d, J = 3.2 Hz, 1H), 7.18 (d, J = 1.2 Hz, 1H), 6.83 (s, 1H), 3.94 (s, 3H), 1.56 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.1, 143.7, 140.1, 140.0, 139.3, 136.4, 132.1, 128.1, 128.1, 128.0, 100.6, 80.9, 35.1, 30.2, 28.1. LC-MS (ESI): m/z = 323.10 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H19ClN4O2S, 379.0990; found 379.1012.
5.6.2. tert-Butyl-3-chloro-5-methyl-6-(thiophen-3-yl)-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl)carbamate (5b). Off white solid, yield: 88%, mp: 183–185.2 °C. 1H NMR (500 MHz, CDCl3) δ 7.59 (s, 1H), 7.52 (dd, J = 4.8, 3.0 Hz, 1H), 7.37 (d, J = 4.9 Hz, 1H), 6.73 (s, 1H), 3.94 (s, 3H), 3.29 (s, 3H), 1.38 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.1, 143.5, 142.1, 139.9, 139.0, 136.5, 131.5, 127.7, 126.9, 125.0, 100.0, 80.9, 35.1, 30.2, 28.1. LC-MS (ESI): m/z = 323.10 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H19ClN4O2S, 379.0990; found 379.0995.
5.7. 3-Chloro-N,5-dimethyl-6-(thiophenyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine derivative (6)
To a solution of tert-butyl 3-chloro-5-methyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl)carbamate (1.0 g, 2.6 mmol) in dichloromethane (30 mL) was added TFA (1.5 mL, 13 mmol) and the resulting solution was stirred at room temperature for 10 min and then refluxed for 3 h. The reaction mixture was basified with NaOH and extracted with DCM. The organic extract was dried over anhydrous MgSO4 and, concentrated in vacuum to dryness to get the desired product.
5.7.1. 3-Chloro-N,5-dimethyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (6a). Light brown solid, yield: 92%. mp: 127.8–130 °C. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 5.1 Hz, 1H), 7.29 (d, J = 3.5 Hz, 1H), 7.18–7.12 (m, 1H), 6.62 (s, 1H), 4.98 (s, 1H), 3.89 (s, 3H), 3.10 (d, J = 1.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 148.1, 136.2, 135.2, 135.2, 133.4, 128.3, 127.9, 126.8, 126.8, 99.2, 30.0, 28.9. LC-MS (ESI): m/z = 279.10 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C12H11ClN4S, 279.0471; found 279.0503.
5.7.2. 3-Chloro-N,5-dimethyl-6-(thiophen-3-yl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (6b). Brown solid, yield: 93.7%, mp: 157–158 °C. 1H NMR (500 MHz, CDCl3) δ 7.48 (dd, J = 2.9, 1.3 Hz, 1H), 7.45 (d, J = 5.0 Hz, 1H), 7.33 (dd, J = 5.0, 1.2 Hz, 1H), 6.55 (s, 1H), 4.96 (s, 1H), 3.85 (s, 3H), 3.10 (d, J = 3.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 147.9, 138.4, 135.3, 134.9, 132.5, 127.9, 127.8, 126.3, 123.6, 98.5, 30.0, 28.9. LC-MS (ESI): m/z = 279.20 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C12H11ClN4S, 279.0471; found 279.0473.
5.8. General procedure for Sonogashira cross-coupling reaction (7)
3-chloro-N,5-dimethyl-6-(thiophen-2-yl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (1.0 g, 3.5 mmol) was dissolved in DMF (10 mL). To that solution, TEA (1.08 g, 10 mmol), CuI (0.068 g, 0.35 mmol), Pd(PPh3)2Cl2 (0.25 g, 0.35 mmol) and 2-ethynylthiophene (0.38 mL, 3.5 mmol) were added, and heated under microwave for 1 h at 90 °C. After cooling the reaction mixture was diluted with water and extracted with EA. The combined organic extract was concentrated in vacuum and the crude residue thus obtained was purified by column chromatography (20% EA: hexane) affording the desired compound.
5.8.1. N,5-Dimethyl-6-(thiophen2-yl)-3-(thiophen-2-ylethynyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (7a). Light brown solid, yield: 78%, mp: 143–145 °C. 1H NMR (500 MHz, CDCl3) δ 7.44 (dd, J = 5.1, 0.7 Hz, 1H), 7.40–7.38 (m, 1H), 7.36 (dd, J = 5.1, 0.8 Hz, 1H), 7.34 (dd, J = 3.6, 0.7 Hz, 1H), 7.16 (dd, J = 5.0, 3.7 Hz, 1H), 7.04 (dd, J = 5.1, 3.7 Hz, 1H), 6.63 (s, 1H), 5.21 (d, J = 4.8 Hz, 1H), 3.93 (s, 3H), 3.12 (d, J = 5.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.2, 138.5, 137.0, 136.5, 133.4, 132.9, 128.3, 127.9, 127.2, 127.1, 127.0, 122.2, 117.6, 99.4, 89.6, 89.1, 29.9, 28.7. LC-MS (ESI): m/z = 351.20 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H14N4S2, 351.0738; found 351.0751.
5.8.2. N,5-dimethyl-6-(thiophen-3-yl)-3-(thiophen-3-ylethynyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (7b). Light brown solid, yield: 83%, mp: 215–217 °C. 1H NMR (500 MHz, CDCl3) δ 7.64 (dd, J = 2.9, 1.0 Hz, 1H), 7.52 (dd, J = 2.9, 1.2 Hz, 1H), 7.45 (dd, J = 5.0, 2.9 Hz, 1H), 7.35 (dd, J = 5.0, 1.2 Hz, 1H), 7.33 (dd, J = 5.0, 3.0 Hz, 1H), 7.27 (dd, J = 5.0, 1.0 Hz, 1H), 6.56 (s, 1H), 5.24 (d, J = 4.8 Hz, 1H), 3.89 (s, 3H), 3.13 (d, J = 5.0 Hz, 3H).·13C NMR (126 MHz, CDCl3) δ 153.2, 140.4, 136.7, 136.5, 132.5, 129.9, 129.7, 127.8, 126.3, 125.6, 123.8, 121.3, 117.7, 98.9, 90.9, 85.6, 29.9, 28.7. LC-MS (ESI): m/z = 351.10 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H14N4S2, 351.0738; found 351.0740.
5.9. General procedure for intramolecular cyclization (8)
To a solution of N,5-dimethyl-6-(thiophen-2-yl)-3-(thiophen-2-ylethynyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (1.0 g, 2.8 mmol) in DMF (10 mL) t-BuOK (0.64 g, 5.7 mmol) was added. The resulting solution was refluxed at 120 °C for 60 min under microwave. The reaction mixture was cooled to ambient temperature and then diluted with DCM and water, organic extract was washed with water and dried over anhydrous MgSO4, filtered and concentrated in vacuum. The crude residue was purified by column chromatography (25% EA: hexane) to give the desired compound.
5.9.1. 1,5-Dimethyl-2,6-di(thiophen-2-yl)-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (8a). Brown solid, yield: 75%, mp: 210–211 °C. 1H NMR (500 MHz, CDCl3) δ 7.46 (dd, J = 5.1, 0.7 Hz, 1H), 7.39–7.37 (m, 1H), 7.21–7.17 (m, 1H), 6.85 (s, 1H), 4.06 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 141.9, 137.4, 134.7, 133.8, 127.9, 126.9, 126.9, 99.5, 29.9. LC-MS (ESI): m/z = 351.10 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H14N4S2, 351.0738; found 351.0745.
5.9.2. 1,5-Dimethyl-2,6-di(thiophen-3-yl)-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (8b). Brown solid, yield: 79%, mp: 248–249 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 (s, 1H), 7.49–7.44 (m, 1H), 7.40 (d, J = 4.8 Hz, 1H), 6.77 (s, 1H), 4.01 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 141.6, 139.3, 134.6, 132.9, 128.0, 126.2, 123.6, 98.8, 30.0. LC-MS (ESI): m/z = 351.20 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H14N4S2, 351.0738; found 351.0746.
6. Conclusions
Two structural isomers of thiophene-flanked dipyrrolopyrazine with electron deficient building unit forming D–A–D architecture were designed and synthesized. The compounds in this work would be of interest as π-functional materials owing to their appealing optical, thermal, and electrochemical properties. UV-vis absorption maximum and the absorption edge red-shifted in the case of 2DT-DPP. The single crystal analysis of 3DT-DPP framework showed that the molecule adopts a perfectly planar geometry. The addition of thiophene substituent as the end group creates shorter contacts between the adjacent molecules enabling 2-D networks. We envision that this synthetic concept can also be applied to prepare a variety of coplanar heteroarene substituted with solubilizing side chains. By investigating the properties of DT-DPP isomers, it can be concluded that the substituents on the pyrrole ring proved to modulate the electronic properties. Both isomers form microscopic self-assembled clusters, and solvents affected the morphology of the clusters. These findings may serve as a sensible molecular design guideline for future electronic property control of self-assembling π-conjugated materials, leading to the development of new electroactive and photoactive materials.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2016R1D1A1B04932654) in Korea.
References
- Z. Liang, Q. Tang, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 1535–1539 CrossRef CAS PubMed.
- P. de Echegaray, M. J. Mancheño, I. Arrechea-Marcos, R. Juárez, G. López-Espejo, J. T. López Navarrete, M. M. Ramos, C. Seoane, R. P. Ortiz and J. L. Segura, J. Org. Chem., 2016, 81, 11256–11267 CrossRef CAS PubMed.
- Y.-I. Park, J.-H. Son, J.-S. Kang, S.-K. Kim, J.-H. Lee and J.-W. Park, Chem. Commun., 2008, 2143–2145, 10.1039/B718873K.
- D. Chen, S.-J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565–9578 RSC.
- C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 5450–5454 CrossRef CAS PubMed.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6281–6291 CrossRef CAS PubMed.
- M. Tasior and D. T. Gryko, J. Org. Chem., 2016, 81, 6580–6586 CrossRef CAS PubMed.
- C. F. Gers-Panther, H. Fischer, J. Nordmann, T. Seiler, T. Behnke, C. Würth, W. Frank, U. Resch-Genger and T. J. J. Müller, J. Org. Chem., 2017, 82, 567–578 CrossRef CAS PubMed.
- X. Lu, G. Zhou, H. Wang, Q. Feng and Z.-S. Wang, Phys. Chem. Chem. Phys., 2012, 14, 4802–4809 RSC.
- P. Henriksson, C. Lindqvist, B. Abdisa, E. Wang, Z. George, R. Kroon, C. Müller, T. Yohannes, O. Inganäs and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2014, 130, 138–143 CrossRef CAS.
- Q. Wang, J. J. van Franeker, B. J. Bruijnaers, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2016, 4, 10532–10541 CAS.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- G. Kim, S.-J. Kang, G. K. Dutta, Y.-K. Han, T. J. Shin, Y.-Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- M.-C. Chen, C. Kim, S.-Y. Chen, Y.-J. Chiang, M.-C. Chung, A. Facchetti and T. J. Marks, J. Mater. Chem., 2008, 18, 1029–1036 RSC.
- I. Osaka, T. Abe, S. Shinamura and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 6852–6860 CrossRef CAS PubMed.
- S. Subramanian, S. K. Park, S. R. Parkin, V. Podzorov, T. N. Jackson and J. E. Anthony, J. Am. Chem. Soc., 2008, 130, 2706–2707 CrossRef CAS PubMed.
- B. Kan, H. Feng, X. Wan, F. Liu, X. Ke, Y. Wang, Y. Wang, H. Zhang, C. Li, J. Hou and Y. Chen, J. Am. Chem. Soc., 2017 DOI:10.1021/jacs.7b01170.
- Y. Geng, J. Wang, S. Wu, H. Li, F. Yu, G. Yang, H. Gao and Z. Su, J. Mater. Chem., 2011, 21, 134–143 RSC.
- P.-L. T. Boudreault, S. Wakim, N. Blouin, M. Simard, C. Tessier, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2007, 129, 9125–9136 CrossRef CAS PubMed.
- S.-i. Kato, K. Watanabe, M. Tamura, M. Ueno, M. Nitani, Y. Ie, Y. Aso, T. Yamanobe, H. Uehara and Y. Nakamura, J. Org. Chem., 2017 DOI:10.1021/acs.joc.7b00084.
- Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2016, 128, 3413–3416 CrossRef.
- U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- J. I. Wu, C. S. Wannere, Y. Mo, P. v. R. Schleyer and U. H. F. Bunz, J. Org. Chem., 2009, 74, 4343–4349 CrossRef CAS PubMed.
- Y.-Y. Liu, C.-L. Song, W.-J. Zeng, K.-G. Zhou, Z.-F. Shi, C.-B. Ma, F. Yang, H.-L. Zhang and X. Gong, J. Am. Chem. Soc., 2010, 132, 16349–16351 CrossRef CAS PubMed.
- K. Liu, C.-L. Song, Y.-C. Zhou, X.-Y. Zhou, X.-J. Pan, L.-Y. Cao, C. Zhang, Y. Liu, X. Gong and H.-L. Zhang, J. Mater. Chem. C, 2015, 3, 4188–4196 RSC.
- Z. Liang, Q. Tang, R. Mao, D. Liu, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef CAS PubMed.
- Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed.
- Z. Cai, Y. Guo, S. Yang, Q. Peng, H. Luo, Z. Liu, G. Zhang, Y. Liu and D. Zhang, Chem. Mater., 2013, 25, 471–478 CrossRef CAS.
- Y. Ren, A. M. Hiszpanski, L. Whittaker-Brooks and Y.-L. Loo, ACS Appl. Mater. Interfaces, 2014, 6, 14533–14542 CAS.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- P. Meti, E.-S. Lee, J.-W. Yang and Y.-D. Gong, RSC Adv., 2017, 7, 18120–18131 RSC.
- H. Meng, F. Sun, M. B. Goldfinger, G. D. Jaycox, Z. Li, W. J. Marshall and G. S. Blackman, J. Am. Chem. Soc., 2005, 127, 2406–2407 CrossRef CAS PubMed.
- C. R. Hopkins and N. Collar, Tetrahedron Lett., 2004, 45, 8087–8090 CrossRef CAS.
- J. Chisaka, M. Lu, S. Nagamatsu, M. Chikamatsu, Y. Yoshida, M. Goto, R. Azumi, M. Yamashita and K. Yase, Chem. Mater., 2007, 19, 2694–2701 CrossRef CAS.
- M. Ashizawa, T. Hasegawa, S. Kawauchi, H. Masunaga, T. Hikima, H. Sato and H. Matsumoto, RSC Adv., 2016, 6, 109434–109441 RSC.
- J. Xu, L. Wen, W. Zhou, J. Lv, Y. Guo, M. Zhu, H. Liu, Y. Li and L. Jiang, J. Phys. Chem. C, 2009, 113, 5924–5932 CAS.
- A. Datar, K. Balakrishnan, X. Yang, X. Zuo, J. Huang, R. Oitker, M. Yen, J. Zhao, D. M. Tiede and L. Zang, J. Phys. Chem. B, 2006, 110, 12327–12332 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete analytical data along with the copies of 1H NMR, 13C NMR, LC/MS, and HRMS spectra of all the synthesized compounds. X-ray crystallographic data of 3DT-DPP in CIF format. CCDC 1554177. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06270b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*