
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P450 monooxygenase ComJ catalyses side chain phenolic cross-coupling during complestatin biosynthesis†
Aurelio Mollo‡
 a,
A. Nikolai von Krusenstiern‡a,
Joshua A. Bulosa,
Veronika Ulrichb,
Karin S. Åkerfeldta,
Max J. Cryle*bcd and
Louise K. Charkoudian*a
a,
A. Nikolai von Krusenstiern‡a,
Joshua A. Bulosa,
Veronika Ulrichb,
Karin S. Åkerfeldta,
Max J. Cryle*bcd and
Louise K. Charkoudian*a
aDepartment of Chemistry, Haverford College, Haverford, PA 19041, USA. E-mail: lcharkou@haverford.edu
bDepartment of Biomolecular Mechanisms, Max Planck Institute for Medical Research, Jahnstrasse 29, 69121 Heidelberg, Germany. E-mail: max.cryle@monash.edu
cEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
dThe Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology, ARC Centre of Excellence in Advanced Molecular Imaging, Monash University, Clayton, Victoria 3800, Australia
First published on 12th July 2017
Abstract
Complestatin is a non-ribosomal heptapeptide belonging to the glycopeptide antibiotic family that displays anti-complement, anti-HIV integrase, neuroprotective, anti-apoptotic, and antibacterial activities. Development of complestatin as a pharmaceutical agent and biological probe has been stymied by the difficulty in isolating the molecule from its natural source, Streptomyces lavendulae, as well as by the uneconomical and environmentally unfriendly nature of current total synthetic routes. Of particular challenge to the organic chemist is the installation of the biaryl and aryl–ether–aryl linkages that provide complestatin with the structural rigidity responsible for its potent pharmacological properties. Herein, we demonstrate that ComJ, a P450 monooxygenase from the complestatin biosynthetic gene cluster, can catalyse phenolic cross-linking of amino acid side chains in vitro. ComJ acts with high efficiency and low substrate stereoselectivity, a finding which paves the way towards the use of ComJ as a biocatalyst for the chemo-enzymatic synthesis of complestatin and other related molecules. The ability of ComJ to accept peptides of alternative stereochemistries raises intriguing questions about the evolutionary origins of glycopeptide antibiotic biosynthesis and the capacity of S. lavendulae to produce different conformations of complestatin.
1. Introduction
Glycopeptide antibiotics (GPAs) are an important class of non-ribosomal peptides (NRPs) used to treat Gram-positive bacterial infections.1 They are assembled independently of the ribosome by large enzyme complexes known as non-ribosomal peptide synthetases (NRPSs). NRPs are typically assembled in an N-to-C fashion, with each amino acid incorporated by its own distinct module comprised of multiple catalytic domains (Fig. 1). The “core” domains include: (1) the adenylation (A) domain, responsible for selecting the correct amino acid for incorporation and activating it as a mixed anhydride in an ATP-dependent reaction; (2) the peptidyl carrier protein (PCP) domain, responsible for nucleophilic attack of the A domain-activated amino acid via its post-translationally-introduced phosphopantetheine (Ppant) arm, resulting in thioesterification of the amino acid and freeing the A domain to activate another amino acid; and (3) the condensation (C) domain, responsible for catalysing peptide bond formation between the newly formed aminoacyl-PCP and an upstream peptidyl-PCP, thus increasing the length of the peptide chain by one amino acid.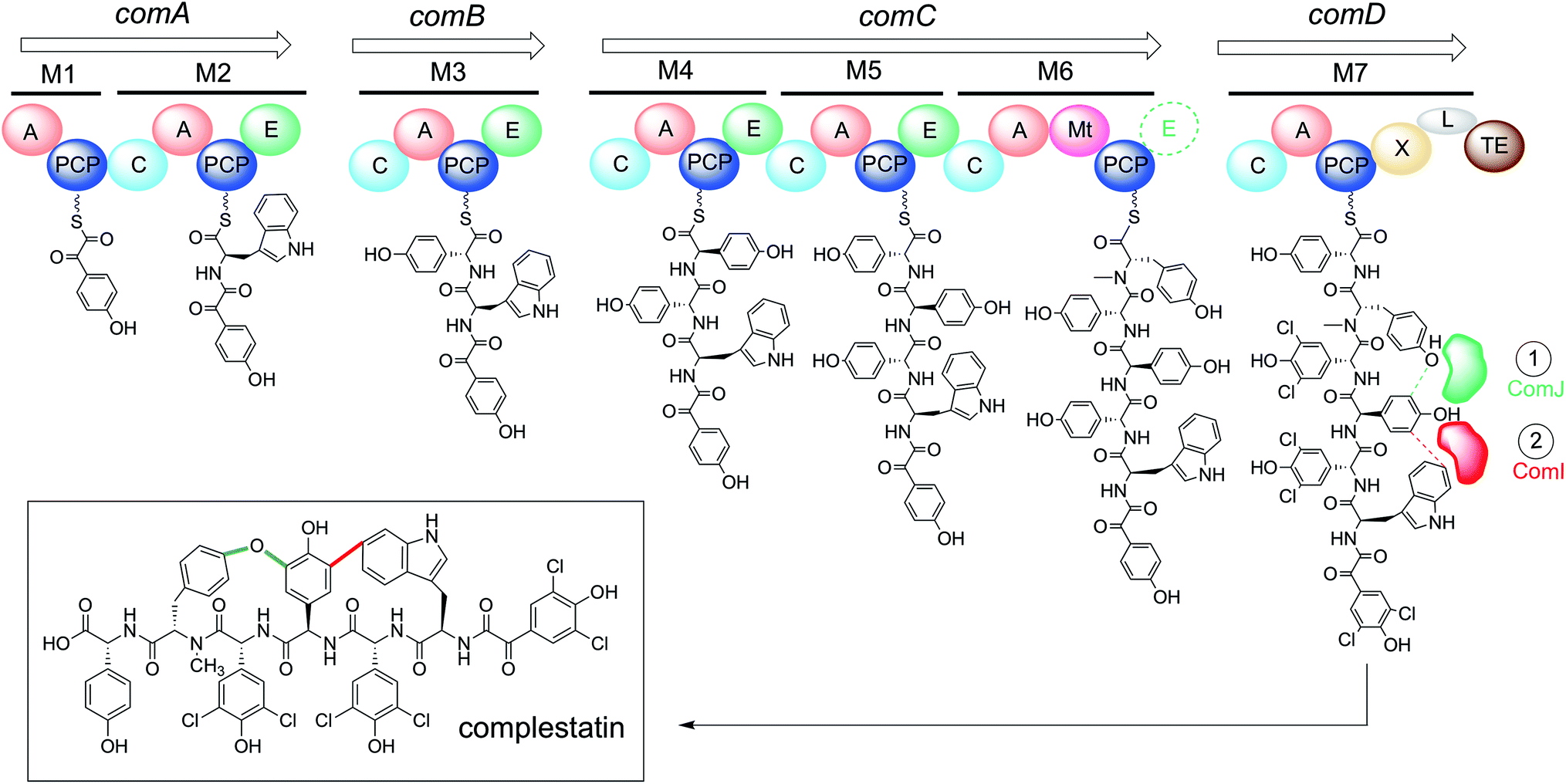 | ||
| Fig. 1 The complestatin NRPS is composed of 29 distinct domains distributed across four proteins (ComA–D). The domains are arranged into seven functional modules (M1–M7), each responsible for the incorporation of one amino acid into the growing chain; putative intermediates found at each module are shown tethered to their cognate carrier protein domains. When the growing chain reaches the terminal module as an α-ketoacyl hexapeptide, it is chlorinated by in trans-acting halogenases (not shown),35 then cyclised by the in trans-acting P450s ComI and ComJ to yield fully active complestatin (boxed). Bioinformatic analysis suggests that M6 contains an additional E domain (dashed) that was overlooked in the original annotation (see text):32 the structure of complestatin is shown as published, but the stereochemistry of the tyrosine residue remains unclear. A = adenylation domain; PCP = peptidyl carrier protein domain; C = condensation domain; E = epimerisation domain; Mt = N-methyltransferase domain; L = extended linker domain; TE = thioesterase domain; M = module. | ||
Among NRPs, GPAs are unique in that they are characterised by a high percentage of non-proteinogenic amino acids as well as synthetically challenging biaryl and biaryl ether linkages.1,2 These unusual cross-links, absent from traditional ribosome-assembled peptides, play important roles in rigidifying the peptide scaffold into potent bioactive conformations. In vivo, the biaryl and biaryl ether linkages are installed stereo- and regioselectively by dedicated P450 monooxygenases (known as “Oxy enzymes”),3 which are recruited to the terminal module of the NRPS by the adjacent X-domain, a conserved domain present in the terminal module of all GPA NRPSs.4 Recent results indicate that the Oxy enzymes undergo a continuous association/dissociation process with the X-domain, only to “lock in” upon recognition of their cognate substrate and thus catalysing side chain cross-linking in a defined order.5 Due to their potential use as biocatalysts, Oxy enzymes from many different GPA systems have been structurally and biochemically characterised to date.6–11 In the future, these enzymes could prove to be useful tools for enabling access to novel structural and chemical diversity, including molecules that contain biaryl motifs that would otherwise be challenging to synthesise via classical approaches.12
Complestatin (Fig. 1) is an atypical GPA isolated in the early 1980s from the soil bacterium Streptomyces lavendulae.13 It possesses a remarkable range of pharmacological properties, including anti-complement,13 anti-HIV integrase,14,15 neuroprotective,16,17 anti-apoptotic,18 and antibacterial19 effects. Because of the presence of an aryl–ether–aryl–aryl moiety cross-linking three of its amino acid side chains, total synthetic approaches towards complestatin remain arduous. Current routes in fact require in excess of 20 steps and involve the use of large amounts of metal catalysts, together rendering these routes economically and environmentally unsustainable for industrial scale productions.2,20–22 A greener, more efficient route towards the synthesis of this molecule and its analogues, which may be used as biological probes or as lead compounds in drug development, is greatly needed.
Complestatin's regulatory and biosynthetic gene cluster (BGC) was first sequenced and annotated in 2001. At the time it was inferred based on homology that two enzymes, called ComI and ComJ, are involved in cyclising the linear heptapeptide to yield fully active complestatin (Fig. 1).23 More recently, it was shown through gene disruption studies and subsequent analysis of cell extracts that ComI and ComJ are indeed responsible for installing the biaryl and phenolic bridges, respectively, with ComJ acting prior to ComI.24 In spite of these advances, however, ComI and ComJ have not been shown to be active in vitro, a result critical to further developing these enzymes into useful biocatalysts. Previous work in the field has suggested that an environmentally friendly route might involve the synthesis of the linear form of complestatin, followed by P450-mediated cross-linking,4,25 thus exploiting the remarkable biosynthetic capability of P450 enzymes to install the synthetically-challenging crosslinks. Towards this goal, we herein demonstrate that ComJ is an active P450 enzyme that catalyses aryl–ether–aryl bond formation in vitro with high efficiency and low substrate stereoselectivity, defining it as a potential biocatalyst for the chemo-enzymatic reconstitution of complestatin and related molecules.
2. Materials and methods
2.1 Chemicals and reagents
All chemicals were purchased from commercial vendors and used without further purification. Solvents used for solid-phase peptide synthesis (SPPS) were purchased as anhydrous reagents. All resins used were purchased from CreoSalus (Advanced ChemTech). Enzymes for molecular cloning work were obtained from New England BioLabs. NMR spectra were recorded on a Varian (Agilent) 500 MHz NMR Spectrometer. Turnover mixtures were analysed using an Agilent 6545 Q-TOF LC/MS.2.2 Cloning
Expression plasmids for ComI, ComJ, ComK (pET28a) and the complestatin NRPS-specific PCP7-Xcom protein (pET24b)26 were constructed via Gibson Assembly using template DNA from S. lavendulae. PCP7com and Xcom were cloned from a synthetic gene optimised for E. coli expression (Eurofins MWG, Ebersberg, Germany) using specific primers; PCP7com and Xcom were then cloned into a modified MBP-containing pET24b vector26 using restriction digestion (NcoI/XhoI) and subsequent ligation as described previously.4,27 See ESI for details.†2.3 Protein expression and purification
All proteins used in this paper were designed to exhibit an N-terminal His6-tag and expressed in BL21(DE3) competent cells. Cultures were grown in LB medium§ at 37 °C, 225 RPM, to an OD of 0.6–0.8 before protein production was induced with 0.25 mM IPTG; the cultures were subsequently incubated overnight (12–16 h) at 18 °C, 100 RPM. In the case of the P450 enzymes (ComI, ComJ), the production culture was supplemented with δ-aminolevulinic acid (0.5 mM) to promote formation of the heme moiety, while for ferredoxins (SpFd, ComK) the culture was supplemented with 50 μM FeSO4 to facilitate the assembly of the [2Fe–2S] cluster. Following harvesting of the production cultures (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 × g, 8 min, 4 °C), the cells were resuspended in lysis buffer (50 mM phosphate pH 7.6, 300 mM NaCl, 10 mM imidazole) and lysed by sonication (8 × 15 s pulses with 30 s rest in between, employing a Misonix Microson XL-2000 Ultrasonic Liquid Processor, Power Setting: 7). The lysates were centrifuged (25000 × g, 50 min, 4 °C) and the supernatant incubated with 2–3 mL of pre-equilibrated Ni-NTA resin (GoldBio) for 2–3 h, then transferred to a large Econo-Pac® column (Bio-Rad). The resin was washed twice (50 mM phosphate pH 7.6, 300 mM NaCl, 30 mM imidazole) and the protein subsequently eluted (50 mM phosphate pH 7.6, 300 mM NaCl, 300 mM imidazole). The identity and purity of the proteins were confirmed by SDS-PAGE (Fig. S1†) and the concentrations measured via sequence-based 280 nm extinction coefficients. After dialysis (Slide-A-Lyzer™ Dialysis Cassette, Thermo Fisher Scientific) into storage buffer (50 mM phosphate pH 7.6, 10% glycerol), all proteins were aliquoted, flash-frozen in liquid N2, and stored at −80 °C (see Fig. S1† for titers).
300 × g, 8 min, 4 °C), the cells were resuspended in lysis buffer (50 mM phosphate pH 7.6, 300 mM NaCl, 10 mM imidazole) and lysed by sonication (8 × 15 s pulses with 30 s rest in between, employing a Misonix Microson XL-2000 Ultrasonic Liquid Processor, Power Setting: 7). The lysates were centrifuged (25000 × g, 50 min, 4 °C) and the supernatant incubated with 2–3 mL of pre-equilibrated Ni-NTA resin (GoldBio) for 2–3 h, then transferred to a large Econo-Pac® column (Bio-Rad). The resin was washed twice (50 mM phosphate pH 7.6, 300 mM NaCl, 30 mM imidazole) and the protein subsequently eluted (50 mM phosphate pH 7.6, 300 mM NaCl, 300 mM imidazole). The identity and purity of the proteins were confirmed by SDS-PAGE (Fig. S1†) and the concentrations measured via sequence-based 280 nm extinction coefficients. After dialysis (Slide-A-Lyzer™ Dialysis Cassette, Thermo Fisher Scientific) into storage buffer (50 mM phosphate pH 7.6, 10% glycerol), all proteins were aliquoted, flash-frozen in liquid N2, and stored at −80 °C (see Fig. S1† for titers).
In preparation for spectral assays, ComI and ComJ were further purified in a two-step process consisting of anion exchange chromatography (AEC) and size-exclusion chromatography (SEC). Initially, the proteins were buffer exchanged with AEC buffer A (50 mM Tris–HCl pH 7.4, 20 mM NaCl) using Illustra NAP-25 columns (GE Healthcare, Chalfont St Giles, UK) and then concentrated using Vivaspin® centrifugal concentrators (30000 MWCO; Sartorius, Göttingen, Germany). AEC was performed using a 6 mL Resource™ Q column (GE 17–1179) equilibrated with AEC buffer A and connected to an Äkta purifier 10 system (GE) at RT. The protein was applied onto the column, which was then washed with 5 column volumes (CV) of AEC buffer A, before the protein was eluted over a gradient from 0 to 100% AEC buffer B (50 mM Tris–HCl pH 7.4, 1 M NaCl) over 20 CV. The elution fractions were analysed by SDS-PAGE, with relevant fractions pooled and concentrated prior to SEC. For SEC, protein was loaded onto a Superose 12300 mL column (GE 17-5173) equilibrated using SEC buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl) and connected to an Äkta purifier 10 system at RT. The protein was eluted using 50 mM Tris–HCl pH 7.4, 150 mM NaCl, and analysed by SDS-PAGE. Relevant fractions were pooled, and the protein portioned into aliquots prior to being flash-frozen in liquid N2 and stored at −80 °C.
2.4 Spectral assays
Spectral analysis of ComI and ComJ was performed using a V-650 UV-vis spectrophotometer (JASCO Germany GmbH; Groß-Umstadt, Germany) and the SpectraManager™ software. Data was recorded (1 cm pathlength) using 2.5 μM cytochrome P450 in 50 mM Tris–HCl buffer pH 8.0 at 30 °C from λ = 390–600 nm in 0.2 nm increments for the ferric protein (as purified) as well as for the protein that had been reduced and saturated with carbon monoxide (ferrous-CO). The protein was reduced through the addition of 10 μL of a saturated Na2S2O4 solution. Directly after protein reduction, saturation with carbon monoxide was achieved through bubbling of 60 mL of carbon monoxide gas through the cuvette filled with protein solution via a syringe.2.5 Analytical size-exclusion chromatography
Experiments were performed with 33.3 μM of cytochrome P450 incubated with 100 μM of the NRPS construct of interest in SEC buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl) in a volume of 100 μL at RT for 30 min. This reaction mixture was analysed by SEC using SEC buffer and a flow rate set at 1 mL min−1 and a Superose 12 10/300 GL column (GE 17-5173) connected to an Äkta purifier 10 system controlled by UNICORN 5.20 AB software (GE Healthcare, Chalfont St Giles, UK). Data were analysed using GraphPad Prism 6. For all cases, individual proteins were analyzed as controls.2.6 Solid-phase peptide synthesis
The target tripeptides (Ac-NH-D-Hpg-D-Hpg-L-Tyr-OH = 1, Ac-NH-D-Hpg-D-Hpg-D-Tyr-OH = 2) were constructed on Wang Resin on a 0.2 mmol scale using the Fmoc/tBu-based SPPS approach developed in previous work.28 The starting material for 1 was L-Tyr-loaded Wang resin, whereas the starting material for 2 was “free” Wang resin.¶ For the synthesis of 2, Wang resin was loaded overnight at ambient temperature using 2.5 eq. of amino acid (Fmoc-NH-D-Tyr(tBu)-OH) in the presence of 5 eq. of the coupling agent N,N′-diisopropylcarbodiimide (DIC), 0.1 eq. of the catalytic base 4-dimethylaminopyridine (DMAP), 2.5 eq. of the racemisation inhibitor hydroxybenzotriazole (HOBt), all in 9:1 N,N-dimethylformamide (DMF): dichloromethane (DCM). Subsequent amide coupling reactions were performed over 40 min at ambient temperature in 8 mL of a 0.1 M triethylamine (TEA) solution in DMF containing 4 eq. of amino acid, 4 eq. of DIC, and 8 eq. of HOBt. Deprotection reactions of the Fmoc protecting group were performed over 2 min with 8 mL of 1% v/v 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in DMF. Finally, to prevent homodimerisation of the peptide during subsequent thioesterification, the reactive N-terminus of the resin-bound, Fmoc-deprotected tripeptide was “capped” with an acetyl group through the addition of 1.1 eq. of acetic anhydride in 8 mL DMF for 1 min. The tripeptide was cleaved from the resin via a 2.5 h incubation with 9 mL trifluoroacetic acid, 0.5 mL thioanisole, 0.3 mL 1,2-ethanedithiol, 0.2 mL anisole, and analysed by NMR spectroscopy (see Fig. S7 and S8†).
2.7 Priming the peptide for PCP loading
In order to prepare the peptide for loading onto PCP7-Xcom, the peptide was first converted to a thiophenol thioester by reacting the cleaved peptide (20 mg) for 40 min with 5 eq. of thiophenol in the presence of 1.2 eq. of (benzotriazol-1-yl-oxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) and 1.2 eq. of N,N-diisopropylethylamine (DIPEA) in 1 mL DMF. Following isolation of the thiophenol conjugate via acetonitrile (ACN) precipitation and centrifugation (see Fig. S9 and S10† for NMR data), the compound (10 mg) was transthioesterified with coenzyme A (CoA, trilithium salt, Sigma Aldrich) via a 2–3 h incubation with 0.875 eq. of CoA in 4 mL of a 2:1 H2O:ACN mixture (50 mM phosphate pH 8.30), and the product purified via solid phase extraction (Strata-X polymeric reversed phase; Phenomenex, Torrance, CA) as detailed elsewhere29 (for NMR data, see Fig. S11 and S12;† for MALDI data, see Fig. S13 and S14†).
2.8 Turnover assays
A PCP stock solution was dialysed overnight at 4 °C against 1 L reaction buffer (50 mM NaCl, 50 mM HEPES, pH 7.00). Each loading reaction (350 μL) consisted of the following: 60 μM PCP, 240 μM peptidyl-CoA, 50 mM MgCl2 and 6 μM Sfp R4-4 in reaction buffer. The loading reactions were incubated for 1 h at 30 °C with gentle shaking (150 RPM), after which they were transferred to pre-equilibrated centrifugal filters (30000 MWCO, Vivaspin® by Sartorius) and the excess of peptidyl-CoA was removed by a concentration dilution (4 × 1:5 dilution) procedure using reaction buffer. The buffer-exchanged peptidyl-PCP was used to set up turnover reactions (210 μL) consisting of: 50 μM peptidyl-PCP, 8 μM Oxy (OxyBvan/ComJ), 10 μM E. coli flavodoxin reductase, 20 μM ferredoxin (SpFd/ComK), 2 mM glucose-6-phosphate, 1 U glucose-6-phosphate dehydrogenase, 2 mM NADPH, all in reaction buffer. The turnover mixtures were incubated for 1 h at 30 °C, 300 RPM, with the caps left open to provide O2 as a reactant. The contents of each reaction tube were then separated into 2 × 105 μL portions, to each of which 15 μL of 40% v/v MeNH2/H2O were added (23000 molar excesses over peptide), followed by a further incubation of 15 min at 30 °C, 300 RPM. Each 120 μL mixture was diluted to 500 μL with H2O, then to 1 mL with 1:50 formic acid (FA):H2O. The mixture was added to an SPE column that had been pre-activated with 1 mL MeOH and pre-equilibrated with 1 mL H2O. The columns were washed with 1 mL 5% MeOH/H2O and the turnover species finally eluted with 500 μL 1% FA/MeOH. The combined elutes were concentrated down to ∼100 μL using a SpeedVac concentrator and analysed by LC/MS with detection in negative mode and simultaneous measurement of 205 nm absorbance (see ESI for details†). The UV and extracted ion count (EIC) data was exported into Prism 7 for visualisation and analysis. The extent of conversion of linear peptide into cyclised product was estimated by quantifying the per cent decrease in 205 nm absorbance of the reactant starting material. All experiments were carried out in triplicate and the difference in means determined by the t-test (parametric and unpaired, no Welch's correction).
3. Results and discussion
3.1 Spectral assays
In order to investigate the potential catalytic competence of ComI and ComJ, a spectral assay was initially performed. The UV-visible spectra of the two proteins presented the following features: a Soret maximum at λmax = 418 nm and β/α bands at λmax = 537 and 572 nm in the case of ComJ; and a Soret maximum at λmax = 418 nm and β/α bands at λmax = 535 and 570 nm in the case of ComI (Fig. 2). These spectra correspond to the characteristic absorption spectra of cytochrome P450 enzymes in the low-spin resting state, with the heme iron in its water-bound ferric form.30 Reduction of the proteins using Na2S2O4 resulted in the reduction of ferric to ferrous heme; subsequent saturation of the protein solution with carbon monoxide (CO) led to the displacement of the water ligand by CO and significant changes in the UV-vis absorption spectra (Fig. 2). In particular, the appearance of a major peak at approximately λmax = 450 nm in the spectrum of each protein suggested that the axial cysteine ligand was present mostly in its deprotonated thiolate state, indicating that ComI and ComJ were catalytically competent cytochrome P450s.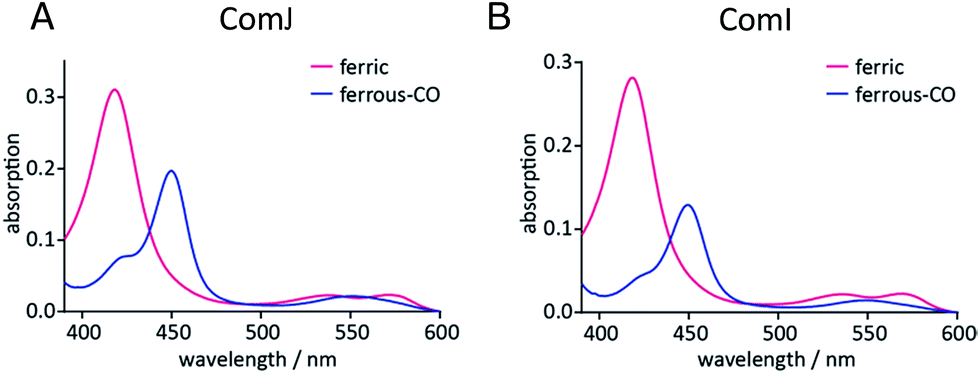 | ||
| Fig. 2 Spectral analyses of (A) ComJ and (B) ComI. The absorption spectra were recorded for ferric (magenta) and ferrous-CO (blue) hemes from 390–600 nm. The appearance of a ∼450 nm peak in the ferrous-CO spectrum of both proteins suggests they are catalytically competent cytochrome P450s. | ||
We next evaluated whether ComI and ComJ interact with PCP domains, X-domains, and/or PCP-X didomains from the 7th module of the complestatin NRPS, thus opening up the possibility of a biomimetic route to synthesis of the molecule.
Monomeric Xcom (X-domain from M7 of complestatin NRPS; see Fig. 1), PCP7com (PCP domain from M7 of complestatin NRPS; see Fig. 1), and heterodimeric PCP7-Xcom constructs were designed and cloned into modified pET vectors, containing maltose-binding protein (MBP) to enhance solubility and expression titers, as detailed in Section 2.2.
Protein–protein interactions were next evaluated by analytical size-exclusion chromatography (SEC) with simultaneous monitoring of the absorption at λ = 280 nm for protein absorption and at λ = 415 nm for the heme-specific (P450) absorption. The results (Fig. 3) indicate that ComI and ComJ both interact tightly with MBP-PCP7-Xcom. In addition to the above-mentioned interaction, both P450s exhibited tight interaction with monomeric Xcom constructs (Fig. S2†) but not with monomeric PCP7com constructs (Fig. S3†). Collectively, these data suggest that interaction of ComI and ComJ with the NRPS assembly line in vivo occurs primarily through the X-domain.5
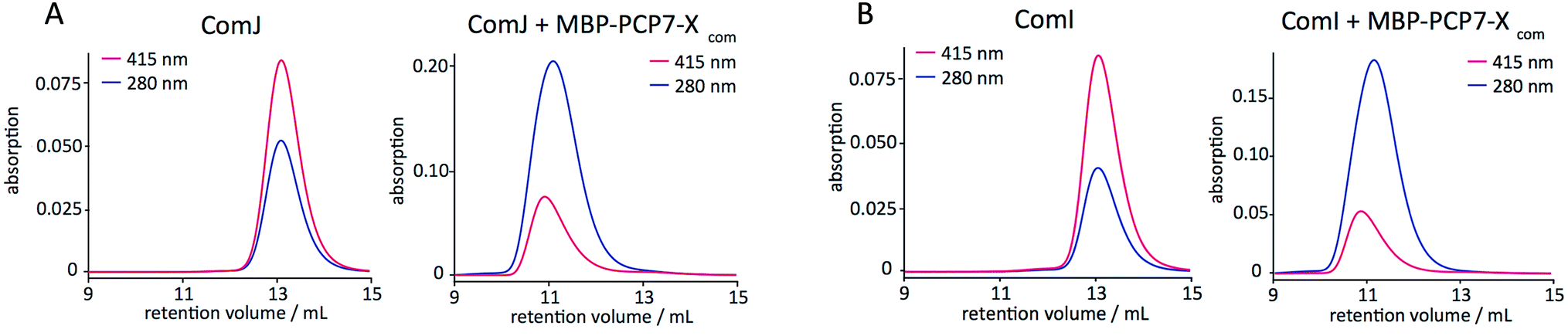 | ||
| Fig. 3 SEC traces for (A) ComJ and (B) ComI, alone (left) and in the presence of MBP-PCP7-Xcom (right). Absorptions at λ = 280 nm (blue; protein-specific) and at λ = 415 nm (magenta; heme-specific) were monitored simultaneously. A shift towards smaller retention volumes is evident upon addition of MBP-PCP7-Xcom to either P450, indicative of complexation. [P450] 33.3 μM, [PCP-X] 100 μM. | ||
Having established the ability of the catalytically competent P450s to dock onto the PCP7-Xcom didomain, we proceeded to testing the ability of ComJ to catalyse a cyclisation reaction (Fig. 4). Previous research by the Robinson Lab found that OxyBvan, the ComJ homologue from the vancomycin biosynthetic pathway, was able to successfully cyclise a tripeptide substrate in vitro representing the core of the cognate heptapeptide.31 Based on this precedent, we investigated the ability of ComJ to cyclise a tripeptide substrate representing the core of the complestatin heptapeptide (Fig. 4).
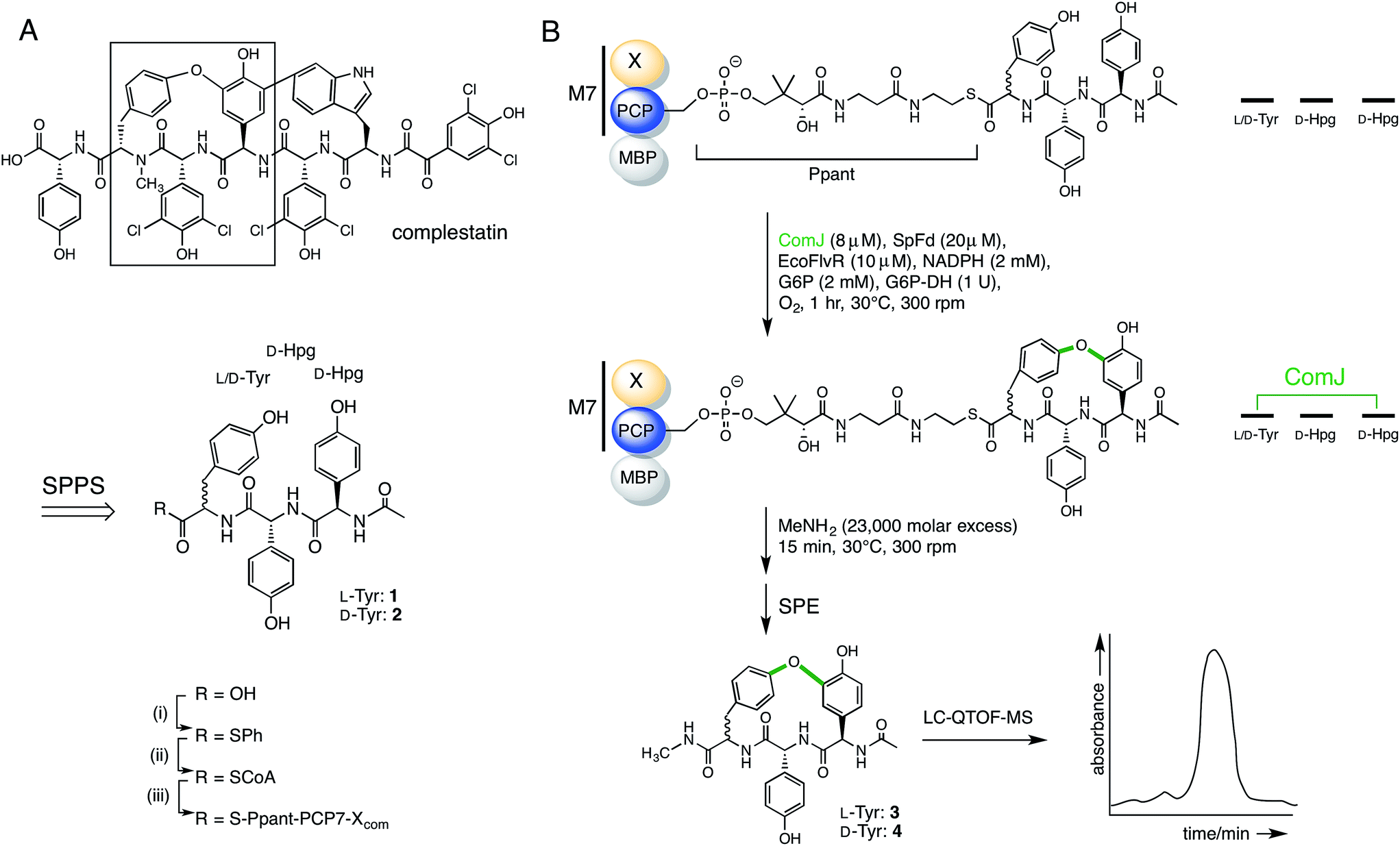 | ||
| Fig. 4 Summary of the reaction flow from starting linear peptide to cyclised product. (A) Complestatin is a bismacrocyclic heptapeptide. Each ring is installed by a separate P450 enzyme (see Fig. 1). Inspired by the left-hand macrocycle, containing a biaryl ether bond (boxed), we constructed epimeric linear tripeptides (1, 2) by solid-phase peptide synthesis (SPPS). Following cleavage from the resin, the peptides (R = OH) were converted to CoA conjugates (R = SCoA) via a thiophenol intermediate (R = SPh), before being loaded onto an MBP fusion construct of the PCP7-X didomain from the complestatin NRPS (R = S-Ppant-PCP7-Xcom; Ppant structure shown in part B). (B) Peptidyl-PCP, shown top, was treated with ComJ and other proteins required to provide a sustained electron flow to the P450 heme. Following biaryl ether bond installation over a 1 h period, the cyclised product was cleaved using methylamine, purified by solid-phase extraction (SPE), and detected by LC-MS. See Section 2.7 for complete procedure. | ||
In addition to testing the cyclisation efficiency of D-Hpg-D-Hpg-L-Tyr (1)—the tripeptide substrate based on the published structure of complestatin—we also evaluated the epimeric tripeptide D-Hpg-D-Hpg-D-Tyr (2). The rationale behind this was two-fold. First, a recent bioinformatics analysis of the complestatin BGC revealed the presence of an additional, predicted epimerisation domain in module 6 of the complestatin NRPS that was not reported in the original annotation32 (Fig. 1), which suggests possible enzymatic conversion of the L-Tyr residue to D-Tyr during biosynthesis. Second, alternative conformations of complestatin and the existence of structurally related natural products from Streptomyces isolates have been reported in the literature.2 Together, these findings inspired us to explore the stereoselectivity of ComJ.
The target peptides were constructed using Fmoc/tBu-based SPPS on the Wang resin. Due to the high susceptibility of Hpg residues to racemisation during peptide synthesis, the Fmoc-deprotection and coupling protocols were optimised to minimise the length and intensity of exposure of the peptides to basic conditions (see Section 2.6).28 The peptides were cleaved from the resin, then converted to their CoA conjugates via a thiophenol intermediate before being loaded onto the PCP7-Xcom didomain in an Sfp-dependent reaction.33 After incubating peptidyl-PCP with the relevant cytochrome P450 enzyme for 1 h at 30 °C, turnover was evaluated via methylamine-assisted cleavage, followed by solid-phase extraction (SPE) and LC-QTOF-MS detection (Fig. 4).
The robustness of the coupled turnover and LC-MS detection protocol was tested by performing a positive control for turnover based on the results of Robinson and co-workers. Thus, the L-Tyr-containing tripeptide (1) was loaded onto MBP-PCP7-Xvan25 and OxyBvan added as the P450. This resulted in 79% turnover (Fig. S4†) in agreement with published results.31
Next, the biocatalytic activity of ComJ was tested by performing turnover of 1 loaded onto MBP-PCP7-Xcom. As shown in Fig. 5, 1 was turned over by ComJ in 48% efficiency, with spinach ferredoxin (SpFd) providing the reducing equivalents to ComJ. This constitutes the first direct observation of ComJ activity in vitro. In addition to turning over 1, ComJ was also found to turn over the D-Tyr-containing tripeptide 2 with a slightly higher efficiency of 60% (P < 0.001, Fig. 5E), suggesting that ComJ may possess a slight preference for D- over L-Tyr. The finding that ComJ can accept an epimerised peptide substrate suggests that an alternative conformation of complestatin is in fact a feasible product of the complestatin gene cluster.
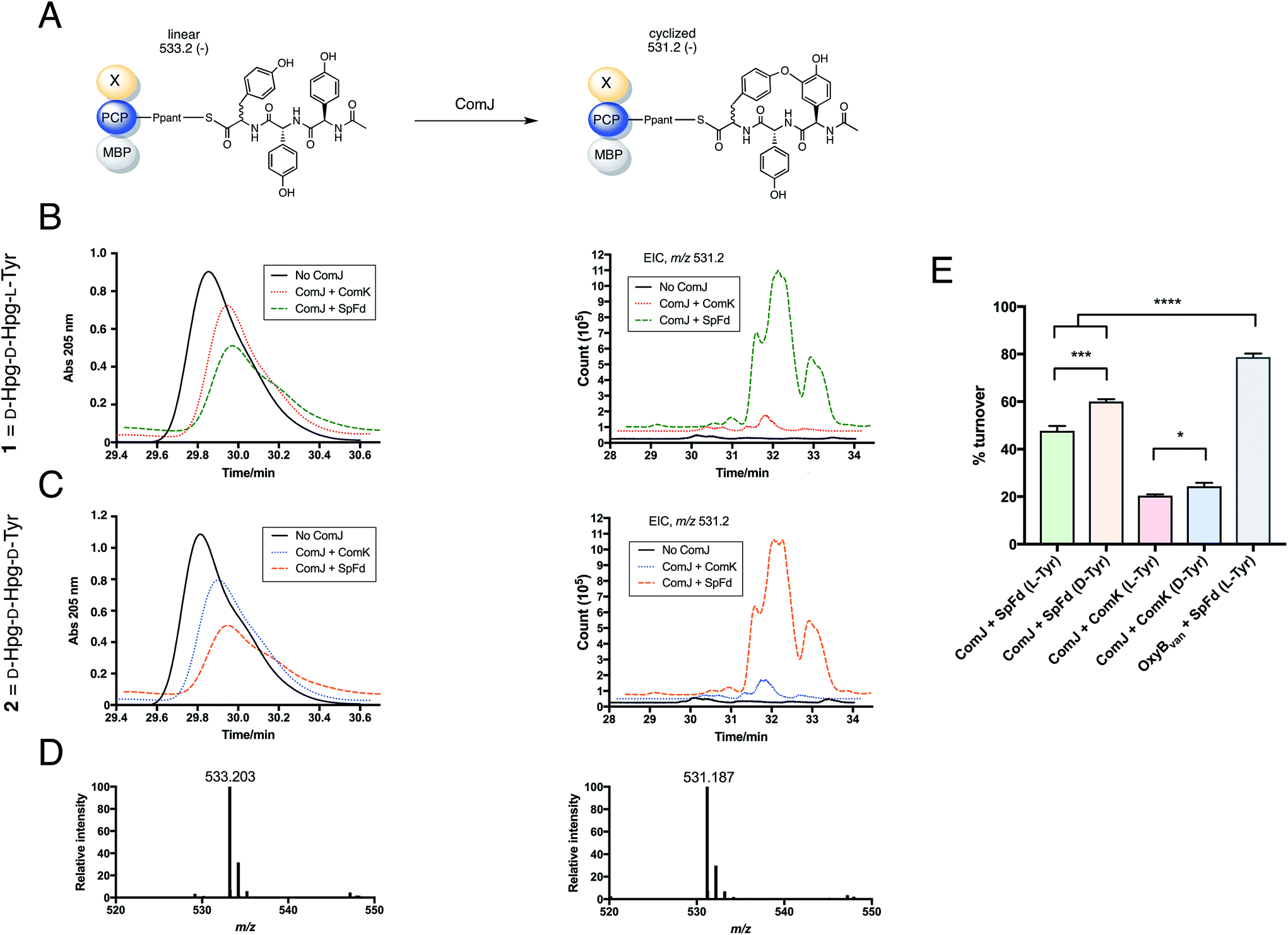 | ||
| Fig. 5 Results of turnover studies on 1 (D-Hpg-D-Hpg-L-Tyr) and 2 (D-Hpg-D-Hpg-D-Tyr). (A) Schematic showing turnover of PCP-X—bound tripeptide by ComJ. The linear form of the peptide (post-methylamine cleavage) has an m/z of 533.2 (negative mode), whereas the cyclised form has an m/z of 531.2, 2 Da lower owing to the loss of two hydrogen atoms. (B) Left: UV trace showing the decrease in linear peptide concentration in the presence of ComJ and either ComK (red dotted line) or SpFd (green dashed line) for 1 loaded on MBP-PCP7-Xcom. Right: extracted ion count (EIC) showing the concomitant increase in cyclised product. (C) Left: UV trace showing the decrease in linear peptide concentration in the presence of ComJ and either ComK (blue dotted line) or SpFd (orange dashed line) for 2 loaded on MBP-PCP7-Xcom. Right: EIC showing the concomitant increase in cyclised product. In both B and C, solid black lines represent negative (no ComJ) controls. (D) Mass spectra for linear peptide (left, m/z = 533.203, expected: 533.204) and cyclised product (right, m/z = 531.187, expected: 531.188). (E) Bar graph summarising the turnover percentages of 1 and 2 by ComJ in the presence of SpFd or ComK; the presenting PCP construct for all ComJ reactions was MBP-PCP7-Xcom. The positive control with OxyBvan as the P450 and MBP-PCP7-Xvan as the peptide presentation platform is shown for comparison (see Fig. S4† for relevant spectra). All experiments were performed in triplicate and turnover percentages computed by quantifying the decrease in linear starting material concentration relative to the negative (no Oxy) controls. Significant differences between means were computed using the unpaired t-test. *P < 0.05, ***P < 0.001, ****P < 0.0001. | ||
We next tested the possibility of a tight ComJ-Xcom interaction hindering catalysis, as we observed slightly lower extent of conversion of the linear starting materials (1, 2) into cyclised products (3, 4) by ComJ compared to the OxyBvan control (P < 0.0001 for both 1 and 2, Fig. 5E). It was recently discovered in fact that the cytochrome P450 enzyme StaH involved in the biosynthesis of the GPA A47934 possesses low catalytic efficiency, and that when Xsta (X-domain from A47934 NRPS) is exchanged with Xtei (X-domain from teicoplanin NRPS), the P450 activity increases dramatically, which suggests that an overly tight interaction between StaH and Xsta hinders catalytic efficiency in vitro.27 However, in our case, exchanging MBP-PCP7-Xcom with MBP-PCP7-Xvan did not result in any significant increase in turnover (Fig. S5†), allowing us to exclude this hypothesis in this particular case.|| Increasing the reaction time for turnover also did not result in more cyclised product (Fig. S5†).
The fully annotated complestatin BGC (GenBank: AF386507.1) encompasses comK, whose protein product was predicted based on homology to be a ferredoxin.23 Thus, we decided to test the relative efficiency of ComK versus SpFd in transferring reducing equivalents to ComJ. Expressing ComK in TB medium supplemented with 50 μM FeSO4 (see Section 2.2) resulted in brown protein fractions, consistent with the presence of a chromophoric [2Fe–2S] cluster absorbing at 422 nm (Fig. S6†).34 As shown in Fig. 5, replacing SpFd with ComK resulted in ∼20% turnover for both L- and D-Tyr-containing tripeptides, with the turnover rate for the latter being slightly higher (P < 0.05, Fig. 5E). This represents the first observation of ComK acting as a ferredoxin, although spinach ferredoxin remains superior with respect to turnover efficiency. One hypothesis suggested by Khosla and co-workers23 is that in vivo, ComK is likely to interact with a specific electron source encoded outside the complestatin BGC. We speculate that the characterisation and subsequent inclusion of this oxidoreductase in the turnover mixture might enable higher rates of turnover, comparable if not superior to those achieved with spinach ferredoxin.
Turnover was also attempted using longer, non-chlorinated heptapeptides, but no turnover could be detected (data not shown). Recent results suggest that chlorine atoms may play a role in helping orient peptide substrates in the polar P450 active site,35 and therefore it is possible that the non-chlorinated heptapeptides were simply too non-polar to interact favourably with the P450 enzyme. With that in mind, future studies should investigate the activity on substrates that more closely resemble the natural substrates for cyclisation.
4. Conclusions
In conclusion, we report the first direct observation of in vitro activity by ComJ, the biaryl ether-installing P450 from the complestatin BGC. ComJ turned over the model tripeptide 1 in 48% yield and 2 in 60% yield, revealing a lack of strong substrate stereoselectivity. The slight preference for 2 (D-Tyr-containing peptide) over 1 (L-Tyr-containing peptide) fuels the question as to whether the complestatin NRPS can make and process a peptide containing a D-Tyr-residue at position 6. Future experiments will be needed to determine the role of the epimerisation domain in module 6 and the capacity of the complestatin gene cluster to biosynthesise an alternative conformation of complestatin.The experiments presented here focused on ComJ as a biaryl ether-installing P450, but the UV/vis and SEC data reveal that ComI also possesses all the characteristics of a competent P450 that is able to interact productively with the NRPS machinery. Future studies will need to investigate ComI activity—especially in conjunction with prior cyclisation by ComJ—thus completing the picture of complestatin biosynthesis and paving the way towards a chemo-enzymatic synthesis of the molecule and potential derivatives.
Conflict of interest
The authors have no conflicts of interest to declare.Abbreviations
| A | Adenylation domain |
| ACN | Acetonitrile |
| AEC | Anion exchange chromatography |
| BGC | Biosynthetic gene cluster |
| C | Condensation domain |
| CoA | Coenzyme A |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DIC | N,N′-Diisopropylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| DMF | N,N-Dimethylformamide |
| E | Epimerisation domain |
| EcoFlvR | E. coli flavodoxin reductase |
| EIC | Extracted ion count |
| FA | Formic acid |
| GPA | Glycopeptide antibiotic |
| G6P | Glucose-6-phosphate |
| G6P-DH | Glucose-6-phosphate dehydrogenase |
| HOBt | Hydroxybenzotriazole |
| L | Extended linker domain |
| M | Module |
| MALDI | Matrix-assisted laser desorption ionisation (mass spectrometry) |
| MBP | Maltose-binding protein |
| Mt | N-Methyltransferase domain |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NRP | Non-ribosomal peptide |
| NRPS | Non-ribosomal peptide synthetase |
| PCP | Peptidyl carrier protein domain |
| Ppant | 4′-Phosphopantetheine |
| PyBOP | (Benzotriazol-1-yl-oxy)tripyrrolidinophosphonium hexafluorophosphate |
| Q-TOF LC/MS | Quadrupole time-of-flight liquid chromatography/mass spectrometry |
| SEC | Size exclusion chromatography |
| SPE | Solid-phase extraction |
| SpFd | Spinach ferredoxin |
| SPPS | Solid-phase peptide synthesis |
| tBu | tert-Butyl (protecting group) |
| TE | Thioesterase domain |
| TEA | Triethylamine |
Acknowledgements
This work was financially supported by the Corporation for Science Advancement's Cottrell College Scholars Award (L. K. C., #2020047) and the Arnold and Mabel Beckman Foundation. M. J. C. is grateful for the support of the Deutsche Forschungsgemeinschaft (Emmy-Noether Program, CR 392/1-1), Monash University and the EMBL Australia program. This research was supported under Australian Research Council's Discovery Projects funding scheme (project number DP170102220) to M. J. C. We thank Agilent Technologies (Santa Clara, CA, USA) for the loan of the Q-TOF LC/MS and Bo Wang (Texas A&M University) for MALDI analyses. We are grateful to Professor John Robinson (University of Zurich) for providing expression plasmids for OxyBvan, spinach ferredoxin, and E. coli flavodoxin reductase, Professor Jun Yin (Georgia State University) for providing the Sfp R4-4 expression plasmid, Dr Clara Brieke (MPIMF Heidelberg) for advice regarding peptide synthesis, and Dr Anthony Macherone (Agilent Technologies) for advice regarding LC/MS analyses. We thank Yang Wu (Haverford College) for technical support, and Professor Chaitan Khosla (Stanford University) and Dr Martin Schnermann (NIH/NCI) for helpful discussions.Notes and references
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00820.
- M. J. Cryle, C. Brieke and K. Haslinger, in Amino Acids, Peptides and Proteins, The Royal Society of Chemistry, 2014, vol. 38, pp. 1–36 Search PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- M. Peschke, K. Haslinger, C. Brieke, J. Reinstein and M. J. Cryle, J. Am. Chem. Soc., 2016, 138, 6746–6753 CrossRef CAS PubMed.
- N. Geib, K. Woithe, K. Zerbe, D. B. Li and J. A. Robinson, Bioorg. Med. Chem. Lett., 2008, 18, 3081–3084 CrossRef CAS PubMed.
- M. Peschke, C. Brieke and M. J. Cryle, Sci. Rep., 2016, 6, 35584 CrossRef CAS PubMed.
- K. Haslinger, E. Maximowitsch, C. Brieke, A. Koch and M. J. Cryle, ChemBioChem, 2014, 15, 2719–2728 CrossRef CAS PubMed.
- V. Ulrich, C. Brieke and M. J. Cryle, Beilstein J. Org. Chem., 2016, 12, 2849–2864 CrossRef CAS PubMed.
- K. Haslinger and M. J. Cryle, FEBS Lett., 2016, 590, 571–581 CrossRef CAS PubMed.
- M. J. Cryle, J. Staaden and I. Schlichting, Arch. Biochem. Biophys., 2011, 507, 163–173 CrossRef CAS PubMed.
- M. Peschke, M. Gonsior, R. D. Süssmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 CrossRef CAS PubMed.
- I. Kaneko, D. T. Fearon and K. F. Austen, J. Immunol., 1980, 124, 1194–1198 CAS.
- K. Matsuzaki, H. Ikeda, T. Ogino, A. Matsumoto, H. B. Woodruff, H. Tanaka and S. Omura, J. Antibiot., 1994, 47, 1173–1174 CrossRef CAS PubMed.
- S. B. Singh, H. Jayasuriya, G. M. Salituro, D. L. Zink, A. Shafiee, B. Heimbuch, K. C. Silverman, R. B. Lingham, O. Genilloud, A. Teran, D. Vilella, P. Felock and D. Hazuda, J. Nat. Prod., 2001, 64, 874–882 CrossRef CAS.
- S. Y. Seo, B. S. Yun, I. J. Ryoo, J. S. Choi, C. K. Joo, S. Y. Chang, J. M. Chung, S. Oh, B. J. Gwag and I. D. Yoo, J. Pharmacol. Exp. Ther., 2001, 299, 377–384 CAS.
- I. D. Yoo, B. S. Yun, I. J. Ryoo, S. Y. Lee, M. H. Shin and S. Oh, Neurochem. Res., 2002, 27, 337–343 CrossRef CAS PubMed.
- E. C. Kim, B. S. Yun, I. J. Ryoo, J. K. Min, M. H. Won, K. S. Lee, Y. M. Kim, I. D. Yoo and Y. G. Kwon, Biochem. Biophys. Res. Commun., 2004, 313, 193–204 CrossRef CAS PubMed.
- Y. J. Kwon, H. J. Kim and W. G. Kim, Biol. Pharm. Bull., 2015, 38, 715–721 CAS.
- J. Garfunkle, F. S. Kimball, J. D. Trzupek, S. Takizawa, H. Shimamura, M. Tomishima and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 16036–16038 CrossRef CAS PubMed.
- H. Shimamura, S. P. Breazzano, J. Garfunkle, F. S. Kimball, J. D. Trzupek and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 7776–7783 CrossRef CAS PubMed.
- Z. Wang, M. Bois-Choussy, Y. Jia and J. Zhu, Angew. Chem., Int. Ed., 2010, 49, 2018–2022 CrossRef CAS PubMed.
- H. T. Chiu, B. K. Hubbard, A. N. Shah, J. Eide, R. A. Fredenburg, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8548–8553 CrossRef CAS PubMed.
- O. K. Park, H. Y. Choi, G. W. Kim and W. G. Kim, ChemBioChem, 2016, 17, 1725–1731 CrossRef CAS PubMed.
- C. Brieke, M. Peschke, K. Haslinger and M. J. Cryle, Angew. Chem., Int. Ed. Engl., 2015, 54, 15715–15719 CrossRef CAS PubMed.
- J. Bogomolovas, B. Simon, M. Sattler and G. Stier, Protein Expression Purif., 2009, 64, 16–23 CrossRef CAS PubMed.
- V. Ulrich, M. Peschke, C. Brieke and M. J. Cryle, Mol. BioSyst., 2016, 12, 2992–3004 RSC.
- C. Brieke and M. J. Cryle, Org. Lett., 2014, 16, 2454–2457 CrossRef CAS PubMed.
- C. Brieke, V. Kratzig, M. Peschke and M. J. Cryle, in Methods in Molecular Biology, Clifton, N.J., 2016, vol. 1401, pp. 85–102 Search PubMed.
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895 CrossRef CAS PubMed.
- K. Woithe, N. Geib, O. Meyer, T. Wörtz, K. Zerbe and J. A. Robinson, Org. Biomol. Chem., 2008, 6, 2861–2867 CAS.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 78 CrossRef PubMed.
- Y. Zou and J. Yin, J. Am. Chem. Soc., 2009, 131, 7548–7549 CrossRef CAS PubMed.
- D. B. Li, K. Woithe, N. Geib, K. Abou-Hadeed, K. Zerbe and J. A. Robinson, in Methods in Enzymology, 2009, vol. 458, pp. 487–509 Search PubMed.
- M. Peschke, C. Brieke, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Biochemistry, 2017, 56, 1239–1247 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06518c |
| ‡ These authors contributed equally to this work. |
| § It was observed that expressing ferredoxins in TB medium resulted in higher yields compared to expressing the same proteins in LB medium. |
| ¶ The decision to start from free Wang resin instead of D-Tyr-loaded Wang resin in the synthesis of 2 was based on the much lower purchase price of free Wang resin compared to the corresponding pre-loaded resin. |
| || Although PCP7van was exchanged for PCP7com, effects from direct PCP-Oxy binding can be excluded given that ComJ does not appear to bind to the PCP7 domain but only to the X-domain (Fig. S2 and S3†). |
| This journal is © The Royal Society of Chemistry 2017 |