
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2-responsive self-healable hydrogels based on hydrophobically-modified polymers bridged by wormlike micelles†
Chunming Xionga,
Kang Pengb,
Xiaofen Tanga,
Zhengrong Yea,
Yang Shia and
Haiyang Yang *b
*b
aResearch Institute of Science and Technology, China National Petroleum Corporation, Beijing, 100083, P. R. China
bCAS Key Laboratory of Soft Matter Chemistry, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: yhy@ustc.edu.cn
First published on 11th July 2017
Abstract
CO2-responsive hydrogels, using CO2 as a “green” trigger, have recently been of considerable interest. Herein, a novel CO2-responsive self-healable hydrogel is fabricated by simply mixing hydrophobically modified polyacrylamide (HMPAM) with sodium dodecyl sulfate (SDS)–N,N,N′,N′-tetramethyl-1,3-propanediamine (TMPDA) surfactant micelles. In the presence of CO2, the SDS–TMPDA spherical micelles transform into long wormlike micelles, serving as multivalent cross-linkages to bridge HMPAM chains based on hydrophobic interactions. The interpenetrating three-dimensional network induces a sol-to-gel transition, accompanied by a 360 and 400 times enhancement of the zero-shear viscosity and storage modulus, G′ (ω = 6.28 rad s−1), respectively. In additon, the sol–gel transition can be repeatedly and reversibly switched by cyclically bubbling and removing CO2 without any harm. Furthermore, the CO2-responsive hydrogel exhibits significant shear-thinning and self-healing properties, suggesting that the hydrogel is injectable and could be used as a potential plug to block CO2 gas breakthrough channels during CO2 flooding. Therefore, we believe that the presented work will enable the development of the design of CO2-responsive self-healable hydrogels and their practical applications.
Introduction
Smart hydrogels that undergo physicochemical changes in response to external stimuli, such as pH,1–3 light,4,5 CO2,6–9 thermal,10,11 ionic strength12,13 and electrical potential,14 have been extensively explored as scaffolds for tissue engineering, carriers for controlled drug release, actuators and sensors in biomedical devices. Among them, CO2-responsive hydrogels are extremely important, and special, due to the “green” properties of CO2. CO2 is a non-toxic, inexpensive, benign and abundant gas.15 Moreover, using CO2 as a trigger can lead to many switching cycles without the accumulation of byproducts.16,17Up to now, CO2-responsive systems are generally categorized into three types based on their CO2-responsive groups: amidine, amine, and carboxyl groups.18 Firstly, Jinying Yuan and co-workers19 designed a specific amidine-containing diblock copolymer PEO-b-PAD to fabricate CO2-responsive polymeric vesicles with a biomimetic “breathing” feature. The size of these vesicles over a wide range was tuned by CO2, which could control the degree of protonation of amidine species. Secondly, the group of Yue Zhao20 demonstrated a novel polymer brush that exhibited CO2-controllable switching between extended (hydrated) and collapsed (dehydrated) chain conformations. Such CO2-swithchable polymer solubility could be repeated many times without contamination of the solution by accumulated salts, unlike the conventional pH change induced by adding acids and bases. Thirdly, Viktor Fischer et al.21 presented the synthesis of molecularly controlled carboxyl-functionalized nanoparticles, which could be coagulated by bubbling CO2.
Although CO2-responsive polymers have gained a rapid development over the last few years, some challenges still remain.22 The amidine group is prone to hydrolysis in aqueous environments. CO2-responsive amphiphilic block copolymers require a demanding synthesis. For some latexes, the effective CO2-responsive behaviors only exist under a low solid content situation. Last but not least, CO2-responsive polymers are generally costly and hence have limitation in various applications. In particular, CO2 flooding has become a significant technology for enhanced oil recovery (EOR). It is crucial to develop CO2-induced hydrogels via industrialization paths to efficiently plug CO2 gas breakthrough channels to improve the sweep volume of CO2.23,24
On the other hand, the group of Yujun Feng reported three kinds of CO2-responsive surfactant-based self-assembled structures (UC22AMPM, ODPTA, and SDS–TMPDA).25–27 After treatment with CO2, free surfactants, vesicles, and spherical micelles switched to worm-like micelles in these systems, respectively. Inspired by previous studies28,29 of the interaction between surfactants and hydrophobically modified polymers, we can apply these surfactant responsers to endow the conventional responseless hydrophobically modified polyacrylamide (HMPAM) with fascinating CO2-responsive and self-healing properties.
Herein, a novel CO2-responsive self-healable hydrogel was fabricated by simply mixing HMPAM with sodium dodecyl sulfate (SDS)–N,N,N′,N′-tetramethyl-1,3-propanediamine (TMPDA) surfactant micelles in aqueous solution. HMPAM was synthesized by free-radical micellar copolymerization of acrylamide, acrylic acid and n-dodecylmethacrylate, representing an important class of water-soluble associative polymers.30,31 The SDS–TMPDA mixture was chosen, because CO2 would protonate the tertiary amine groups the TMPDA molecules to induce a sphere-to-worm transition of SDS.27 As shown in Fig. 1, SDS–TMPDA spherical micelles dissolved the HMPAM hydrogel in the absence of CO2. After bubbling CO2 into the aqueous mixture, wormlike micelles formed, serving as multivalent cross-linkages to bridge HMPAM chains based on the hydrophobic interaction. The interpenetrating three-dimensional network induced a sol-to-gel transition, accompanied by 360 and 400 times the enhancement of zero-shear viscosity and storage modulus G′ (ω = 6.28 rad s−1), respectively. The sol–gel transition could be repeatedly and reversibly switched by cyclically bubbling and removing CO2 without any residual chemicals such as salts which might be harmful to the performance of the hydrogel. More importantly, the CO2-responsive hydrogel exhibited significant shear-thinning and self-healing properties, suggesting that the hydrogel was injectable and could be used as the potential plug to block CO2 gas breakthrough channels during CO2 flooding. We believe that the presented work will provide a versatile and simple strategy for the design of CO2-responsive self-healable hydrogels, fostering their use in a wide range of applications such as enhanced oil recovery (EOR).
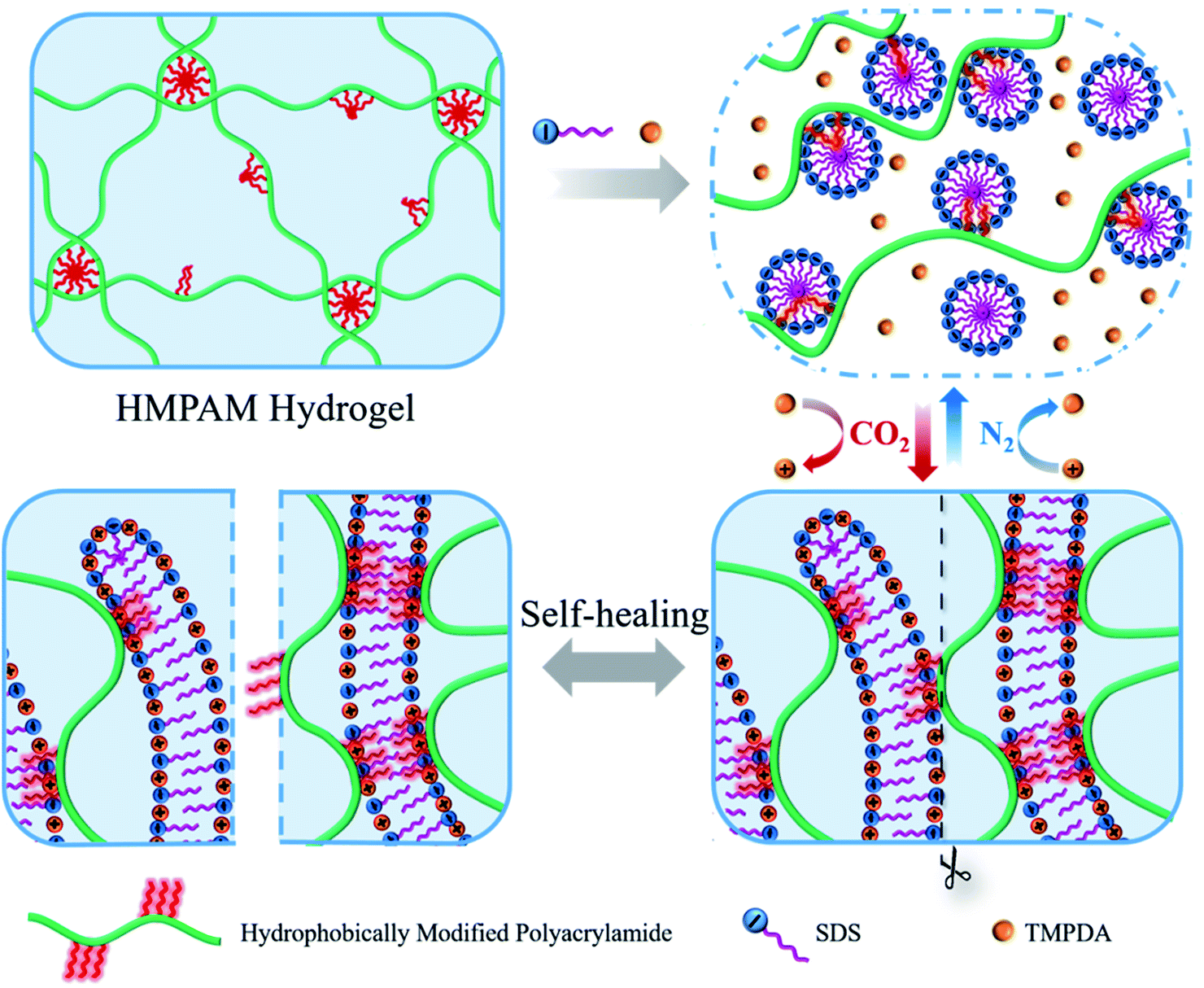 | ||
| Fig. 1 Schematic illustration of the CO2-responsive sol–gel transition and the self-healing behavior of the hydrogels based on SDS–TMPDA wormlike micelles bridged HMPAM. | ||
Experimental section
Materials
Acrylamide (AM), acrylic acid (AAc), sodium dodecyl sulfate (SDS), N,N,N′,N′-tetramethyl-1,3-propanediamine (TMPDA) and ammonium persulfate (APS) were purchased from Sinopharm Chemical Reagent CO. Ltd. n-Dodecylmethacrylate (C12) was obtained from Aldrich. All the materials were used as received without further purification and experiments were performed using deionized (DI) water.Synthesis of hydrophobically modified polyacrylamide (HMPAM)
HMPAM was synthesized according to a previously reported method.29 Briefly, 4.50 g SDS and 0.21 g C12 was dissolved in 100.0 mL water to obtain an optically transparent solution. Then adding and dissolving 9.23 g AM and 0.50 g AAc with stirring for several minutes. Finally, 0.05 g APS initiated the copolymerization at 50 °C for 12 h under nitrogen. In the monomer feed, the molar fractions of AM, AAc and C12 were 94.4%, 5.0%, and 0.6%, respectively. After polymerization, the polymer was twice precipitated in a methanol/acetone mixture and dried in a vacuum at room temperature for 2 days.Sample preparation
Appropriate amounts of HMPAM were dissolved in DI water under vigorous stirring for 3 h. Then SDS–TMPDA was mixed with the HMPAM hydrogel for 1 h at room temperature. The molar ratio of SDS to TMPDA was fixed at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. And the concentrations of SDS–TMPDA were given as the concentrations of TMPDA. After determining the phase behavior by visual inspection to have clear one-phase solutions, CO2 was bubbled into the solutions at 25 °C with a fixed flow rate of 0.1 L min−1 for 10 min under a pressure of 0.1 MPa for the gelation. Then the sample was equilibrated in a sealed vial for 24 h prior to subsequent tests. The formed hydrogel was converted to the sol state after removing CO2 by bubbling N2 at the same flow rate as for CO2 at 75 °C for 30 min.
:1. And the concentrations of SDS–TMPDA were given as the concentrations of TMPDA. After determining the phase behavior by visual inspection to have clear one-phase solutions, CO2 was bubbled into the solutions at 25 °C with a fixed flow rate of 0.1 L min−1 for 10 min under a pressure of 0.1 MPa for the gelation. Then the sample was equilibrated in a sealed vial for 24 h prior to subsequent tests. The formed hydrogel was converted to the sol state after removing CO2 by bubbling N2 at the same flow rate as for CO2 at 75 °C for 30 min.
Self-healing behavior
The hydrogel sample after preparation and equilibrium was cut in the middle, and then the two halves were stained with Rhodamine B (red dye) and Indigo carmine (blue dye), respectively. After merging the two pieces together for 60 s, the healed hydrogel could support its own weight.Rheological measurements
Rheological measurements were conducted on an ARG2 stress controlled rheometer (TA AR-G2) at 25 °C using a cone-plate of 40 mm diameter with a cone angle of 4°. The viscosity was measured as a function of shear rate for determining the zero-shear viscosity. The storage modulus (G′) and loss modulus (G′′) were measured as a function of frequency within a linear viscoelastic regime, which was determined from the prior stress-sweep test.To investigate the self-healing properties of the samples in response to applied shear forces, the samples were placed between the para-plate and the platform with special care. The following programmed procedure (applied shear force, expressed in terms of strain; duration in parentheses) was used: 1% (400 s) → 3500% (300 s) → 1% (400 s) → 3500% (300 s) → 1% (400 s) → 3500% (300 s) → 1% (400 s).
During all measurements, a solvent trap was used to minimize water evaporation.
Results and discussion
CO2 responsiveness of the mixture of HMPAM and SDS–TMPDA
In our study, hydrophobically modified polymers consisted of a water-soluble poly(AM-co-AAc) backbone onto which a small number of hydrophobic C12 alky chains were attached. Generally, the block distributed hydrophobic moieties from different polymer chains can associate and build a transitory three-dimensional network above the concentration of overlapping the coils.30,32 Therefore, we firstly studied the effect of polymer concentrations on the HMPAM hydrogel. As depicted in Fig. 2a, the zero-shear viscosity (which was determined from the curve of viscosity versus shear rate in Fig. S1†) of HMPAM hydrogels showed an obvious increment with the polymer weight percentage increasing. Furthermore, from the curves of linear viscoelastic modulus, G′(ω) and G′′(ω), for the HMPAM hydrogel with various polymer weight percentages, it could be observed that the G′ values had a substantial elastic response and were always larger than the G′′ values over the entire range of frequencies when the polymer weight percentage exceeded 0.3 wt% (Fig. S2†). These results indicated that more cross-linkages were constructed at a higher polymer concentration. Here, the polymer weight percentage was set as 2 wt% to obtain a hydrogel structure, as determined in Fig. 2b.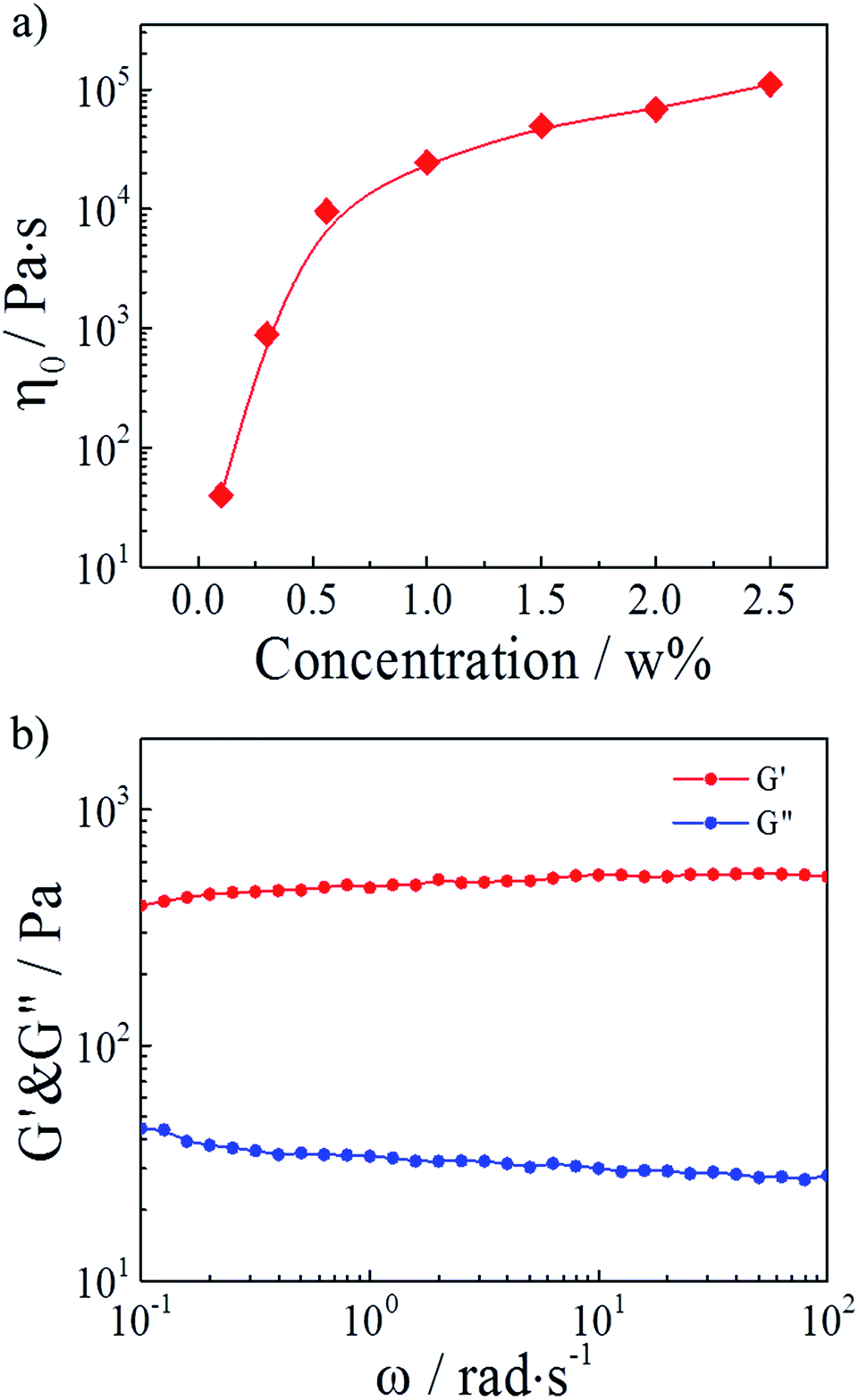 | ||
| Fig. 2 (a) Zero-shear viscosity of HMPAM hydrogels as a function of the polymer weight percentage. (b) Linear viscoelastic modulus, G′(ω) and G′′(ω), for a 2 wt% HMPAM hydrogel. | ||
Then, the combination with SDS–TMPDA micelles would endow the conventional responseless HMPAM hydrogel with fascinating CO2-responsive properties. The frequency dependencies of the storage modulus G′ and the loss modulus G′′ were presented in Fig. 3a for the HMPAM hydrogel with various concentrations of SDS–TMPDA before bubbling CO2. When the concentration of SDS–TMPDA was relatively low (20 mM), the storage modulus G′ exceeded the loss modulus G′′ in the whole frequency range, still indicative of a hydrogel structure. Increasing the concentration of SDS–TMPDA to 30 mM, G′ and G′′ intersected, exhibiting viscoelastic behavior. Above 35 mM, G′′ was dominant over G′, implying that the HMPAM hydrogel was converted to the sol state. Meanwhile, the zero-shear viscosity dropped 3 orders of magnitude as increasing the concentration of SDS–TMPDA from 20 mM to 40 mM (Fig. S3†), which is consistent with the results on G′ and G”. The gel-to-sol transition of HMPAM by mixing with SDS–TMPDA spherical micelles was attributed to the spherical micellar solubilisation of the hydrophobes.
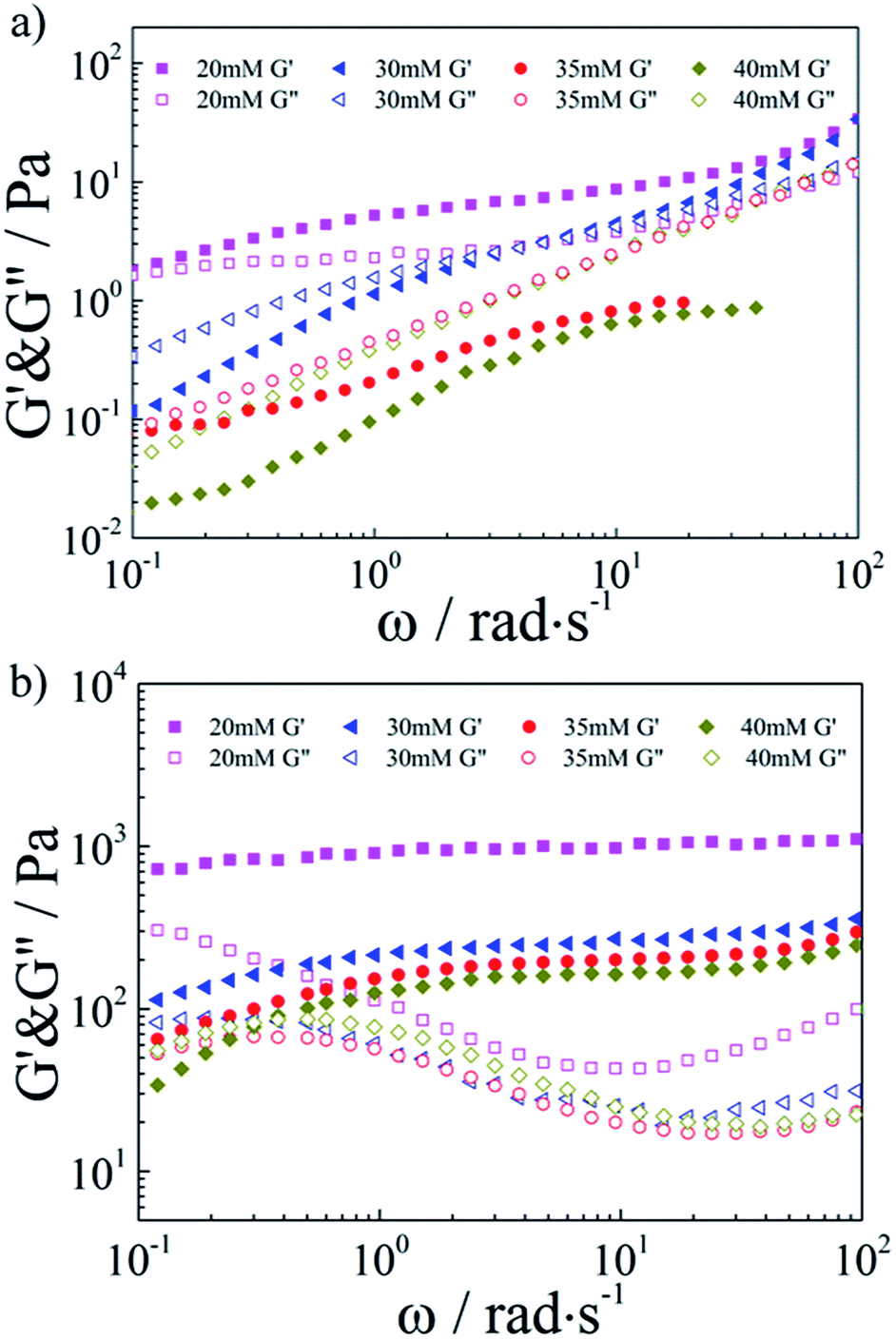 | ||
| Fig. 3 Linear viscoelastic modulus, G′(ω) and G′′(ω), for the HMPAM hydrogel with various concentrations of SDS–TMPDA (a) before and (b) after bubbling CO2. | ||
After bubbling CO2, the frequency dependencies of the storage modulus G′ and the loss modulus G′′ were presented in Fig. 3b. For all the concentrations of SDS–TMPDA, CO2 induced stronger mechanical properties. Specifically speaking, G′ was increased by 2 to 3 orders of magnitude and behaved weakly frequency dependent in the whole frequency range investigated, when the concentration of SDS–TMPDA was 20 mM. For 30 mM and 35 mM, G′ was dominant over G′′ over the entire frequency range, indicative of a predominantly elastic network rather than a viscous sol state. This elastic nature manifested that HMPAM chains were bridged by wormlike micelles based on the hydrophobic interaction. In other words, bubbling CO2 would protonate the TMPDA molecules, leading to the formation of wormlike micelles which served as multivalent cross-linkages. However, for 40 mM, G′ and G′′ intersected at a low frequency. As the concentration of SDS–TMPDA increasing, more wormlike micelles formed. It became less probable that an individual wormlike micelle contained side chains from several chains, suggesting the decrease in cross-linking density. Therefore, the mechanical properties of the hydrogel were correspondingly weakened. In Fig. S4,† the curves of viscosity versus shear rate for the HMPAM hydrogel with various concentrations of SDS–TMPDA after bubbling CO2 also provided evidence of the interpretation.
Considering all the above results, the mixture of 2 wt% HMPAM and 35 mM SDS–TMPDA was selected as a representative sample for subsequent tests. Photographs demonstrating the CO2-responsive sol–gel transition were presented in Fig. 4a. After bubbling CO2 into the aqueous mixture, wormlike micelles formed, serving as multivalent cross-linkages to bridge HMPAM chains based on the hydrophobic interaction. The interpenetrating three-dimensional network induced a sol-to-gel transition. Then the formed hydrogel could be converted to the initial sol state after removing CO2 by bubbling N2.
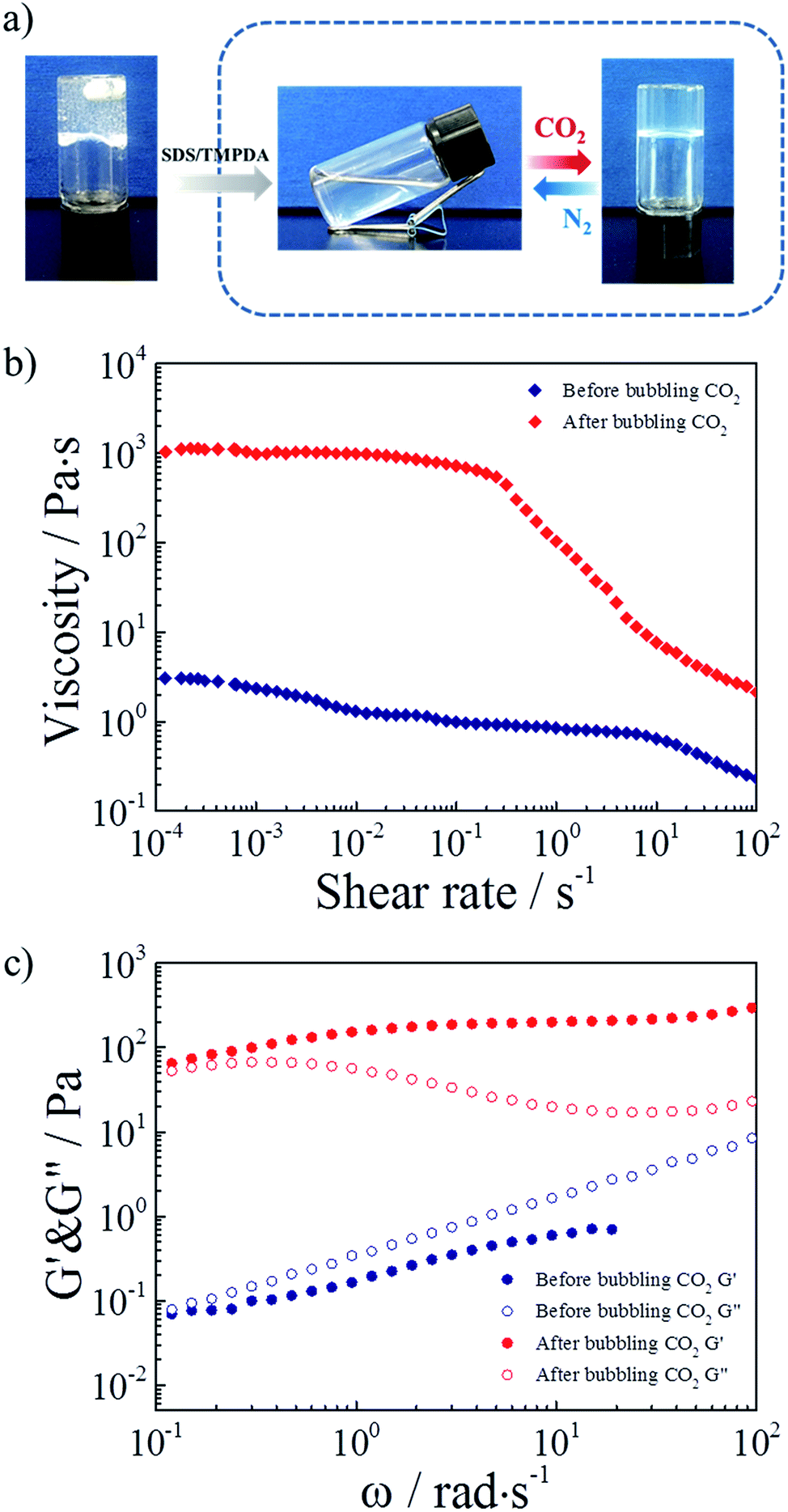 | ||
| Fig. 4 (a) Photographs demonstrating the CO2-responsive sol–gel transition of the hydrogel based on SDS–TMPDA wormlike micelles bridged HMPAM. (b) Viscosity versus shear rate plots for the representative sample before and after bubbling CO2. (c) Linear viscoelastic modulus, G′(ω) and G′′(ω), for the representative sample before and after bubbling CO2. | ||
As depicted in Fig. 4b, 360 times the enhancement of zero-shear viscosity was observed in the presence of CO2. In addition, the CO2 induced hydrogel displayed a significant shear-thinning phenomenon. The viscosity remained almost constant up to a shear rate of 0.2 s−1, and then decreased more than 2 orders of magnitude with increasing shear rate. The dramatic change of viscosity indicated that the hydrophobes of HMPAM were pulled out of wormlike micelles under a high shear rate and the non-covalent interactions were considerably broken. For comparison, the curves of viscosity versus shear rate for the 35 mM SDS–TMPDA solution before and after bubbling CO2 were measured in Fig. S5.†.
As depicted in Fig. 4c, 400 times the enhancement of storage modulus G′ (ω = 6.28 rad s−1) was observed in the presence of CO2. The sol-to-gel transition was determined by the frequency-sweep test. Generally, G′ > G′′ indicated a solid-like response while G′ < G′′ indicated a liquid-like response. Meanwhile, the frequency dependence was mainly attributed to the relative weak physical interaction of the supramolecular network. It was noteworthy that the 35 mM SDS–TMPDA solution still possessed a predominantly viscous nature after bubbling CO2 (Fig. S6†). The G′ and G′′ of the SDS–TMPDA solution before bubbling CO2 were beyond the detection limit of the instrument.
Reversibility of CO2-induced gelation
As a green trigger, CO2 can lead to many switching cycles without the accumulation of byproducts. Thus, the reversible switchability in rheological behaviors was evaluated next. As shown in Fig. 5a, the zero-shear viscosity of the hydrogel could be repeatedly and reversibly switched upon alternately bubbling CO2 and N2. For more than four cycles, the change in the zero-shear viscosity was detected without any deterioration. Referring to the study of the research group of Yujun Feng, this finding could be explained that releasing CO2 from the hydrogel by bubbling N2 at 70 °C reconverted TMPDA to its initial non-ionic state, resulting in the collapse of wormlike micelles and the following destruction of the physically cross-linked network. On the other hand, Fig. 5b showed variation of the G′ and G′′ values of the invertible hydrogel during four cycles, further proving the perfect switchable ability.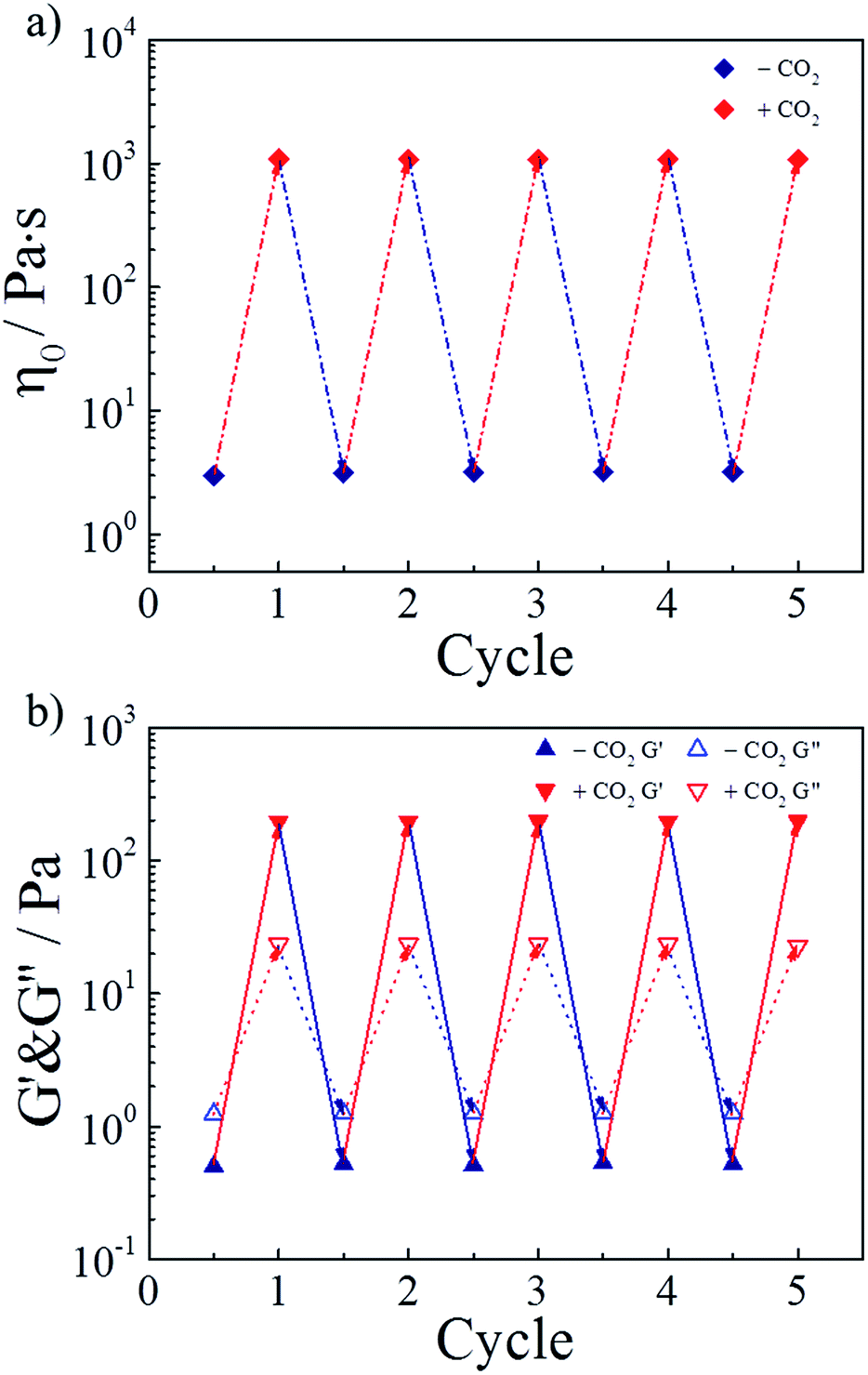 | ||
| Fig. 5 Reversible switchability of (a) zero-shear viscosity and (b) G′ and G′′ (ω = 6.28 rad s−1) for the representative sample upon alternate treatment of CO2 and N2. | ||
Self-healing study
Polymeric hydrogels capable of autonomous healing upon damage have numerous potential applications such as in drug delivery systems, artificial molecular tissues and oil industries.33–38 SDS–TMPDA wormlike micelles not only endowed the HMPAM hydrogel with CO2-responsive properties but also the fascinating self-healing ability. It could be seen that the CO2-induced hydrogel sample merged into a whole without any cracks, after the equilibration in a sealed vial for 24 h, from the photograph in Fig. 4a. It was also observed that a large amount of cracks still appeared in the initial HMPAM hydrogel. Therefore, the self-healing property of the representative hydrogel sample was studied by following tests.The self-healing ability was firstly demonstrated visually in Fig. 6a. Two pieces of hydrogel which were stained with different colors could self-repair into one integral piece within 60 s. And the healed hydrogel could support its own weight.
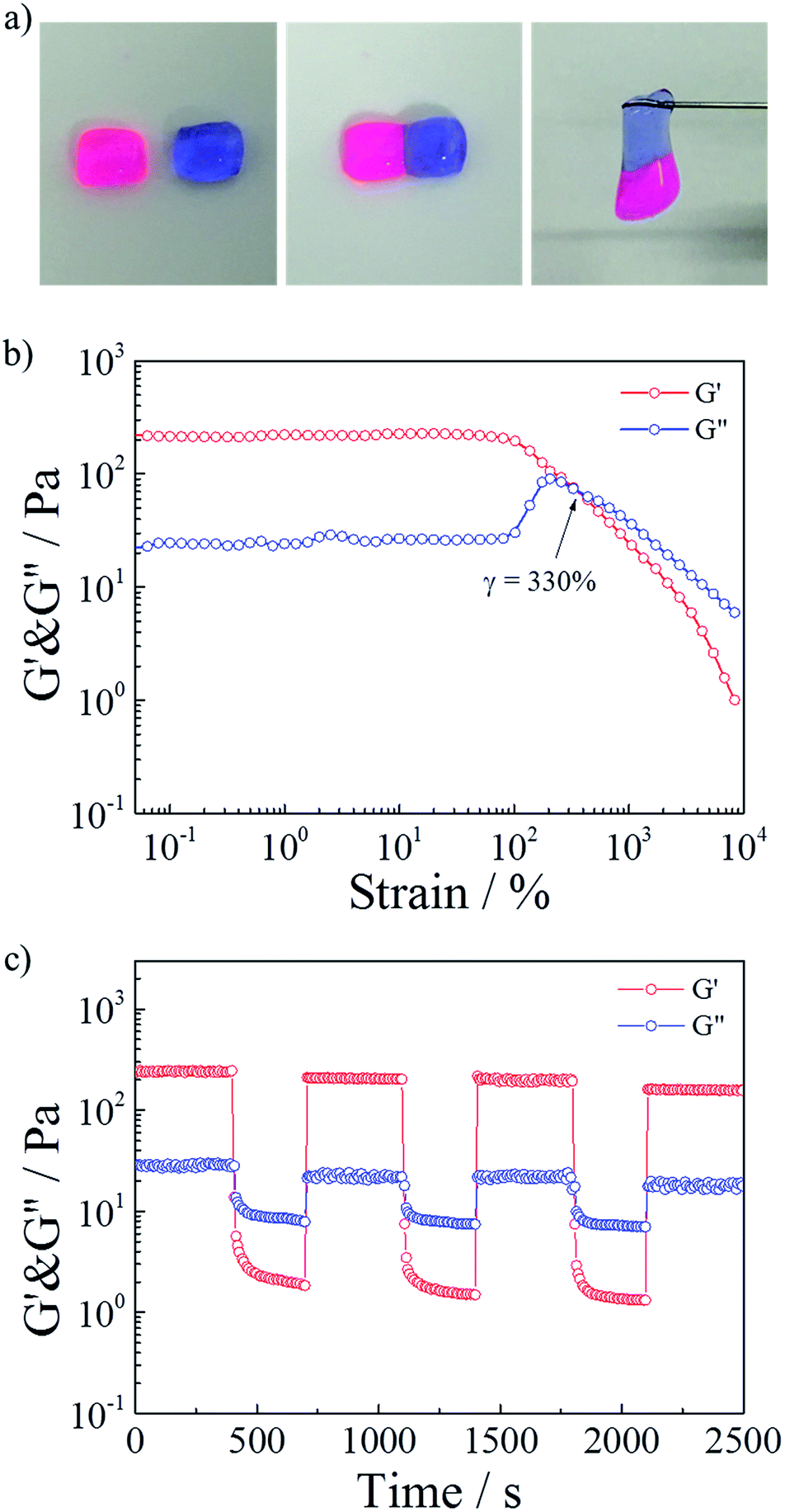 | ||
| Fig. 6 (a) Two pieces of hydrogel, one was stained with Rhodamine B (red dye), one was stained with Indigo carmine (blue dye). The hydrogel were brought together for 60 s, and the healed hydrogel could support its own weight. (b) Storage modulus G′ and loss modulus G′′ of a hydrogel as a function of strain. (c) Repeated dynamic strain step tests (γ = 1% or 3500%). | ||
Then a strain sweep measurement was conducted on the presentative sample to test the influence of the strain. As depicted in Fig. 6b, G′ was larger than G′′ (elastic-dominating) under small strain (γ < 100%). And the values of G′ and G′′ kept constant, suggesting that the hydrogel network remained unaffected due to the intact cross-linkages. While the strain kept increasing, a gel to sol transition point (γ = 330%) occurred, implying that the hydrogel network was destroyed due to the disassociation of the cross-linkages at a high deformation strain.
Finally, repeated dynamic strain step tests (γ = 1% or 3500%) were carried out in Fig. 6c. It could be seen that a 3500% strain could completely inverted the G′ and G′′ values, indicative of the deconstruction of the network. By returning the strain to 1%, G′ and G′′ recovered their original values rapidly, indicating the quick recovery of the inner network of the hydrogel. During the cyclic tests, this recovery behavior was significantly reversible. When SDS–TMPDA wormlike micelles were present, the hydrophobic moieties of the network chains could be solubilized and dynamically cross-linked.39,40 After fracture, the hydrophobic units reversibly disengaged from the associations and easily found their partners in the other cut surface due to the hydrophobic interactions together with the help of the internal dynamics.31,41–43 Therefore, the hydrogel exhibited the significant self-healing property within a short period of time.
Conclusions
In conclusion, a novel CO2-responsive self-healable hydrogel was fabricated by simply mixing HMPAM with SDS–TMPDA surfactant micelles in aqueous solution. After bubbling CO2, wormlike micelles formed, serving as multivalent cross-linkages to bridge HMPAM chains based on the hydrophobic interaction. The interpenetrating three-dimensional network induced a sol-to-gel transition. Then removing CO2 could reconvert the hydrogel to its initial sol state. The sol–gel transition was repeatedly and reversibly switched by cyclically bubbling and removing CO2 without any harm. In addition, the CO2-responsive hydrogel exhibited significant shear-thinning and self-healing properties, which allowed it to withstand repeated deformation and quickly recover its mechanical properties and structure. Therefore, we believe that the presented work should undoubtedly offer a universal and simple strategy to design CO2-responsive self-healable hydrogels and provide new opportunities with regard to their practical applications such as smart fluids, intelligent delivery systems and a potential CO2 plugging agent for enhanced oil recovery (EOR) performed by CO2 flooding.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 51273189), the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05016), and the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05046).References
- M. AngeláAleman-Garcia, Chem. Sci., 2015, 6, 4190–4195 RSC.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed.
- W. Xu, X. He, M. Zhong, X. Hu and Y. Xiao, RSC Adv., 2015, 5, 3157–3167 RSC.
- K. Shi, Z. Liu, Y.-Y. Wei, W. Wang, X.-J. Ju, R. Xie and L.-Y. Chu, ACS Appl. Mater. Interfaces, 2015, 7, 27289–27298 CAS.
- Y.-Y. Xiao, X.-L. Gong, Y. Kang, Z.-C. Jiang, S. Zhang and B.-J. Li, Chem. Commun., 2016, 52, 10609–10612 RSC.
- D. Han, O. Boissiere, S. Kumar, X. Tong, L. Tremblay and Y. Zhao, Macromolecules, 2012, 45, 7440–7445 CrossRef CAS.
- Z. Guo, Y. Feng, Y. Wang, J. Wang, Y. Wu and Y. Zhang, Chem. Commun., 2011, 47, 9348–9350 RSC.
- D. M. Rudkevich and H. Xu, Chem. Commun., 2005, 2651–2659 RSC.
- L. Zhang, J. Qian, Y. Fan, W. Feng, Z. Tao and H. Yang, RSC Adv., 2015, 5, 62229–62234 RSC.
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Angew. Chem., 2015, 127, 7484–7488 CrossRef.
- L. Yu, W. Fu and Z. Li, Soft Matter, 2015, 11, 545–550 RSC.
- Z. Sun, F. Lv, L. Cao, L. Liu, Y. Zhang and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 7944–7948 CrossRef CAS PubMed.
- Z. Tao, K. Peng, Y. Fan, Y. Liu and H. Yang, Polym. Chem., 2016, 7, 1405–1412 RSC.
- B. P. Lee and S. Konst, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- Y. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958–960 CrossRef CAS PubMed.
- D. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2011, 1, 57–61 CrossRef.
- S. Lin and P. Theato, Macromol. Rapid Commun., 2013, 34, 1118–1133 CrossRef CAS PubMed.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., 2011, 123, 5025–5029 CrossRef.
- S. Kumar, X. Tong, Y. L. Dory, M. Lepage and Y. Zhao, Chem. Commun., 2013, 49, 90–92 RSC.
- V. Fischer, K. Landfester and R. Munoz-Espi, ACS Macro Lett., 2012, 1, 1371–1374 CrossRef CAS.
- H. Liu, S. Lin, Y. Feng and P. Theato, Polym. Chem., 2017, 8, 12–23 RSC.
- G. Yuncong, Z. Mifu, W. Jianbo and Z. Chang, Pet. Explor. Dev., 2014, 41, 88–95 CrossRef.
- D.-X. Li, L. Zhang, Y.-M. Liu, W.-L. Kang and S.-R. Ren, Pet. Sci., 2016, 13, 247–258 CrossRef CAS.
- Y. Zhang, Z. Chu, C. A. Dreiss, Y. Wang, C. Fei and Y. Feng, Soft Matter, 2013, 9, 6217–6221 RSC.
- Y. Zhang, Y. Feng, J. Wang, S. He, Z. Guo, Z. Chu and C. A. Dreiss, Chem. Commun., 2013, 49, 4902–4904 RSC.
- Y. Zhang, Y. Feng, Y. Wang and X. Li, Langmuir, 2013, 29, 4187–4192 CrossRef CAS PubMed.
- X. Hao, H. Liu, Y. Xie, C. Fang and H. Yang, Colloid Polym. Sci., 2013, 291, 1749–1758 CAS.
- X. Hao, H. Liu, Z. Lu, Y. Xie and H. Yang, J. Mater. Chem. A, 2013, 1, 6920–6927 CAS.
- F. Candau and J. Selb, Adv. Colloid Interface Sci., 1999, 79, 149–172 CrossRef CAS.
- D. C. Tuncaboylu, A. Argun, M. Sahin, M. Sari and O. Okay, Polymer, 2012, 53, 5513–5522 CrossRef CAS.
- E. K. Penott-Chang, L. Gouveia, I. J. Fernández, A. J. Müller, A. Díaz-Barrios and A. E. Saez, Colloids Surf., A, 2007, 295, 99–106 CrossRef CAS.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- U. Gulyuz and O. Okay, Soft Matter, 2013, 9, 10287–10293 RSC.
- H. Meng, P. Xiao, J. Gu, X. Wen, J. Xu, C. Zhao, J. Zhang and T. Chen, Chem. Commun., 2014, 50, 12277–12280 RSC.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- H. Liu, C. Xiong, Z. Tao, Y. Fan, X. Tang and H. Yang, RSC Adv., 2015, 5, 33083–33088 RSC.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- D. C. Tuncaboylu, M. Sahin, A. Argun, W. Oppermann and O. Okay, Macromolecules, 2012, 45, 1991–2000 CrossRef CAS.
- A. Argun, M. P. Algi, D. C. Tuncaboylu and O. Okay, Colloid Polym. Sci., 2014, 292, 511–517 CAS.
- U. Gulyuz and O. Okay, Macromolecules, 2014, 47, 6889–6899 CrossRef CAS.
- G. Akay, A. Hassan-Raeisi, D. C. Tuncaboylu, N. Orakdogen, S. Abdurrahmanoglu, W. Oppermann and O. Okay, Soft Matter, 2013, 9, 2254–2261 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06418g |
| This journal is © The Royal Society of Chemistry 2017 |