
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of micro-shell Cu–Al porous ceramometals as catalysts for the water–gas shift reaction
S. Tikhov *a,
T. Minyukovaa,
K. Valeeva,
S. Cherepanovaa,
A. Salanova,
V. Kaichevab,
A. Saraevab,
A. Andreeva,
O. Lapinaab and
V. Sadykovab
*a,
T. Minyukovaa,
K. Valeeva,
S. Cherepanovaa,
A. Salanova,
V. Kaichevab,
A. Saraevab,
A. Andreeva,
O. Lapinaab and
V. Sadykovab
aBoreskov Institute of Catalysis SB RAS, Lavrentieva Ave., 5, 630090 Novosibirsk, Russia. E-mail: tikhov@catalysis.ru
bNovosibirsk State University, Pirogova Str., 2, 630090 Novosibirsk, Russia
First published on 1st September 2017
Abstract
XRD, NMR, SEM with mapping, and XPS were used to examine the structure and microstructure of porous ceramometals, CuAlO/CuAl, synthesized by mechanochemical alloying of a mixture of Cu + Al (Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al = 87:13 wt%) followed by hydrothermal treatment and heat treatment in air. The egg-shell nature of the microstructure was revealed: the metallic cores consisting of copper and aluminum alloys are surrounded by the oxide matrix containing copper oxides and X-ray amorphous mixed oxides of copper and aluminum. Catalytic activity of ceramometals in the water–gas shift reaction (WGSR) estimated for their fine fractions at 240 °C and expressed as efficient first-order rate constants is lower than that of a CuZnAl oxide catalyst due to the lower (approximately by an order of magnitude) specific surface area of the ceramometals. The specific activity of ceramometals expressed as the specific rate constant related to the unit surface area of metallic copper exceeds that for the CuZnAl oxide catalyst and correlates with the content of crystallized intermetallics. Mechanical, textural and thermophysical properties of ceramometals were assessed. The presence of ultramacropores with sizes up to tens of microns was shown to be typical for ceramometals. As a result, the activity of granulated ceramometal catalysts in the WGSR, due to a high diffusion permeability, is comparable with that of granulated CuZnAl oxide. A mild leaching substantially increases the activity of cermet granules.
:Al = 87:13 wt%) followed by hydrothermal treatment and heat treatment in air. The egg-shell nature of the microstructure was revealed: the metallic cores consisting of copper and aluminum alloys are surrounded by the oxide matrix containing copper oxides and X-ray amorphous mixed oxides of copper and aluminum. Catalytic activity of ceramometals in the water–gas shift reaction (WGSR) estimated for their fine fractions at 240 °C and expressed as efficient first-order rate constants is lower than that of a CuZnAl oxide catalyst due to the lower (approximately by an order of magnitude) specific surface area of the ceramometals. The specific activity of ceramometals expressed as the specific rate constant related to the unit surface area of metallic copper exceeds that for the CuZnAl oxide catalyst and correlates with the content of crystallized intermetallics. Mechanical, textural and thermophysical properties of ceramometals were assessed. The presence of ultramacropores with sizes up to tens of microns was shown to be typical for ceramometals. As a result, the activity of granulated ceramometal catalysts in the WGSR, due to a high diffusion permeability, is comparable with that of granulated CuZnAl oxide. A mild leaching substantially increases the activity of cermet granules.
1 Introduction
The water–gas shift reaction (WGSR) is one of the primary industrial steps of hydrogen production. In recent decades, many works devoted to kinetics and properties of catalysts for this process have been published1–5 and summarized in a series of reviews.6–9 These catalysts are usually comprised of Cu, Zn and Al oxide composites prepared by coprecipitation or impregnation. Among the key properties of a catalyst (support) for WGSR is its ability to ensure a high dispersion of copper. According to data reported in ref. 2, specific activity of metallic copper in the WGS remains nearly constant in a broad range of dispersions, while structure sensitivity is claimed in other works.1 Hence, variation of conditions of catalytic testing makes it difficult to compare the results. Another main problem of the low temperature WGSR reaction is a low activity of the conventional granulated CuZnAl catalyst per the unit volume of the catalyst bed8–10 explained by a low loading density and poor access of reagents into the bulk of granules with a low porosity. A possible way to increase such a volume activity is the use of ceramometal catalysts with enhanced real and loading density possessing also a developed system of transport macropores and mesopores. Such catalysts could also prevent emergence of temperature gradients within the catalytic layer due to a high thermal conductivity provided by the presence of metal particles in their bulk. Cermet catalysts synthesized from Al–Cu–Fe alloys were applied earlier for the steam reforming of methanol.11,12 These catalysts possess a complex microstructure comprised of the metal cores surrounded by the oxidic shell with segregated nanoparticles of metallic Cu. The specific activity of copper was found to change non-monotonously with its dispersion.11 The aim of the present work was development and detailed characterization of a new ceramometal CuAlO/CuAl catalyst prepared through the step of mechanical alloying of Cu–Al powders. Main characteristics of these catalysts precursors were reported in the previous work.132 Experimental
2.1. Preparation
Aluminum powder (PAP-2, GOST 5494-95, Russia) with platelet-shaped particles (typical sizes 10/40 microns) and electrolytic copper powder (PMS-1, GOST 4960-75, Russia) of dendritic shape were used as starting materials.(a) Mechanical activation (MA) of the mixture containing (wt%) 87Cu + 13Al was used to synthesize the powdered precursor. MA was carried out in a high-power planetary ball mill APF (the diameter of milling steel balls 5 mm; ball-to-powder mass ratio 8:1; powder weight 100 g; balls acceleration 65 g). The activation time was 3–12 min. The increase of MA time decreases the average particle size and increases the alloying degree. The vials and milling balls were made of steel. The volume of the milling vial was 1 L. The content of Fe incorporated into the powder mixtures as contamination was very low (0.14 ry0.02 wt%). To prevent sticking of the powder to balls and vial walls, ethanol was used as a process control agent. It was added into the vial at 30 s before the start of milling.
(b) The product of mechanical activation was loaded into a stainless steel die specially designed to ensure a free access of water and hydrogen release. Variation of the internal shape of the die where a powder was loaded provides a broad variation of the shape of monoliths.
(c) For the hydrothermal treatment (HTT), the loaded die was placed into the vessel with boiling water and kept there for five hours. This step provides formation of strong monoliths due to conjugation of the red-ox reaction of aluminum oxidation and creation of contacts between the particles due to generation of aluminum hydroxides. Similar to the MA step, oxidation of Al at the HTT step diminishes the extent of the exothermic reaction of Me–Al interaction with air at the calcination step.
(d) The monolithic product of HTT was removed from the die, dried for 1 hour at 120 °C and calcined under air at 550 °C. This yields a mechanically strong material and develops the mesoporous structure of cermets. The density of granules increased with decreasing the time of mechanical activation from 3.87 (12 min) to 4.24 g cm−3 (3 min).
The reference sample was represented by the CuZnAl bulk oxide catalyst (similar to the commercial one) synthesized by the heat treatment of mixed Cu–Zn–Al hydroxide (CZA). The hydroxide was obtained via coprecipitation from 10% aqueous solutions of metal nitrates by a sodium carbonate solution with subsequent washing and drying according to the procedure reported in a patent.14
2.2. Characteristics of samples
X-ray diffraction (XRD) patterns of powders were recorded by means of a D8 Advance powder diffractometer (Bruker AXS) equipped with a one-dimensional Lynx-Eye detector using Cu Kα radiation. XRD patterns were recorded in the 2θ range 15–95° with a step of 0.05° and a counting time of 3 s per step. The quantitative analysis was carried out using Rietveld refinement in TOPAS software (Bruker AXS GmbH). The average size of the crystallites was also determined using TOPAS taking into account the instrumental peak broadening. The instrumental peak broadening was determined from the XRD pattern of a reference sample (SRM676, corundum) by the approximation method using a convolution of Lorentzian and Gaussian functions. The aluminum content in Cu1−xAlx solid solutions was calculated using Vegard's law.Scanning electron microscopy (SEM) studies of powders were performed on a JSM 6460SV microscope (JEOL Ltd.) equipped with an energy-dispersive spectroscopy unit EDX INCA (Oxford Inst.). Samples for these studies were prepared either by spreading the powders over a substrate or by fixing particles in an organic binder followed by polishing. The latter technique allowed to study the microstructure of the powder particles. Transmission electron microscopy (TEM) studies of powders were carried out using JEM-2010 and JEM-2200FS (JEOL Ltd.) microscopes; High-Angle Annular Dark Field Scanning Transmission Electron Mode (HAADF STEM) and Energy Dispersive X-ray Spectroscopy (EDX) were utilized. The EDX in the STEM mode was used for the elemental mapping of samples by Al K (E = 1.486 keV) and Cu K (E = 8.040 keV) radiation and semi-quantitative analysis of areas as small as 1 nm in diameter. To prepare samples for TEM studies, powders were dispersed in ethanol and suspensions were subjected to ultrasonic treatment at 35 kHz. The suspension was poured onto a holey carbon substrate on a Mo grid and dried at 20 °C.
Al NMR experiments were carried out on a Bruker Avance 400 MHz spectrometer with a magnetic field of 9.4 T. Spectra were recorded at resonance frequencies of 27Al and 63Cu in this magnetic field, i.e. 104.31 and 106.12 MHz, respectively. Due to rather broad resonance lines and experimental dead time, the spectra were acquired by spin echo technique with two equivalent pulses p1 – τ – p1 – τ. The pulse duration p1 of 27Al NMR experiments was 0.5 μs (π/8), and for 63Cu NMR spectra p1 was 1 μs (π/6). The interpulse delay τ was 15 μs for static experiments, and it was synchronized with the sample spinning rate in MAS (magic angle spinning) experiments. The experiments were carried out with different MAS probes of 4, 2.5 and 1.3 mm outer rotor diameter providing sample rotation at 15, 30, and 60 kHz. The static spectra were obtained with the 4 mm probe. The spectral width used in experiments was 5 MHz, and the sequence repetition delay varied from 0.1 to 2 s since all samples contained intermetallic and paramagnetic compounds which decrease T1 relaxation times. The chemical shifts were externally referenced to diluted Al(OH)63+ at 0 ppm in 27Al experiments, and to copper wire at 2396 ppm.15–17 The decomposition of static spectra was done on Lorentzian and Gaussian lines to obtain the best fit.
The chemical analysis of the catalyst surface was carried out using X-ray photoelectron spectroscopy (XPS). The XPS measurements were performed on a SPECS's Surface Nano Analysis GmbH (Germany) photoelectron spectrometer equipped with a PHOIBOS-150-MCD-9 hemispherical electron energy analyzer, a FOCUS-500 X-ray monochromator, and an XR-50M X-ray source with a double Al/Ag anode. The spectrometer was also equipped with a high-pressure cell (HPC), which enables heating samples before analysis in gaseous mixtures at pressures up to 0.5 MPa.18 The core-level spectra were obtained using the monochromatic AlKα radiation (hν = 1486.74 eV) under ultrahigh vacuum conditions. The charge correction was performed by setting the Al 2p peak at 74.5 eV corresponding to aluminum in Al2O3. Before the XPS analysis, the catalysts were treated in 1 bar O2 at 270 °C for 1 h in HPC. After treatment of samples in the HPC the intensity of C 1s peak was very low, which did not allow this peak to be used for calibration of the binding energy scale. Relative element concentrations were determined from the integral intensities of XPS peaks using the cross-sections according to Scofield.19 For detailed analysis, the spectra were fitted into several peaks after the background subtraction by the Shirley method. All the spectra were analyzed using the CasaXPS software.18 The line shape used for fitting the Al 2p and Cu 3p spectra was the product of Lorentzian and Gaussian functions. An asymmetric, so-called “LF” line shape was used for approximation of the Cu 2p3/2 spectra of copper.20 Note that the asymptotic form of the LF line shape is equivalent to the asymptotic form of the theoretical Doniach-Sunjic asymmetric line shape. However, this new line shape describes the asymmetric XPS peak more correctly than the conventional Doniach-Sunjic function does because the LF function is finite. After the XPS analysis of the oxidized samples, the catalysts were treated in 1 bar H2 at 270 °C for 1 h, and the XPS analysis of the reduced catalysts was carried out. XPS data were used to estimate the surface concentration of copper (ΘCu = Cu/(Cu + Al)) and the surface area of metallic copper SCu ≈ SBET·ΘCu.
The macropore volume was assessed from the difference between the volumes of micro/mesopores and the total pore volume. The total pore volume of the monolith was estimated from the values of true and apparent densities. True density was measured using a helium pycnometer – Autopycnometer 1320 (Micromeritics). The fraction of micropores and mesopores (called for brevity the “mesopores) as well as the specific surface area (SSA) were determined from adsorption isotherms of nitrogen recorded at 77 K using an ASAP-2400 Micromeritics instrument. The macropore volume was estimated from the difference of the total and mesopore volume.
Mechanical properties (compressive strength) were estimated as the limiting pressure that can be applied to the circle side of cylindrical pellets or granules before its destruction.21,22 Thermal expansion coefficient was measured on a DIL-402C (Netzsch) horizontal dilatometer. Samples were examined as prisms with a section of 6 × 6 mm2 and a length of 10 mm in the range from 50 to 500 °C in air at a heating rate of 2° min−1.
Catalytic properties in WGSR were studied in a laboratory flow setup with the gas chromatographic analysis of the reaction mixture under the same conditions as earlier used for studies of the commercial Cu–Zn–Al–O catalyst.9 The catalysts were activated in the mixture of 5% H2 in He at 3000–4000 h−1 with a temperature ramp of 2° min−1 up to 270 °C for 2 h. Activity was measured at a pressure of 1 bar in the mixture of CO:H2O:H2 = 8:42:50 at a steam/gas ratio of 0.6–0.7; a catalyst (0.14–0.25 mm fraction) mixed with quartz sand of the same particle size was used. The height of the catalyst bed diluted with quartz was 3 cm, and the reactor diameter was 20 mm. The temperature was continuously controlled with the aid of a thermocouple placed in a pocket of the reactor at the center of the catalyst bed. Catalyst was characterized by the apparent rate constant of the forward reaction, which was determined taking into account the reversibility of the reaction in an approximation of the first order with respect to CO. The reaction rate constant was calculated under the assumption that the reactor operated in the plug flow regime. All catalytic data are given for the catalysts that reached a steady state activity (after continuous operation for at least 25 h). The bulk density of the fraction of catalysts without quartz varied from 1 to 2.3 g cm−3. Activity of granules having a prismatic shape and dimensions 3.2 × 3.2 × 5 mm3 was also studied in the special Temkin single-row reactor.23 Due to a considerable variation of the bulk density at close weights, contact time also changed significantly, from 0.02 to 0.05 s. Thermal stability (kstab) was estimated from the ratio of k at 240 °C after 20 h of operation to the initial k (without overheat).
3 Results and discussion
3.1. Phase composition according to XRD and NMR data
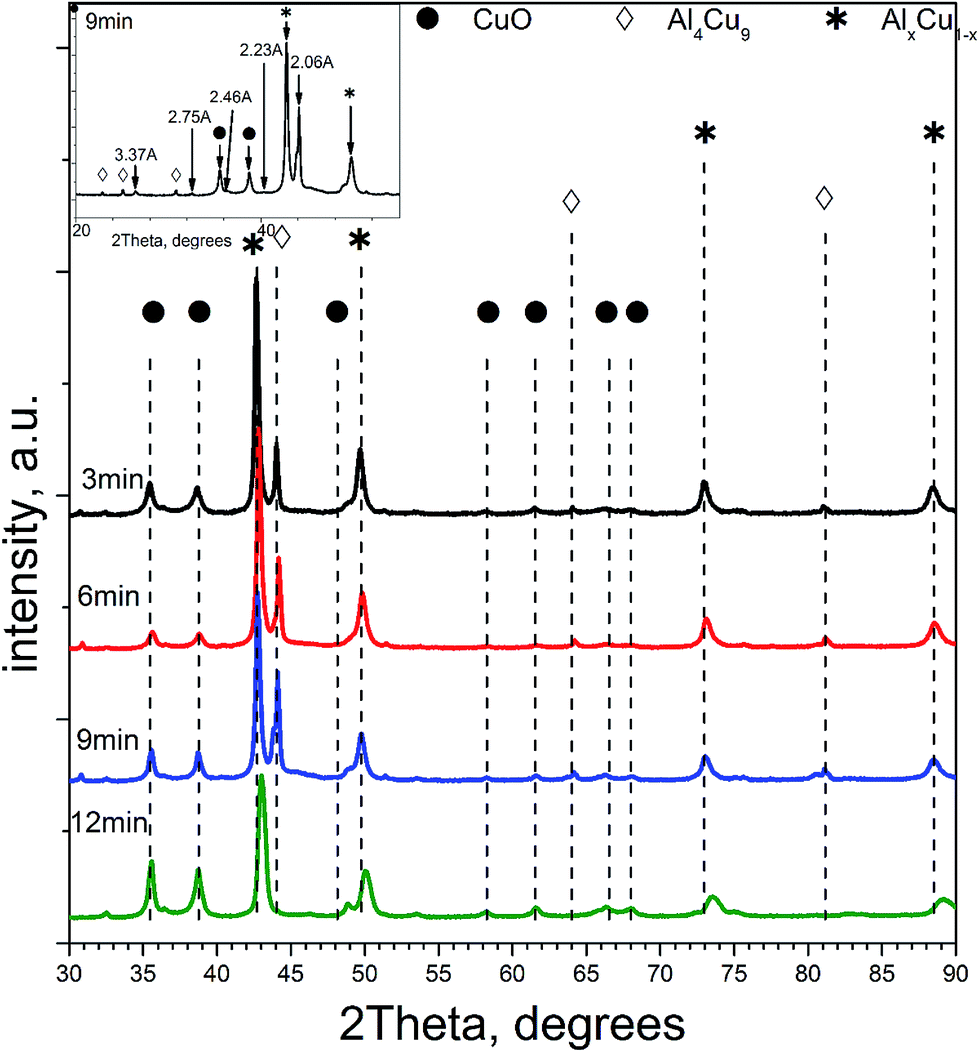 | ||
| Fig. 1 Diffraction patterns of ceramometals at different MA times. Symbols denote peaks corresponding to the following phases: * – a solid solution of aluminum in copper, AlxCu1, ◊ – Al4Cu9 intermetallic [24-0003], ● – CuO [05-0661]. The inset displays a fragment of the diffraction pattern of the new phase. | ||
Table 1 lists qualitative and quantitative results of XRD phase analysis of ceramometals corresponding to diffraction patterns in Fig. 1. The new phase is not presented in Table 1 because it has a low (ca. 1 wt%) concentration.
| 3 min | 6 min | 9 min | 12 min | |
|---|---|---|---|---|
| Cu1−xAlx solid solution | 49% (x = 0.14) | 62% (x = 0.10) | 48% (x = 0.12) | 53.0% (x = 0.06) |
| Al4Cu9 intermetallic | 12% | 20% | 27% | 0% |
| CuO | 39% | 18% | 25% | 47% |
As seen in Table 1, the concentration of Al4Cu9 intermetallic goes through the maximum with increasing the time of preliminary MA of the copper–aluminum powder. Such changes in the intermetallic concentration were observed earlier by XRD for the MA products13 that were further used as precursors for the synthesis of cermets. A similar trend is typical also for other MA conditions for CuAl-containing systems.24,25 The concentration of the solid solution is nearly constant, except for the sample activated 6 min. The concentration of aluminum in the solid solution decreases monotonically with the time of activation (excluding 6 min point), while CuO concentration goes through the minimum. Remarkable is the absence of crystallized forms of mixed copper–aluminum oxides of the spinel type. It should be noted that XRD could not detect X-ray amorphous components in cermets as well as in MA products.12,13 Hence, variation of the content of aluminum-containing components in the X-ray amorphous state could not be followed by this method.
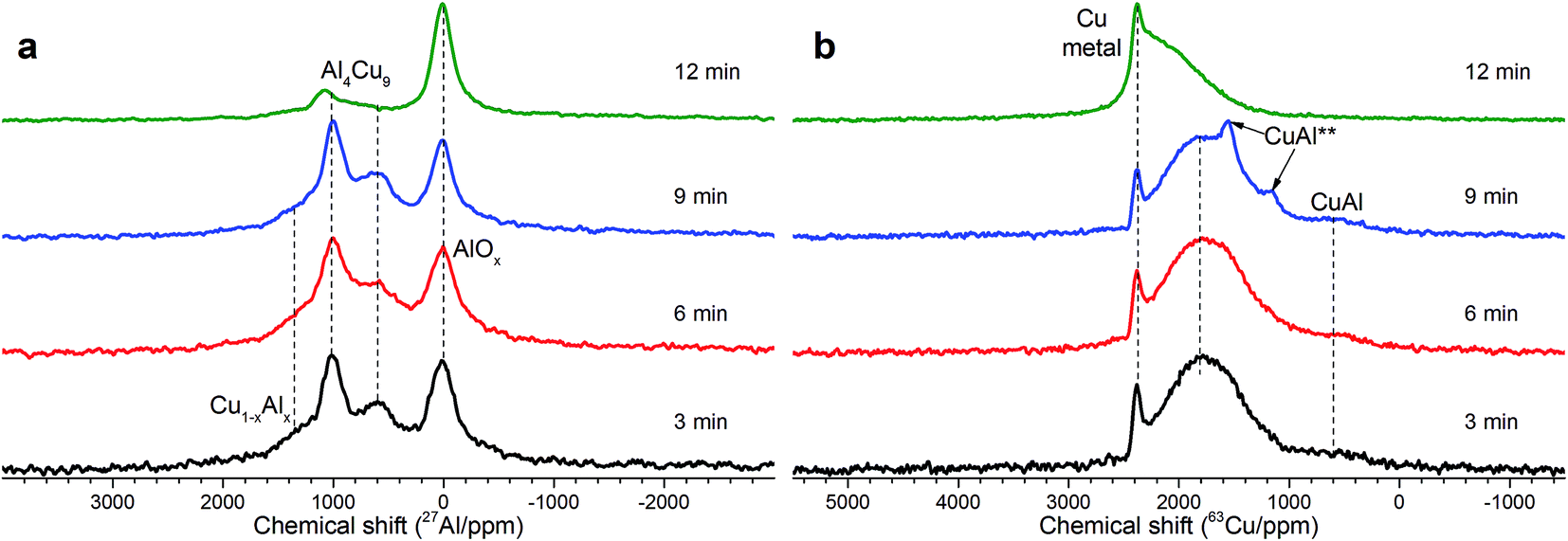 | ||
| Fig. 2 (a) Static 27Al NMR spectra of Cu–Al cermets (85:13 wt%) in dependence on the time of mechanical activation (down-top): 3, 6, 9 and 12 min. The intensity is normalized to maxima in the spectra. (b) Static 63Cu NMR spectra of Cu–Al cermets (85:13 wt%) in dependence on MA time (down-top): 3, 6, 9 and 12 min. The intensity is normalized to maxima in the spectra. | ||
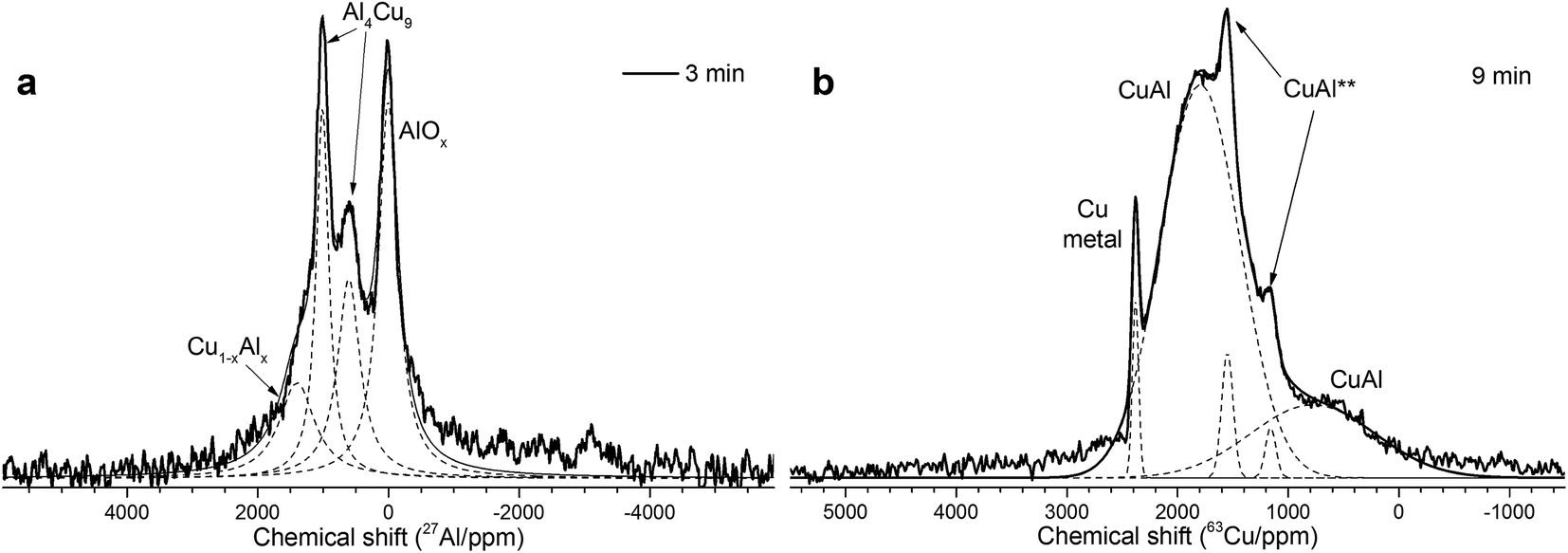 | ||
| Fig. 3 (a) An example of 27Al spectrum decomposition for the Cu–Al cermet (85:13 wt%) with 3 min activation time; (b) an example of 63Cu spectrum decomposition for the Cu–Al cermet (85:15 wt%) with 9 min activation time. | ||
As follows from our previous study of ball milled source precursors,13 lines at ∼870 and ∼590 ppm can be related to Al4Cu9 intermetallic compound, which for cermet are located at 1000 and 600 ppm (Fig. 2a). The line at ∼1400 ppm is assigned to Cu1−xAlx solid solutions with various stoichiometry.13,17 Therefore, these results are in the qualitative agreement with XRD data, while aluminum oxide is detected only by NMR. Due to the skin effect, not all aluminum atoms are excited equivalently.
MAS spectra in Fig. 4 show a better resolution of the lines displayed in Fig. 2a. The shoulder at 1400 ppm disappears since its width is not averaged under MAS conditions. First, the Al oxide part is resolved to ensure the separation of octahedral (∼0 ppm) and tetrahedral (∼60 ppm) Al sites. Another feature of 15 kHz 27Al spin echo MAS spectra is the emergence of a rather narrow line near ∼900 ppm. Such a small width may correspond to a well-crystallized Cu–Al** compound containing less than 1 at% of the observed Al. It can be concluded from the data shown in Fig. 4 that concentration of the Cu–Al** compound increases with MA time up to 9 min and completely disappears after 12 min of activation.
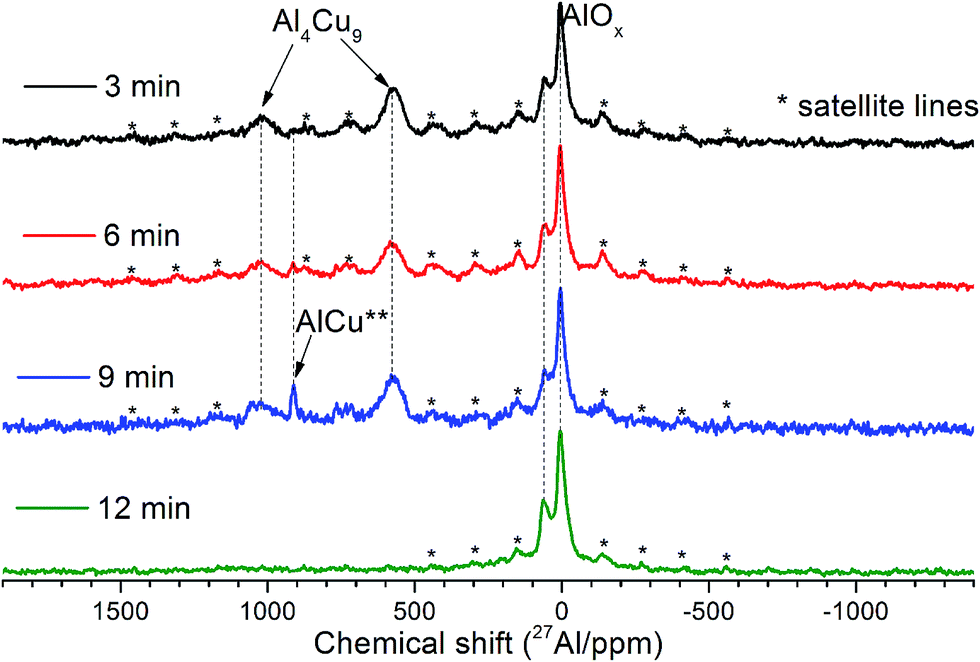 | ||
| Fig. 4 15 kHz spin echo 27Al MAS spectra of Cu–Al cermets for the sample activated for 3, 6, 9 and 12 min. | ||
The simulation of AlT and AlO sites of 15 kHz echo MAS 27Al NMR spectra by simple Czjeck model gives AlO/AlT ratios from 1.1 to 0.6. Neither of the ratios corresponds to a certain alumina that generally has AlO/AlT in the range between 2 and 37.26,27 This observation coupled with a broad basement under main AlOx peaks and abnormally intensive satellite lines suggests that samples composition could be close to CuAl2O4 which is a partially inversed spinel; moreover, samples may contain delafossite or CuxAlyOz structures.28,29
In general, according to XRD and NMR data, the content of Cu–Al phases (Al4Cu9, CuAl**, etc.) passes through the maximum with increasing the MA time, whereas CuO oxide shows an opposite behavior. Ceramometals prepared at all times of mechanical activation contain a noticeable amount of oxides, most likely in the form of mixed Cu–Al oxides. However, these compounds are very dispersed and XRD amorphous.
3.2. Microstructure and space distribution of oxide and metal phases in the bulk of Cu–Al–O/CuAl cermets
Fig. 5 displays the microstructure of cermets as revealed by SEM. As seen in this Figure and confirmed by EDX data (Fig. 6), all cermets consist of metallic cores surrounded with the oxide layer, thus forming a microshell structure. Macropores observed as dark patches in Fig. 5 are situated between stacked cermet particles. The oxide layer forms a framework that integrates all the components into a monolith. The composition of the metallic core is close to that of the initial blend. The averaged composition of the oxide layer, according to the mapping data presented as a diagram (Fig. 6), is close to Cu2AlO3–4. Such a composition has an excess of copper in comparison with copper–aluminum spinel. Oxide fragments with the stoichiometry CuAlO2+x (corresponds to the structure of rhombohedral or hexagonal modifications of delafossite)30 and CuO were often observed in samples activated for 9 and 12 min. The thickness of the oxide layer decreases with increasing the MA time. Sample activated for 9 min has oxide “streaks” which may result from cracking of brittle copper–aluminum alloys. Sample activated for 12 min has many fine particles which are completely oxidized upon calcination in air. Black spots correspond to epoxy resin penetrating through ultramacropores of cermets to a depth of 500 μm. Thus, the oxide part of ceramometals after calcination in air concentrates mostly at the external surface of cermet macropores, sometimes as “streaks” due to cracking of metallic cores. | ||
| Fig. 5 Electron microscopy images of Cu–Al–O cermet sections obtained from MA products at different alloying time: (a) 3 min, (b) 6 min, (c) 9 min, (d) 12 min. | ||
 | ||
| Fig. 6 Phase diagram according to EDX mapping of Cu–Al–O cermet sections obtained from MA products at different alloying time: (a) 3 min, (b) 6 min, (c) 9 min, (d) 12 min. | ||
The ratio of the oxide and metal parts of ceramometals can be qualitatively estimated from the phase diagrams obtained by the elemental mapping. As seen in Fig. 6, the intensity of the metallic part (Fig. 6a, a spot near the Cu–Al line with the oxygen content close to zero) in the sample activated for 3 min is much higher as compared to the oxide one. The 12 min sample (Fig. 6d) shows an opposite pattern, in spite of a lower thickness of the oxide layer. The latter may be caused by the dispersion of the Cu–Al precursor during its mechanochemical synthesis.13,24 This increases the total Me/air interfacial area, thus facilitating an increase in the oxide amount even at a smaller thickness of the oxide layer.
The presence of aluminum oxide in the surface layer of ceramometals was additionally confirmed by XPS analysis. X-ray photoelectron spectroscopy was used to examine a series of catalysts treated in oxygen and hydrogen at 270 °C. Their typical spectra are displayed in Fig. 7. One can see that the spectrum recorded after oxidative treatment has the Al 2p peak with the binding energy of 74.5 eV (Fig. 7a). This position of the peak is typical for Al3+ ions in Al2O3 oxide (74.2–74.6 eV).31–34 Additional peaks in the region of high binding energies correspond to the Cu 3p3/2–Cu 3p1/2 doublet. A shift of the Cu 3p3/2–Cu 3p1/2 doublet to lower binding energies and increase in their relative intensity indicate reduction of copper cations on the surface.
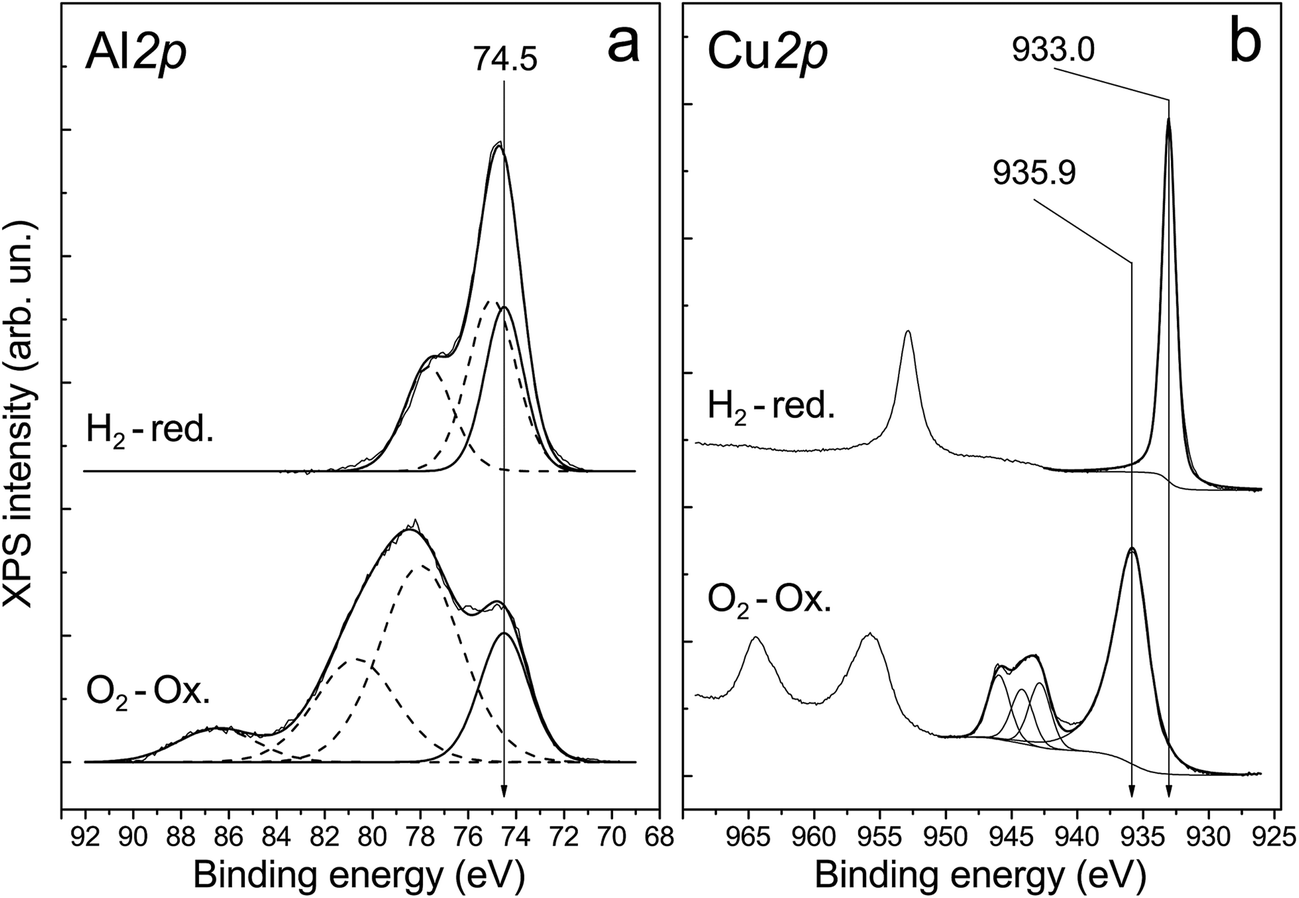 | ||
| Fig. 7 Core-level XPS spectra obtained after treatment of the catalyst in 1 bar O2 at 270 °C (O2–Ox) and in 1 bar H2 at 270 °C (H2-red) directly in the high pressure cell of the photoelectron spectrometer. | ||
The analysis of Cu 2p spectra revealed that the chemical (and, probably, structural) state of the surface layers of ceramometals is very complicated (Fig. 7b). On the one hand, according to XPS data, the Cu:Al atomic ratio is ∼1:1.4/1.5 (3 and 12 min of MA) and 1:1.0/1.1 (9 and 6 min of MA). It means that the cationic stoichiometry in the surface layers is closer to that of CuAlO2 delafossite30 or other structures of a mixed oxide in which the oxidation state of copper is 1+. On the other hand, copper in the surface layers of the oxidized catalyst is mostly in the Cu2+ state. This statement is evidenced by the binding energy of Cu 2p3/2 equal to 935.9 eV, the intensity ratio of the main peak to shake-up satellite (0.46), and the Auger parameter equal to 1851.4 eV.33–35 CuO oxide is usually characterized by a lower binding energy of Cu 2p3/2, in the range of 933.6–934.6 eV.34,36–43 This phenomenon can be attributed to the presence of subnanosized Cu–O clusters in the surface layers of ceramometals. In such clusters, the initial oxide state is close to Cu2+; however, they can easily loose oxygen even in a weakly reducing medium up to the Cu0 state which is active in the WGSR.34 Indeed, after treatment in hydrogen, the peaks of shake-up satellites typical of Cu2+ disappear from the Cu 2p spectrum, and the Auger parameter becomes equal to 1851.3, which indicates a complete reduction of copper to the metallic state.
Hence, CuAlO/CuAl ceramometal catalysts have a complex arrangement. Their metallic cores are comprised of the solid solution of aluminum in copper and the intermetallic, which prevent a complete oxidation of the metal component upon calcination in air. The oxide component, which has a thickness of several microns, is X-ray amorphous and forms a joint framework. This component includes at least the mixed oxides of copper and aluminum and, possibly, copper–oxygen clusters. As the time of preliminary MA of the Cu–Al precursor is increased, concentration of the solid solution goes through the maximum, and that of CuO – through the minimum for sample 6 min of MA, while concentration of intermetallics (Al4Cu9 and unknown phase) goes through the maximum for sample with 9 min of MA (Table 1, Fig. 7). An increase in the concentration of the oxide component with time of MA is related also to deagglomeration of the brittle Cu(Al) solid solution, which forms already at the MA step,13 thus increasing the metal/gas interfacial area.
3.3. Specific activity in WGSR and the nature of active surface sites
Temperature dependences of CO conversion for ceramometal catalysts are given in Fig. 8 in comparison with known commercial analog of ceramic catalyst (CZA).10 At close space velocities (4200–4400 h−1 for CuAl ceramometals and ∼5000 h−1 for CZA catalyst) CO conversions are apparently higher for CZA catalyst. Conversions clearly increase with the time of MA of Cu–Al mixture, even exceeding those for CZA catalyst in the case of sample activated for 12 min.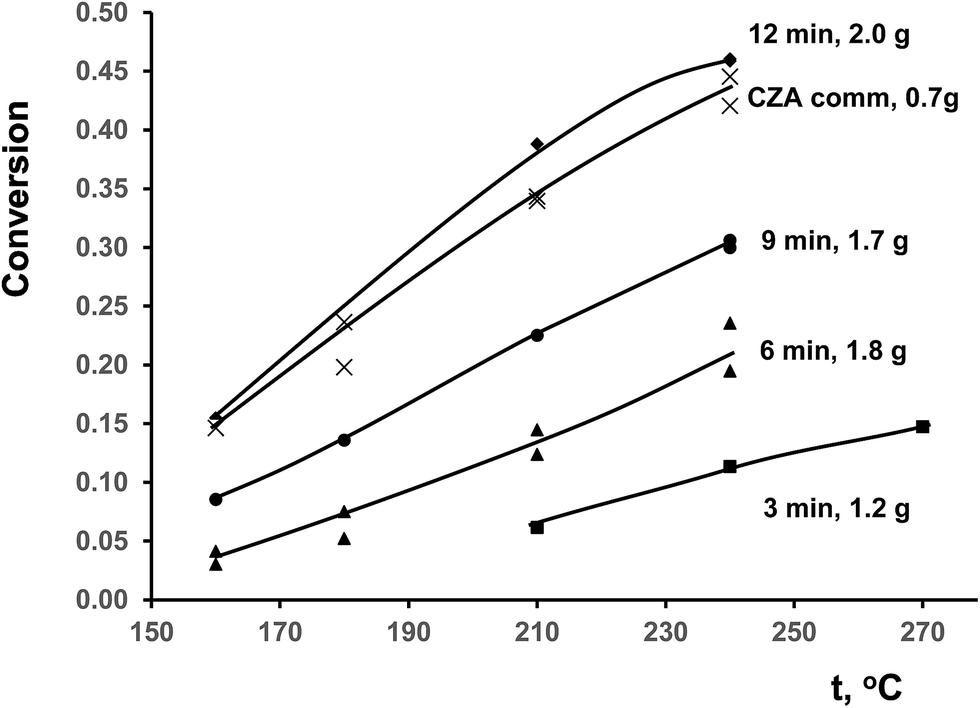 | ||
| Fig. 8 Temperature dependence of CO conversion in WGSR for studied catalysts. The weights of samples were varied due to difference of loading density. | ||
Due to different specific densities of ceramometals a proper comparison of their catalytic activity requires estimation of specific rate constants listed in Table 2. When the rate constant is estimated using the equation for the plug-flow reactor, an important parameter is the contact time, which at the same weight hourly space velocity depends on the volume of the catalyst bed. In the case of ceramometal catalysts, their much higher density due to presence of metallic particles should be taken into account. This results in a substantial decrease in the volume of the catalytic layer at comparable catalyst weights (Table 2). The estimated rate constant (k, s−1) at 240 °C monotonically increases with the time of preliminary MA approximately by a factor of 2.5, but remains lower as compared to that for the porous oxide catalyst (Table 2). Stability of the catalysts changes in a similar way, while stability of CZA is better. Obviously, the key factor determining the catalyst activity is the specific surface area; on the average, its value for the ceramic analog exceeds that for ceramometal catalysts by an order of magnitude (Table 2).
| Sample | m, g | δgran, g cm−3 | δcat.bed, g cm−3 | k, s−1 | Stability, arb.un. | SBET, m2 g−1 | k/(m × SBET), s−1 m−2 | Cu/(Cu + Al) | k/(m × SCu), s−1 m−2 Cu |
|---|---|---|---|---|---|---|---|---|---|
| a δcat.bed – catalyst bed density; Cu/(Cu + Al) – from XPS data. | |||||||||
| CuAl(3) | 1.24 | 4.24 | ∼2.3 | 2.8 | 0.6 | 14.4 | 0.2 | 0.59 | ∼0.3 |
| CuAl(6) | 1.79 | 4.22 | ∼2.2 | 3.3 | 0.8 | 9.9 | 0.2 | 0.51 | ∼0.4 |
| CuAl(9) | 1.71 | 4.00 | ∼2.1 | 4.8 | 0.8 | 9.0 | 0.3 | 0.52 | ∼0.6 |
| CuAl(12) | 2.00 | 3.87 | ∼2.0 | 7.8 | 0.9 | 15.7 | 0.2 | 0.59 | ∼0.3 |
| CZA comm | 0.70 | 2.00 | ∼1.0 | 9.1 | >0.9 | 121 | 0.1 | 0.39 | ∼0.3 |
However, the specific (per the unit surface area) activity of ceramometal catalysts is 2–3 times higher as compared to ceramic copper–zinc–aluminum one (see Table 2: k/(m × S)). It means that the concentration of active sites (known to be copper atoms)1,2 on the surface of ceramometal catalysts is substantially higher.
In principle, this observation is supported by XPS data, according to which the concentration of copper on the surface of all ceramometal catalysts after catalytic testing is higher in comparison with ceramic CZA one (Table 2). It should be noted that reduction of these catalysts decreases the surface copper concentration, but the general tendency is preserved. In a similar way, the surface concentration of copper in various oxides with the perovskite structure was found to be approximately twofold higher as compared to copper catalysts for WGSR.44
A more detailed analysis showed that changes in the copper concentration on the surface (∼30%) are not so pronounced as changes in specific activity (∼200%) (Table 2). Thus, it can be assumed that differences between the catalysts are caused not only by the concentration of active sites but also by a considerable difference in specific activity of these sites. This is confirmed also by the values of constants referred to the unit surface area of metallic copper, which were estimated form XPS data and the total specific surface area of the catalysts. For most of ceramometal catalysts, the indicated values are higher than for the oxide catalyst (Table 2). Thus, ceramometal catalysts have sites with a higher specific activity as compared to the conventional catalyst containing mixed oxides. The concentration of such sites on the surface of ceramometals goes through the maximum as the time of preliminary MA is increased (Table 2). An essential difference in the activity per the unit surface area of metallic copper was observed in earlier works as well.1,44,45
As shown by XRD, samples of ceramometal catalysts after catalytic testing have nearly the same phase composition as the initial samples, except for CuO, which turns into metallic copper (Fig. 9). With the increase of the time of preliminary mechanical alloying of Cu–Al powder the average crystallite size of the solid solution decreases from 25–34 nm to ∼11 nm, and that of the Al4Cu9 intermetallic – from 33–46 to 14–19 nm. The size of metallic copper particles changes non-monotonically. First it decreases from 17–24 (3 min MA) to 7–10 nm for the 6 min sample, and then increases to 28–40 nm for the 12 min sample. However, we should stress that XRD is not able to distinguish particles of metallic copper formed on the surface of oxide microshell envelope and its crystallites located within the metal core of ceramometals. Moreover, XRD is not able to detect nanoparticles with the size below 2 nm. Thus, according to NMR data, in the initial ceramometals metallic copper is present, while it is not revealed by XRD (vide supra Fig. 1–3). Hence, analysis of X-ray particle sizes for metallic copper in CuAlO/CuAl ceramometals is apparently not informative as compared with XPS data.
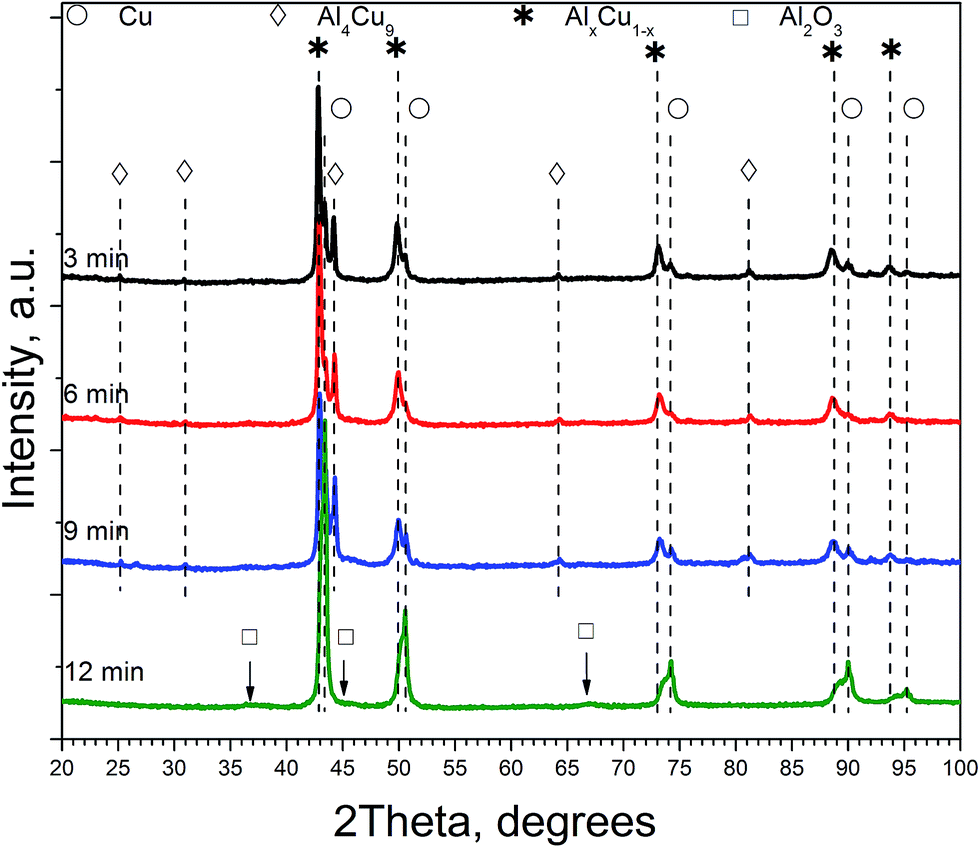 | ||
| Fig. 9 Diffraction patterns of cermets at different MA times after reduction in hydrogen (270 °C) and catalytic experiments. Symbols denote peaks corresponding to the following phases: * – solid solution of aluminum in copper, AlxCu1, ◊ – Al4Cu9 intermetallic [24-0003], ○ – Cu [04–0836], □ – Al2O3. | ||
A comparison of the values of constants referred to the unit surface area of metallic copper by XPS data with the content of the Al4Cu9 intermetallic in cermets revealed quite a good correlation of these values (Fig. 10). Note that the unknown intermetallic phase also has a maximum concentration in the sample activated for 9 min (Fig. 4) with the highest activity per the surface site. This suggests that the highly active sites of metallic copper are located on the surface of various copper and aluminum alloys, or more exactly, on the surface of the oxide substrate that forms a microshell envelope on the surface of these cores after HTT and calcination in air. The activity of such sites is much higher as compared to the known sites, so their structure should have some distinctive features. Most likely, these sites are associated with copper in a high dispersion having a clustered subnanosized structure. According to the literature, the specific surface area of metallic copper particles tested in WGSR does not exceed 35 m2 g−2; so in a spherical approximation, this gives the particle size not smaller than 19 nm. In our case, copper clusters on the surface of ceramometals seem to have a much smaller size, which could make them much more active.
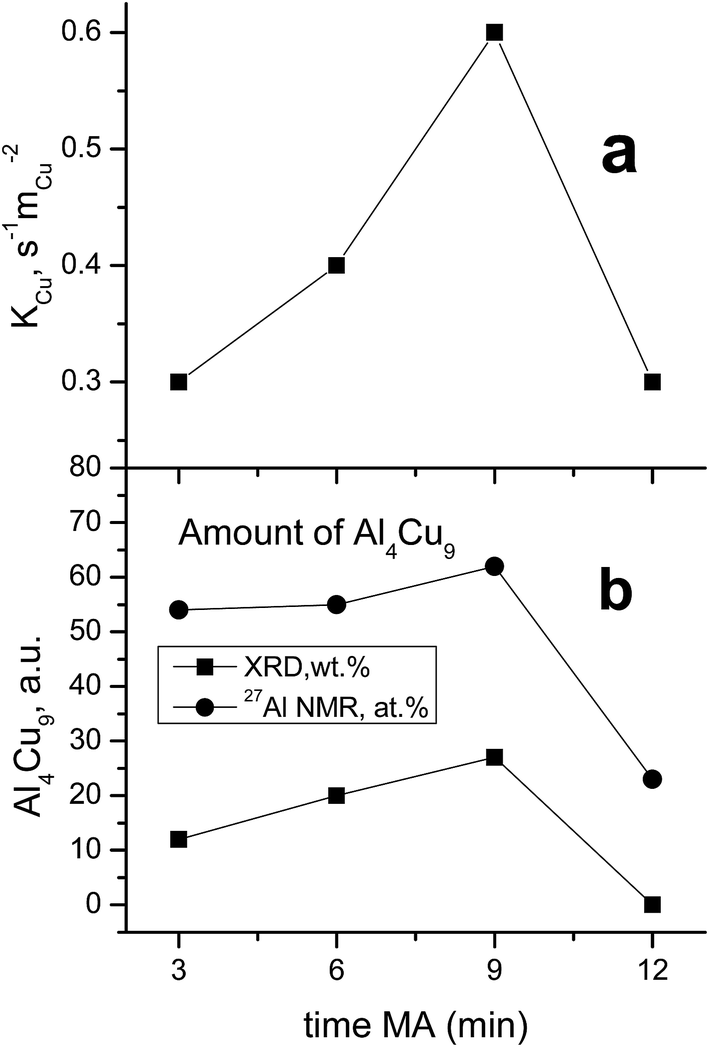 | ||
| Fig. 10 The effect of the time of preliminary MA of the Cu–Al precursor on specific catalytic activity of surface copper (a) and a relative content of Al4Cu9 intermetallic according to XRD and NMR data (the content of aluminum in cermets with respect to that in other phases) (b). | ||
In an earlier study on steam reforming of methanol over the catalysts synthesized from Cu–Al–Fe alloys, a conclusion was made on the presence of “dispersed copper particles which cannot be observed by XRD”.11 Later, the presence of dispersed particles was associated with the formation of a thin oxide layer on the surface of Cu–Al–Fe alloy, which stabilizes the dispersed copper particles.12 The effect of a substrate on the properties of such subnanoparticles (clusters) is caused not only by stabilization of subnanosizes but also by a partial distortion of the structure and variation of the chemical properties due to a strong interaction with support.35 The surface structure of the Al4Cu9 intermetallic, which was described in detail in,46 can be used as an example to analyze the effect of such alloys on the formation of subnanodispersed structures of metallic copper on their surface. Thus, for the (110) face, aluminum actually ‘decorates’ the surface of metallic copper, separating it into the aggregates including from two to some tens of copper atoms. It is well known that the bulk oxidation of aluminum alloys with transition metals is hindered by the formation of a dense oxide layer. The formation of the subsurface layer during HTT and calcination in air may be accompanied by the emergence of oxide subnanostructures differing from the structures of bulk oxides observed by structural methods. In the course of reductive treatments, copper will change its oxidized state to the metallic one, whereas aluminum will not do this. Overall, this should stabilize copper subnanoparticles (clusters), which possess an increased catalytic activity. Similar stable nanocomposites Cu/Al2O3, which are formed upon reduction of copper and aluminum mixed oxides with the delafossite structure by hydrogen are reported in the literature.30
More coarse particles of metallic copper observed by XRD in oxidized cermets are formed most likely from copper oxide.12 The absence of a strict correlation between concentration of intermetallic or copper on the surface and specific activity may be caused by changes in the ratio of nanodispersed and more active subnanodispersed copper stabilized on the surface of intermetallic in dependence on the properties of the precursor.
3.4. Pore structure and its effect on the strength and activity
The volume of micro- and mesopores in ceramometal catalysts is not high (0.01–0.04 cm3 g−1) (Table 3). It is much lower as compared to the bulk oxide analog (0.25 cm3 g−1). This determines a much higher specific surface of the latter (Table 3). However, the macropore volume (>0.1 μm) of ceramic catalyst estimated from the difference between the total pore volume and mesopore volume is close to zero, whereas for ceramometal catalysts it is equal to 0.04–0.07 cm3 g−1 (Table 3). Macropores of ceramometals with the size up to tens of microns are clearly seen as black spots on the electron microscopy images (Fig. 6). These macropores are located between cermet particles comprised of the metal cores with the oxide shell, while mesopores are situated in the oxide shells. The macropore volume changes non-monotonically and contrariwise to the strength of cermets. Sample with 9 min of MA has a minimum strength (Table 3). However, irrespective of the developed macropore structure, which usually results in the decrease of the catalyst strength, the total strength of ceramometals is comparable with the strength of ceramic catalyst (8 MPa).| Sample | Time MA, min | SSA, m2 g−1 | Vmeso, cm3 g−1 | Vmacro, cm3 g−1 | Compress. strength, MPa |
|---|---|---|---|---|---|
| CuAl(3) | 3 | 14.4 | 0.02 | 0.05 | 10.5 |
| CuAl(6) | 6 | 9.9 | 0.01 | 0.06 | 9.9 |
| CuAl(9) | 9 | 9.0 | 0.02 | 0.07 | 7.7 |
| CuAl(12) | 12 | 15.7 | 0.04 | 0.04 | 9.7 |
| CZA | — | 122 | 0.25 | <0.001 | 8.0 |
The developed macropore structure should enhance the diffusion permeability of the granules which is a traditional shape of industrial catalysis.44 A high diffusion permeability will equalize the activity of the coarse fraction of ceramometal catalysts and ceramic catalyst. Indeed, the activity of a small fraction of cermet (12 min MA) was lower as compared to that of the oxide catalyst, whereas an opposite trend was observed for the coarse fraction (Fig. 11): activity of the 3 × 5 mm2 cermet granules (k = 5.1 s−1) exceeded by more than 20% the activity of granulated oxide catalyst (k = 4.4 s−1). Thus, when going from a fine to a coarse fraction, activity of the cermet decreases by 35%, and activity of the oxide catalyst by 52%. Note also that the density of ceramometals (∼4 g cm−3) is substantially higher than that of CZA (∼2 g cm−3), while for known oxide catalysts the density is even lower (∼1 g cm−3),10 which compensate their high activity per the mass unit. These factors are important arguments demonstrating that ceramometal catalysts are promising for WGSR.
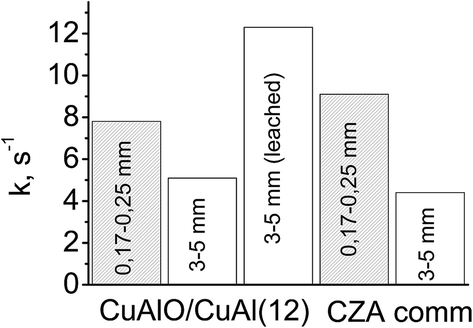 | ||
| Fig. 11 Activity of small and coarse fractions of different catalysts in WGSR (240 °C, CO:H2O:H2 = 8:42:50). | ||
Leaching that promotes formation of additional nanostructures based on copper oxide can be an additional factor increasing the concentration of surface active sites.47 In steam reforming of methanol over the catalysts synthesized from Cu–Al–Fe alloys, a mild leaching results in the substantial increase of activity.11,12 In our study, a similar treatment of cermet granules also gave more than a twofold increase in the catalyst activity (Fig. 11).
This effect can be also related to the presence of aluminum-enriched layers on the surface of Cu–Al alloys.46 After oxidative treatments, such a layer will be oxidized and block the surface of metallic copper. Upon leaching of aluminum, blocking of the surface will decrease and activity will increase. It cannot be ruled out that the decreased activity for 3 and 6 min samples, irrespective of a relatively high concentration of the intermetallic (Fig. 10), is related exactly to the high concentration of aluminum in the surface layer, which decreases with increasing the MA time to 9 and 12 min.
For the most active ceramometal CuAlO/CuAl (MA time 12 min), the effective linear thermal expansion coefficient for the temperature range of 25–500 °C was equal to ∼10 × 10−6 (deg−1). For other cermets, it varied within a range from 9 to 28 × 10−6 (deg−1) in dependence on the MA time for a powdered precursor, affecting both the metal/oxide ratio and the pore volume of ceramometals. Overall, the variation range of thermal expansion coefficient makes it possible to select such conditions of the synthesis which would protect ceramometal coatings against cracking and peeling from the metal tubes or other metallic substrates (microchannel plates, foils, etc.). Thus, ceramometal catalysts are promising for use in compact devices for the production of hydrogen.
4 Conclusions
The evolution of the structure and microstructure of CuAlO/CuAl ceramometals synthesized by mechanochemical activation of the Cu + Al mixture (Cu:Al = 87:13 wt%) with subsequent hydrothermal treatment leading to integration of the powder into a monolith, drying and calcination was studied in dependence on the MA time. It was shown that such ceramometals have the egg-shell microstructure where the metallic cores containing aluminum and copper alloys are surrounded by the oxide matrix containing copper oxides and X-ray amorphous mixed oxides of copper and aluminum. The study revealed non-monotonic changes in the concentration of Al4Cu9 intermetallic in cermets to occur with variation of the MA time from 3 to 12 min and a similar change in the activity referred to the unit surface area. A high concentration as well as a high specific activity of metallic copper species located in the oxide shell covering intermetallic nuclei appear to be due to their subnanodispersed character.
The activity and textural–mechanical properties of cermets were compared with those for the conventional oxide catalyst containing mixed oxides of copper, zinc and aluminum. The activity of a fine (0.14–0.25 mm) fraction of ceramic catalyst was shown to exceed that of ceramometal catalysts due to the developed microporous structure, which provides nearly an order of magnitude greater specific surface area as compared to ceramometals. However, the macropore structure of such a catalyst is developed insufficiently. As a result, the activity of a coarse (3–5 mm) fraction of the ceramometal catalyst decreases to a smaller extent as compared to ceramic one, so their activities become comparable. In the average ceramometal granules possess twice as higher specific density as compared with that of ceramic catalysts. Mild leaching produces an additional increase in the activity of granulated cermets.
Large amount of metal particles provides a high thermal conductivity of ceramometals and makes it possible to construct a reactor with a controllable temperature profile along the catalyst bed, which is very important for providing a maximum efficiency of the process, not only of WGSR but any process running with a great thermal effect and having restrictions on the equilibrium or selectivity.
Conflicts of interest
There are no conflict to declare.Acknowledgements
This work was in part supported by FP7 Project BIOGO.References
- G. C. Chinchen and M. S. Spencer, Catal. Today, 1991, 10, 293–301 CrossRef CAS
.
- P. Kowalik, M. Konkol, K. Antoniak, W. Próchniak and P. Wiercioch, J. Mol. Catal. A: Chem., 2014, 392, 127–133 CrossRef CAS
- T. Utaka, K. Sekizawa and K. Eguchi, Appl. Catal., A, 2000, 194–195, 21–26 CrossRef CAS
- X. Lin, Y. Zhang, L. Yin, C. Chen, Y. Zhan and D. Li, Int. J. Hydrogen Energy, 2014, 39, 6424–6432 CrossRef CAS
- A. Jha, D.-W. Jeong, W.-J. Jang, Y.-L. Lee and H.-S. Roh, Int. J. Hydrogen Energy, 2015, 40, 9209–9216 CrossRef CAS
- A. Budiman, M. Ridwan, S. M. Kim, J.-W. Choi, C. W. Yoon, J.-M. Ha, D. J. Suh and Y.-W. Suh, Appl. Catal., A, 2013, 462–463, 220–226 CrossRef CAS
- R. T. Figueiredo, H. M. C. Andrade and J. L. G. Fierro, J. Mol. Catal. A: Chem., 2010, 318, 15–20 CrossRef CAS
- C. Ratnasamy and J. P. Wagner, Catal. Rev.: Sci. Eng., 2009, 51, 325–440 CAS
- N. A. Baronskaya, T. P. Minyukova, A. A. Khassin, T. M. Yurieva and V. N. Parmon, Russ. Chem. Rev., 2010, 79, 1027–1046 CrossRef CAS
- D. J. Moon and J. W. Ryu, Catal. Lett., 2004, 92, 17–24 CrossRef CAS
- T. Tanabe, S. Kameoka and A. P. Tsai, Catal. Today, 2006, 111, 153–157 CrossRef CAS
- T. Tanabe, S. Kameoka and A. P. Tsai, J. Mater. Sci., 2011, 46, 2242–2250 CrossRef CAS
- D. V. Dudina, O. I. Lomovsky, K. R. Valeev, S. F. Tikhov, N. N. Boldyreva, A. N. Salanov, S. V. Cherepanova, V. I. Zaikovskii, A. S. Andreev, O. B. Lapina and V. A. Sadykov, J. Alloys Compd., 2015, 629, 343–350 CrossRef CAS
- T. M. Yurieva, T. P. Minyukova, L. P. Davidova, O. V. Makarova, L. M. Plyasova and V. F. Anufrienko, Russian patent #2046656, 1995
- B. Li and K. Xiao, J. Appl. Phys., 1992, 71, 3917–3921 CrossRef CAS
- G. S. Carter, L. H. Bennett and D. J. Kahan, in Metallic Shifts in NMR, A review of theory and comprehensive critical data compilation of metallic materials (Progress in materials science, v. 20), ed. B. Chalmers, J. W. Christian and T. B. Massalski, Pergamon Press, Oxford, New York, Toronto, Sidney, Paris, Frankfurt, 1977, vol. 20, pp. 3–21 Search PubMed
- L. Bai-qin and N. Chu, Acta Phys. Sin., 2000, 49, 155–159 Search PubMed
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef
- N. Fairley, www.casaxps.com.
- Z. R. Ismagilov, R. A. Shkrabina and N. A. Koryabkina, Catal. Today, 1999, 47, 51–71 CrossRef CAS
- B. S. M. Seeber, U. T. Gonzenbach and L. J. Gauckler, J. Mater. Res., 2013, 28, 2281–2287 CrossRef CAS
- O. A. Kungurova, A. A. Khassin, S. V. Cherepanova, A. A. Saraev, V. V. Kaichev, N. V. Shtertser, G. K. Chermashentseva, E. Yu. Gerasimov, E. A. Paukshtis, O. V. Vodyankina, T. P. Minyukova and G. Abou-Jaoudé, Appl. Catal., A, 2017, 539, 48–58 CrossRef CAS
- K. Wieczorek-Ciurowa, K. Gamrat and Z. Sawłowicz, J. Therm. Anal. Calorim., 2005, 80, 619–623 CrossRef CAS
- R. Besson, M.-N. Avettand-Fènoël, L. Thuinet, J. Kwon, A. Addad, P. Roussel and A. Legris, Acta Mater., 2015, 87, 216–224 CrossRef CAS
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid State NMR of Inorganic Materials, Pergamon/Elsevier, Oxford, 2002, vol. 6 Search PubMed
- S. F. Tikhov, Y. V. Potapova, V. A. Sadykov, V. B. Fenelonov, I. V. Yudaev, O. Lapina, A. N. Salanov, V. I. Zaikovkii and G. S. Litvak, Mater. Res. Innovations, 2005, 9, 431–446 CAS
- E. Ghanti and R. Nagarajan, Dalton Trans., 2010, 39, 6056 RSC
- H. S. C. O'Neill, M. James, W. A. Dollase and S. A. T. Redfern, Eur. J. Mineral., 2005, 17, 581–586 CrossRef
- M. Kracum, A. Kundu, M. P. Harmer and H. M. Chan, J. Mater. Sci., 2015, 50, 1818–1824 CrossRef CAS
- J. van den Brand, P. C. Snijders, W. G. Sloof, H. Terryn and J. H. W. de Wit, J. Phys. Chem. B, 2004, 108, 6017–6024 CrossRef CAS
- N. Kosova, E. Devyatkina, A. Slobodyuk and V. Kaichev, Solid State Ionics, 2008, 179, 1745–1749 CrossRef CAS
- J. Mendialdua, R. Casanova, F. Rueda, A. Rodriguez, J. Quinones, L. Alarcon, E. Escalante, P. Hoffmann, I. Taebi and L. Jalowiecki, J. Mol. Catal. A: Chem., 2005, 228, 151–162 CrossRef CAS
- B. R. Strohmeier, D. E. Levden, R. S. Field and D. M. Hercules, J. Catal., 1985, 94, 514–530 CrossRef CAS
- S. F. Tikhov, V. A. Sadykov, G. N. Kryukova, E. A. Paukshtis, V. V. Popovskii, T. G. Starostina, G. V. Kharlamov, V. F. Anufrienko, V. F. Poluboyarov and V. A. Razdobarov, J. Catal., 1992, 134, 506–524 CrossRef CAS
- L. Fu and H. Yang, J. Phys. Chem. C, 2014, 118, 14299–14315 CAS
- J. Batista, A. Pintar, D. Mandrino, M. Jenko and V. Martin, Appl. Catal., A, 2001, 206, 113–124 CrossRef CAS
- V. I. Bukhtiyarov, V. V. Kaichev and I. P. Prosvirin, Top. Catal., 2005, 32, 3–15 CrossRef CAS
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS
- J. C. Otamiri, S. L. T. Andersson and A. Andersson, Appl. Catal., 1990, 65, 159–174 CrossRef CAS
- S. Poulston, P. M. Parlett, P. Stone and M. Bowker, Surf. Interface Anal., 1996, 24, 811–820 CrossRef CAS
- M. Richter, M. Fait, R. Eckelt, M. Schneider, J. Radnik, D. Heidemann and R. Fricke, J. Catal., 2007, 245, 11–24 CrossRef CAS
- A. Wollner, F. Lange, H. Schmelz and H. Knozinger, Appl. Catal., A, 1993, 94, 181–203 CrossRef
- S. S. Maluf, P. A. P. Nascente, C. R. M. Afonso and E. M. Assaf, Appl. Catal., A, 2012, 413–414, 85–93 CrossRef CAS
- A. Boisen, T. V. W. Janssens, N. Schumacher, I. Chorkendorff and S. Dahl, J. Mol. Catal. A: Chem., 2010, 315, 163–170 CrossRef CAS
- É. Gaudry, A. K. Shukla, T. Duguet, J. Ledieu, M.-C. DeWeerd, J.-M. Dubois and V. Fournée, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 085411 CrossRef
- J. Li, Z. Yang, T. Wang, N. Yu, L. Sun, C. Nie, H. Teng, C. Jin, X. Chen and H. Geng, RSC Adv., 2015, 5, 75044–75054 RSC
| This journal is © The Royal Society of Chemistry 2017 |