
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neoansamycins from Streptomyces sp. LZ35†
Mengyujie Liua,
Chunhua Lua,
Ruocong Tangb,
Shanren Lia,
Haoxin Wangb and
Yuemao Shen *ab
*ab
aKey Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, No. 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China. E-mail: yshen@sdu.edu.cn; Tel: +86-531-88382108
bState Key Laboratory of Microbial Technology, Shandong University, Jinan, Shandong 250100, P. R. China
First published on 17th July 2017
Abstract
Previously, activation of the cryptic nam gene cluster led to three new naphthalenic ansamycins with unprecedented n-pentyl and n-butyl side chains from the mutant Streptomyces sp. SR201nam1OE strain. In this study, we further characterized the products of the mutant strain and six new neoansamycin congeners, namely neoansamycins D–I (1–6), were elucidated. Among them, compounds 1–3 feature the conserved skeleton of neoansamycins B and C but with n-hexyl side chains, and 4–6 are modified neoansamycins with n-pentyl side chains, illustrating the biosynthetic plasticity and diverse post-PKS modifications of neoansamycins.
Introduction
Ansamycins are a family of macrolactam antibiotics with remarkable potential for druggability, including the RNA polymerase inhibitor rifamycin,1 the Hsp90 inhibitor geldanamycin,2 and the potent microtubule inhibitors maytansinoids.3 Over the past ten years, we have focused on discovering ansamycin-producing strains, and obtained twenty-six 3-amino-5-hydroxyl benzoic acid (AHBA) synthase gene-positive strains from plant-associated and marine-derived actinomycetes through PCR screening of AHBA synthase genes.4 From these AHBA synthase gene-positive strains, we isolated dozens of ansamycins, including hygrocins,5–7 divergolides,8,9 juanlimycins,10 ansatrienes,11,12 and streptovaricin derivatives ansavaricins A–I.13,14Recently, Streptomyces sp. LZ35 was activated to produce novel naphthalenic ansamycins (neoansamycins A–C) by constitutive overexpression of a LuxR family transcriptional regulatory gene,15 and further ten new benzenic ansamycins (5,10-seco-neoansamycins A–J) were obtained by disrupting the nam7 gene in the SR201nam1OE strain.16 The intriguing structure diversity of the neoansamycins A–C and 5,10-seco-neoansamycins A–J encouraged us to search for more new congeners of neoansamycins. In this study, six new analogues of neoansamycins, namely neoansamycins D–I (1–6), were isolated from the fermentation products of the strain S. sp. SR201nam1OE. Their structures were elucidated on the basis of 1D-, 2D-NMR, HRESIMS analysis and X-ray single crystal diffraction. Further, the cytotoxicity and antibacterial activities of compounds 1–6 were also evaluated in this study.
Results and discussion
The strain S. sp. SR201nam1OE was cultured on ISP3 agar media for 14 days at 28 °C. The 100 L fermented agar was diced and extracted three times with EtOAc/MeOH (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v) at room temperature. The crude extract was partitioned between ddH2O and EtOAc. The EtOAc extract was further partitioned between petroleum ether (PE) and 95% aqueous MeOH. The MeOH extract (40 g) was subjected to column chromatography over Sephadex LH-20, reversed-phase (RP) C18 silica gel and finally semi-preparative HPLC to yield compounds 1–6 (Fig. 1).
:20, v/v) at room temperature. The crude extract was partitioned between ddH2O and EtOAc. The EtOAc extract was further partitioned between petroleum ether (PE) and 95% aqueous MeOH. The MeOH extract (40 g) was subjected to column chromatography over Sephadex LH-20, reversed-phase (RP) C18 silica gel and finally semi-preparative HPLC to yield compounds 1–6 (Fig. 1).
 | ||
| Fig. 1 Structures of compounds 1–6. | ||
The molecular formula of neoansamycin D (1) was assigned as C31H39NO6 on the basis of high resolution ESIMS data (m/z 522.2848 for [M + H]+) (ESI Fig. S13†). Interpretation of the NMR data (Tables 1 and 2) revealed that 1 had similar structure as that of neoansamycin B,15 except for the substitute of a hexyl at C-20 instead of an amyl side chain, and which was further confirmed by the 1H–1H COSY and HMBC correlations (ESI Table S1 and Fig. S1†). The NOESY correlations (ESI Table S1†) from H-19 to H-18a and H-20a revealed the relative configurations of 1. Finally, the absolute configurations of neoansamycin D were fully confirmed by the X-ray diffraction analysis (CCDC 1481243†) (Fig. 2).
| Pos. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 3 | 2.46, d (9.4) | 2.29, d (9.6) | 2.46, d (10.9) | 2.83, d (10.2) | 3.11, s | |
| 2.72, d (10.9) | 3.05, d (10.8) | 2.76, d (10.9) | 3.10, d (11.7) | |||
| 7 | 7.75, d (8.4) | 7.86, d (8.4) | 7.64, d (8.4) | 7.89, d (8.6) | 8.29, d (8.6) | 7.85, d (8.6) |
| 8 | 6.96, d (8.4) | 6.96, d (8.4) | 7.01, d (8.2) | 7.33, d (8.7) | 7.15, d (8.7) | 7.22, d (8.6) |
| 12 | 2.21, t (6.9) | 2.35, m | ||||
| 13 | 2.12, m | 2.01, m | 2.00, m | 2.16, m | 1.49, m | 1.51, m |
| 2.91, m | 2.80, m | 1.78, m | ||||
| 14 | 1.68, m | 1.62, m | 1.59, m | 2.01, m | 1.83, m | 1.45, m |
| 1.80, m | 2.00, m | |||||
| 15 | 1.98, m | 1.95, m | 1.19, m | 1.47, m | 2.05, m | 1.21, m |
| 1.43, m | 1.93, t (13.9) | |||||
| 2.04, m | ||||||
| 16 | 5.02, d (6.0) | 4.26, br s | 4.81, d (8.0) | 5.00, d (7.4) | 4.79, d (9.0) | 3.24, d (8.3) |
| 18 | 2.16, m | |||||
| 19 | 2.25, d (9.8) | 2.27, d (8.4) | 2.04, d (9.6) | 2.28, d (10.0) | 2.10, m | 2.33, m |
| 20 | 2.66, m | 2.75, m | 2.64, m | 2.68, m | 2.82, m | 2.68, m |
| 12a | 2.49, m | 2.52, m | 2.48, m | 2.61, m | 1.76, m | 1.46, m |
| 2.86, m | 2.75, m | 1.94, m | 2.00, m | |||
| 12b | 1.12, t (7.3) | 0.93, t (7.8) | 1.06, t (7.3) | 2.72, m | 0.80, t (7.3) | 0.89, t (7.4) |
| 1.19, t (7.4) | ||||||
| 16a | 3.37, s | 3.37, s | 3.11, s | 3.38, s | 3.39, s | 3.40, s |
| 18a | 1.08, s | 1.41, s | 0.95, s | 1.09, s | 0.96, s | 0.92, d (6.8) |
| 20a | 1.91, m | 1.70, m | 1.19, m | 1.89, m | 1.53, m | 1.49, m |
| 1.72, m | ||||||
| 20b | 1.55, m | 1.27, m | 1.19, m | 1.29, m | 1.27, m | 1.19, m |
| 1.51, m | 1.26, m | |||||
| 20c | 1.32, m | 1.45, m | 1.23, m | 1.37, m | 1.24, m | 1.22, m |
| 20d | 1.29, m | 1.29, m | 1.22, m | 1.32, m | 1.30, m | 1.25, m |
| 1.28, m | ||||||
| 20e | 1.33, m | 1.30, m | 1.28, m | 0.92, t (7.1) | 0.89, t (6.7) | 0.86, t (7.2) |
| 20f | 0.91, t (6.7) | 0.91, t (6.0) | 0.86, t (7.1) | |||
| N–H | 8.63, s | 7.41, s | ||||
| 1′ | 5.42, d (7.6) | |||||
| 2′ | 3.63, m | |||||
| 3′ | 3.55, m | |||||
| 4′ | 3.67, m | |||||
| 5′ | 4.18, d (9.6) | |||||
| 6′ | ||||||
| 6′a | 3.77, s |
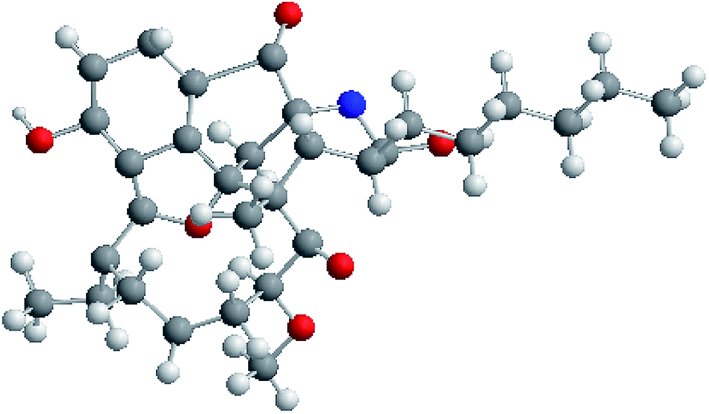 | ||
| Fig. 2 Single-crystal X-ray structure for neoansamycin D (1). | ||
The high-resolution of neoansamycin E (2) gave a quasi-molecular ion at m/z 540.2952 [M + H]+ (ESI Fig. B19†) consistent with the molecular formula C31H41NO7. After careful comparison the NMR data of 1 and 2, we found compound 2 is as an analogue of 1. The chemical shifts of C-11 (δC 150.3 s) and C-12 (δC 118.7 s) in 1 moved to downfield of C-11 (δC 209.5 s) and upfield of C-12 (δH 2.19; δC 49.6 d) in 2, which suggest a keto-enol tautomerization of C-11 and C-12 in 1 and 2 incurred by the ketalization between C-4 hydroxyl and C-12 keto groups. The molecular weight of 2 was more 18 D than that of 1, which further demonstrated the hydrolytic cleavage of the ether bond between C-4 and C-11 in 2. The relative configurations of 2 at C-12, C-16 and C-20 were assigned to be identical to that of 5,10-seco-neoansamycin H14 on the basis of the same biosynthetic origin. Likewise, the relative configurations of 2 at C-18 and C-19 were assigned identical to those of neoansamycin D (1) and B13 on the basis of the same biosynthetic origin.
Neoansamycin F (3) with a quasi-molecular ion at m/z of 554.2748 [M + H]+ was determined to have the molecular formula C31H39NO8. The 1H and 13C NMR spectra revealed 31 signals, corresponding to four CH3, nine CH2, six CH and twelve quaternary C-atoms. The HSQC, 1H–1H COSY and HMBC correlations (ESI Table S3 and Fig. S3†) revealed that the structure of 3 was similar to that of 1. After careful comparison, we found apparent differences including the chemical shifts at C-3 [(δC 91.2 s) in 3 and (δH 2.46 d, 2.72 d, δC 49.9 t) in 1], which demonstrated the location of hydroxyl at C-3 in 3 instead of methylene in 1. The NOESY correlations (ESI Table S3†) from H-18a to H-19 and H-20a revealed the relative configurations at C-18, C-19 and C-20 of compound 3. Considering the identical biosynthetic origin, rest of the configurations of 3 were suggested to be identical to those of neoansamycin D (1).
Neoansamycin G (4) was determined to have molecular formula C37H47NO12 on the basis of HRESIMS (m/z 698.3172 [M + H]+) (ESI Fig. S32†). The 1H and 13C NMR data with the aid of HSQC, 1H–1H COSY and HMBC experiments revealed that 4 was partially identical to neoansamycin B.15 The presence of a β-glucuronic acid moiety was revealed by the 1H NMR signals at δH 5.42 (d, J = 7.6 Hz), 3.63 (m), 3.55 (m), 3.67 (m) and 4.18 (d, J = 9.6 Hz) (Table 1), and the 13C NMR signals at δC 101.0 d, 74.3 d, 77.8 d, 72.7 d, 76.9 d and 170.7 s (Table 2). These assignments were further confirmed by 1H–1H COSY and HMBC correlations. The HMBC correlation from H-6′a (δ 3.77, s, 3H) to C-6′ revealed the presence of 6-O-methyl β-glucuronic acid, which was located at C-9 based on the HMBC correlation from the anomeric proton H-1′ to C-9. Thus, the structure of 4 was established to be neoansamycin B-9-O-β-glucuronide 6′-methyl ester. The relative configurations of 4 were determined identical to those of 1 on the basis of the same biosynthetic origin.
| Pos. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 194.2, C | 195.3, C | 190.3, C | 194.4, C | 194.2, C | 195.3, C |
| 2 | 70.0, C | 70.1, C | 73.9, C | 70.2, C | 69.7, C | 71.2, C |
| 3 | 49.9, CH2 | 51.3, CH2 | 91.2, C | 50.1, CH2 | 52.2, CH2 | 70.2, CH |
| 4 | 89.9, C | 95.2, C | 94.9, C | 90.1, C | 84.6, C | 195.1, C |
| 5 | 157.1, C | 156.9, C | 153.0, C | 156.71, C | 150.7, C | 134.5, C |
| 6 | 119.0, C | 119.2, C | 116.3, C | 121.4, C | 121.3, C | 129.3, C |
| 7 | 129.3, CH | 131.8, CH | 128.2, CH | 129.6, CH | 138.0, CH | 129.9, CH |
| 8 | 119.3, CH | 119.0, CH | 109.0, CH | 117.2, CH | 119.9, CH | 122.4, CH |
| 9 | 159.1, C | 161.4, C | 158.7, C | 156.72, C | 166.7, C | 160.8, C |
| 10 | 118.6, C | 121.6, C | 117.9, C | 121.8, C | 114.3, C | 124.5, C |
| 11 | 150.3, C | 209.5, C | 149.5, C | 149.8, C | 203.8, C | 207.9, C |
| 12 | 118.7, C | 49.6, CH | 115.7, C | 120.9, C | 85.8, C | 53.5, CH |
| 13 | 30.0, CH2 | 29.8, CH2 | 28.4, CH2 | 30.5, CH2 | 26.5, CH2 | 29.6, CH2 |
| 14 | 25.14, CH2 | 26.0, CH2 | 24.5, CH2 | 25.1, CH2 | 19.8, CH2 | 25.4, CH2 |
| 15 | 32.4, CH2 | 30.0, CH2 | 30.4, CH2 | 32.4, CH2 | 35.1, CH2 | 30.5, CH2 |
| 16 | 88.7, CH | 92.5, CH | 85.4, CH | 88.7, CH | 87.1, CH | 82.6, CH |
| 17 | 215.9, C | 214.2, C | 211.1, C | 215.9, C | 214.8, C | 88.8, C |
| 18 | 61.9, C | 60.1, C | 57.7, C | 61.9, C | 63.0, C | 40.6, CH |
| 19 | 60.6, CH | 61.2, CH | 56.5, CH | 60.7, CH | 61.1, CH | 56.6, CH |
| 20 | 46.8, CH | 45.6, CH | 44.6, CH | 46.9, CH | 45.9, CH | 42.3, CH |
| 21 | 180.4, C | 180.7, C | 178.7, C | 180.4, C | 180.2, C | 178.2, C |
| 12a | 25.08, CH2 | 24.6, CH2 | 23.3, CH2 | 25.6, CH2 | 35.0, CH2 | 27.1, CH2 |
| 12b | 15.3, CH3 | 13.2, CH3 | 14.9, CH3 | 15.0, CH3 | 8.6, CH3 | 11.3, CH3 |
| 16a | 58.8, CH3 | 58.4, CH3 | 56.7, CH3 | 58.9, CH3 | 57.5, CH3 | 61.9, CH3 |
| 18a | 31.2, CH3 | 25.4, CH3 | 31.1, CH3 | 31.2, CH3 | 27.5, CH3 | 10.2, CH3 |
| 20a | 31.8, CH2 | 31.5, CH2 | 30.9, CH2 | 31.9, CH2 | 31.2, CH2 | 32.6, CH2 |
| 20b | 27.2, CH2 | 26.8, CH2 | 25.3, CH2 | 27.0, CH2 | 26.6, CH2 | 24.4, CH2 |
| 20c | 30.7, CH2 | 23.5, CH2 | 29.1, CH2 | 33.4, CH2 | 33.2, CH2 | 31.4, CH2 |
| 20d | 23.7, CH2 | 33.3, CH2 | 31.7, CH2 | 23.7, CH2 | 23.6, CH2 | 22.0, CH2 |
| 20e | 32.9, CH2 | 23.6, CH2 | 22.1, CH2 | 14.4, CH3 | 14.5, CH3 | 14.0, CH3 |
| 20f | 14.4, CH3 | 14.4, CH3 | 14.0, CH3 | |||
| 1′ | 101.0, CH | |||||
| 2′ | 74.3, CH | |||||
| 3′ | 77.8, CH | |||||
| 4′ | 72.7, CH | |||||
| 5′ | 76.9, CH | |||||
| 6′ | 170.7, C | |||||
| 6′a | 53.0, CH3 |
Neoansamycin H (5) was determined as an analogue of 1 with the molecular formula C30H37NO7 (HRESIMS m/z 524.2642 [M + H]+) (ESI Fig. S39†). The 1D, 2D NMR data (ESI Table S5 and Fig. S5†) revealed that 5 was similarly identical to neoansamycin B.15 The chemical shifts of C-11 (δC 203.8 s) and C-12 (δC 85.8 s) in 5 and C-11 (δC 148.6 s) and C-12 (δC 116.0 s) in neoansamycin B suggested the tautomerization of enol-form and keto-form at C-11 and C-12 in neoansamycin B and 5, and the downfield shift of C-12 in 5 demonstrated the oxygenation of C-12 and the formation position of the ether bond in 5 is C-4–O–C-12 instead of C-4–O–C-11. The NOESY correlations (ESI Table S5†) from H-12a to H-18a, H-18a to H-19, and H-20a to H-19 revealed the relative configurations of compound 5 at C-12, C-18, C-19 and C-20. The relative configuration of 5 at C-16 was assigned to be identical to that of 5,10-seco-neoansamycin H14 on the basis of the same biosynthetic origin.
The molecular formula of neoansamycin I (6) was determined to be C30H39NO7 on the basis of HRESIMS (m/z 526.2798 [M + H]+). The planer structure was deduced by analysis of its HMQC, 1H–1H COSY and HMBC correlations (ESI Table S6 and Fig. S6†). The presence of dihydronaphthoquinone ring and the formation of a five membered ring (C-2/3/17/18/19) were confirmed by the changes of chemical shifts of C-3 (δC 70.2 d; δH 3.11 s), C-4 (δC 195.1 s), C-17 (δC 88.8 s) and C-18 (δC 40.6 d; δH 2.16 m) in 6 and C-3 (δC 48.6 t; δH 2.31 d & 2.72–2.69 m), C-4 (δC 88.6 s), C-17 (δC 213.5 s) and C-18 (δC 59.9 s) in neoansamycin B.15 The keto-form of C-11 was deduced on the basis of the chemical shift of C-11 (δC 207.9 s) in 6. The relative configurations of 6 were assigned to be identical to those of neoansamycin D (1) on the basis of the same biosynthetic origin.
The antimicrobial activities of 1–6 were measured against Bacillus subtilis 86315, Staphylococcus aureus ATCC 25923, Mycobacterium smegmatis mc2 155, and Candida albicans 5314 by the paper disc diffusion assay (20 μg per disc). Only compounds 1, 5 and 6 exhibited modest activity against B. subtilis 86315 (diameters of inhibitory zones 13, 11, and 9 mm, respectively). In addition, 2 exhibited weak activity against Staphylococcus aureus ATCC 25923 with inhibitory zone of 10 mm. These activities are similar to those of neoansamycins A–C.15
Ansamycins are type I polyketide macrolactams.17 Their highly diversified structures mostly resulted from diverse post-PKS modifications.12,18 In particular, post-PKS modifications are critical in determining their bioactivities.3,19 The structures of neoansamycins D–I mirror diverse and interesting post-PKS modifications. However, their antimicrobial activities are moderate unlike other ansamycins usually are potent, implying that broad screening assays are required for exploiting the bioactivities of this class of novel ansamycins.
Experimental
General experimental procedures
Optical rotations were carried out using an Anton Paar MCP200 automatic polarrimeter. The UV spectra were obtained on a TU-1810 spectrophotometer (Beijing Purkinje General Instrument Co., LTD). NMR spectra were recorded on Bruker DRX-600 NMR spectrometer (Bruker Daltonics Inc., Billerica, MA, USA) with tetramethylsilane (TMS) as an internal standard. HRESIMS were carried out on an LTQ-Orbitrap XL. Sephadex LH-20 was obtained from GE Amersham Biosciences (25–100 μm; Piscataway, New Jersey) and LiChroprep RP-18 were used for column chromatography (CC) from Merck (40–63 μm; Darmstadt, Germany). Semi-preparative HPLC were performed on an Agilent 1200 equipped with a ZORBAX Eclipse XDC18 5 μm column (9.4 × 250 mm).Fermentation and eextraction
The mutant strain SR201nam1OE was cultured on ISP3 medium (oatmeal 30 g, saline salt 1 mL, agar 20 g, pH 7.2) for 14 days at 28 °C. The culture (a total volume of 100 liters) was chopped, diced and extracted three times overnight with an equal volume of EtOAc/MeOH 80:20 (v/v) at room temperature and partitioned between ddH2O and EtOAc until the EtOAc layer was colorless. Then, the EtOAc extract was dried with Na2SO4, and the solvent was removed under vacuum at 38 °C. The EtOAc extract was partitioned with petroleum ether (PE) and MeOH until the PE layer was colorless. The MeOH solution was concentrated under vacuum at 38 °C to obtain MeOH extract (40 g).
Isolation and ppurification of compounds 1–7
The MeOH extract (40 g) was subjected to column chromatography over Sephadex LH-20 (120 g) eluted with MeOH to obtain 5 fractions (Fr. 1–5). Fr. 3 (11 g) was chromatographed over Sephadex LH-20 (120 g) eluted with MeOH to obtain 3 fractions, Fr. 3a–3c. Fr. 3b (6.7 g) was subjected to MPLC over RP-18 silica gel (80 g), and 20 subfractions with 200 mL for each gradient were obtained from the elution with 30, 50, 60, 70, 75, 80 and 100% MeOH, respectively. In accordance with TLC results, 4 fractions, Fr. 3b1–3b4 were obtained. Fr. 3b2 (3.1 g) was subjected to CC over Sephadex LH-20 (120 g) eluted with MeOH to obtain 4 fractions, Fr. 3b2a–3b2d. Fr. 3b2c (984 mg) was further subjected to MPLC over RP-18 silica gel (80 g) eluted with 30%, 40%, 50%, 55%, 60% and 100% CH3CN to obtain Fr. 3b2c1–3b2c4. Fr. 3b2c2 (105.2 mg) were purified by semi-preparative HPLC (eluted with 50% acetonitrile, 4 mL min−1, UV 320 nm) to yield 4 (3 mg). Fr. 3b2c3 (213 mg) were purified by semipreparative HPLC (eluted with 55% acetonitrile in 0.05% formic acid, 4 mL min−1, UV 320 nm) to yield 3 (1.7 mg), and 6 (1.2 mg).Fr. 3b3 (2.93 g) was subjected to CC over Sephadex LH-20 (120 g) eluted with MeOH to obtain 3 fractions, Fr. 3b3a–3b3c. Fr. 3b3b (1.03 g) was further subjected to MPLC over RP-18 silica gel (80 g) eluted with 30%, 40%, 50%, 55%, 60%, 70% and 100% CH3CN to obtained Fr. 3b3b1–3b3b4. Fr. 3b3b3 (153 mg) were purified by semi-preparative HPLC (eluted with 65% acetonitrile in 0.05% formic acid, 4 mL min−1, UV 320 nm) to yield 7 (6.0 mg), 1 (3.0 mg), 2 (2.3 mg) and 5 (8.2 mg).
ε): 306 (2.23), 275 (2.55) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS: m/z 522.2848 [M + H]+ (calcd for C31H40NO6+, 522.2850).ε): 303 (2.28), 277 (2.58) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS: m/z 540.2952 [M + H]+ (calcd for C31H42NO7+, 540.2956).ε): 306 (2.25), 277 (2.58) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS: m/z 554.2748 [M + H]+ (calcd for C31H40NO8+, 554.2748).ε) 311 (2.16), 280 (2.50) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS: m/z 698.3172 [M + H]+ (calcd for C37H48NO12+, 698.3170).ε) 304 (2.07), 275 (2.38) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS: m/z 524.2642 [M + H]+ (calcd for C30H38NO7+, 524.2643).ε) 302 (2.10), 270 (2.43) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS: C30H39NO7, m/z 526.2798 [M + H]+ (calcd for C30H40NO7+, 526.2799).X-ray crystallographic analysis
The single-crystal X-ray diffraction intensity data of 1 were collected with a Bruker SMART-1000 diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) by the ω-scan technique [scan width 0–180°, 2θ ≤ 50°] at 293 (2) K.Crystal data of neoansamycin D (1): C31H39NO6, Mr = 521.67, orthorhombic, a = 8.5601 (9) Å, b = 14.9864 (13) Å, c = 23.2722 (18) Å, α = 90°, β = 90°, γ = 90°, V = 2985.5 (5) Å3, space group P2 (1) 2 (1) 2 (1), Z = 4, Dx = 1.232 mg m−3, μ (Mo Kα) = 0.698 mm−1, and F(000) = 1192. Crystal dimensions: 0.32 × 0.28 × 0.27 mm3. Independent reflections: 4528 (Rint = 0.0495). Theta range for data collection: 3.51 to 66.19°. The final R1 = 0.0778, wR2 = 0.1356 (I > 2σ(I)). CCDC number: 1481243.†
Antimicrobial assay
The antimicrobial activities of compounds 1–6 against Candida albicans 5314, Staphylococcus aureus ATCC 25923, Mycobacterium smegmatis mc2 155, Pseudomonas aeruginosa PA01, Bactllus subtilis 86315 or Salmonella enterica serovar Typhimurium UK-1 χ8956 were measured with a paper disc diffusion assay.20 Kanamycin and amphotericin were used as positive control.Conclusions
In summary, we isolated and characterized six neoansamycin congers, namely neoansamycins D–I (1–6), with unusual extender units and diverse post-PKS modifications from the Streptomyces sp. SR201nam1OE strain. In particular, although various unusual extender units have been found in other polyketide-derived structures,21 this study is the first example that n-hexylmalonyl-CoA was used in the biosynthesis of ansamycins. The promiscuity of neoansamycin PKS could facilitate the bioengineering of novel ansamycins.11,22Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (81373304, 81530091, U1405223), and the Independent Innovation Foundation of Shandong University (IIFSDU, 2014JC027) and Program for Changjiang Scholars and Innovative Research Team in University (IRT_17R68).Notes and references
- H. G. Floss and T. W. Yu, Chem. Rev., 2005, 105, 621–632 CrossRef CAS PubMed.
- L. Whitesell, E. G. Mimnaugh, B. De Costa, C. E. Myers and L. M. Neckers, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8324–8328 CrossRef CAS.
- J. M. Cassady, K. K. Chan, H. G. Floss and E. Leistner, Chem. Pharm. Bull., 2004, 52, 1–26 CrossRef CAS PubMed.
- H. X. Wang, Y. Y. Chen, L. Ge, T. T. Fang, J. Meng, Z. Liu, X. Y. Fang, S. Ni, C. Lin, Y. Y. Wu, M. L. Wang, N. N. Shi, H. G. He, K. Hong and Y. M. Shen, J. Appl. Microbiol., 2013, 115, 77–85 CrossRef CAS PubMed.
- C. Lu, Y. Li, J. Deng, S. Li, Y. Shen, H. Wang and Y. Shen, J. Nat. Prod., 2013, 76, 2175–2179 CrossRef CAS PubMed.
- S. Li, C. Lu, J. Ou, J. Deng and Y. Shen, RSC Adv., 2015, 5, 83843–83846 RSC.
- S. Li, H. Wang, Y. Li, J. Deng, C. Lu, Y. Shen and Y. Shen, ChemBioChem, 2014, 15, 94–102 CrossRef CAS PubMed.
- G. Zhao, S. Li, Z. Guo, M. Sun and C. Lu, RSC Adv., 2015, 5, 98209–98214 RSC.
- S. R. Li, G. S. Zhao, M. W. Sun, H. G. He, H. X. Wang, Y. Y. Li, C. H. Lu and Y. M. Shen, Gene, 2014, 544, 93–99 CrossRef CAS PubMed.
- J. Zhang, Z. Qian, X. Wu, Y. Ding, J. Li, C. Lu and Y. Shen, Org. Lett., 2014, 16, 2752–2755 CrossRef CAS PubMed.
- G. Shi, N. Shi, Y. Li, W. Chen, J. Deng, C. Liu, J. Zhu, H. Wang and Y. Shen, ACS Chem. Biol., 2016, 11, 876–881 CrossRef CAS PubMed.
- X. Li, J. Zhu, G. Shi, M. Sun, Z. Guo, H. Wang, C. Lu and Y. Shen, RSC Adv., 2016, 6, 88571–88579 RSC.
- Z. Zhang, X. Wu, R. Song, J. Zhang, H. Wang, J. Zhu, C. Lu and Y. Shen, RSC Adv., 2017, 7, 14857–14867 RSC.
- Z. Zhang, J. Zhang, R. Song, Z. Guo, H. Wang, J. Zhu, C. Lu and Y. Shen, RSC Adv., 2017, 7, 5684–5693 RSC.
- S. Li, Y. Li, C. Lu, J. Zhang, J. Zhu, H. Wang and Y. Shen, Org. Lett., 2015, 17, 3706–3709 CrossRef CAS PubMed.
- J. Zhang, S. Li, X. Wu, Z. Guo, C. Lu and Y. Shen, Org. Lett., 2017, 19, 2442–2445 CrossRef CAS PubMed.
- H. G. Floss, J. Nat. Prod., 2006, 69, 158–169 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, L. V. Ponomareva, Q. Qiu, R. Woodcock, S. I. Elshahawi, X. Chen, Z. Zhou, B. E. Hatcher, J. C. Hower, C.-G. Zhan, S. Parkin, M. K. Kharel, S. R. Voss, K. A. Shaaban and J. S. Thorson, Angew. Chem., Int. Ed., 2017, 56, 2994–2998 CrossRef CAS PubMed.
- P. Spiteller, L. Bai, G. Shang, B. J. Carroll, T. W. Yu and H. G. Floss, J. Am. Chem. Soc., 2003, 125, 14236–14237 CrossRef CAS PubMed.
- D. Raahave, Antimicrob. Agents Chemother., 1974, 6, 603–605 CrossRef CAS PubMed.
- N. Quade, L. Huo, S. Rachid, D. W. Heinz and R. Müller, Nat. Chem. Biol., 2012, 8, 117–124 CrossRef CAS PubMed.
- Y. Tian, N. Jiang, A. H. Zhang, C. J. Chen, X. Z. Deng, W. J. Zhang and R. X. Tan, Org. Lett., 2015, 17, 1457–1460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and other relevant information for compounds 1–6. CCDC 1481243. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06339c |
| This journal is © The Royal Society of Chemistry 2017 |