
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhthalide derivatives from Ligusticum chuanxiong†
Xu Zhang,
Bing Han,
Zi-Ming Feng,
Ya-Nan Yang ,
Jian-Shuang Jiang and
Pei-Cheng Zhang*
,
Jian-Shuang Jiang and
Pei-Cheng Zhang*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, People′s Republic of China. E-mail: pczhang@imm.ac.cn
First published on 31st July 2017
Abstract
Eleven new phthalide derivatives (1–11) have been isolated from the rhizome of Ligusticum chuanxiong. In particular, 1 and 2 contain a mercaptopropionic acid moiety in the phthalide derivatives. All the structures, including their absolute configurations, were determined by UV, IR, HRESIMS, 1D and 2D NMR spectroscopic measurements, and by a comparison of the experimental and calculated electronic circular dichroism (ECD) spectra. Results of a bioassay showed that compound 4 has a moderate neuroprotective activity on human neuroblastoma SH-SY5Y cell injury induced by H2O2 and oxygen glucose deprivation (OGD).
Introduction
The rhizome of Ligusticum chuanxiong Hort. (syn. Ligusticum striatum DC.) (Apiaceae), named Chuanxiong in traditional Chinese medicine, is commonly used for treating atherosclerosis, cardiovascular disease, anemia and hypertension.1 This plant is mainly distributed in the Sichuan province but also in Yunnan, Guizhou, and Guangxi provinces in China. So far, the phenolic acids, alkaloids, and phthalides2–16 contained in it have been investigated. Among these compounds, tetramethylpyrazine and ferulic acid were considered to be the active constituents.17–24 The phthalides from L. chuanxiong have attracted particular attention due to their vasodilative,17,25,26 neuroprotective,27 antiproliferative,28,29 anti-inflammatory24 and antibacterial effects.30 In particular, Z-ligustilide, a phthalide isolated from L. chuanxiong, displays various pharmacological activities including neuroprotective, anti-inflammatory, antiproliferative and vasorelaxation effects.31–34 It is considered a major bioactive compound relevant to the therapeutic effects and has attracted great interest in recent years. Overall, more than 40 phthalide lactones have been obtained from the rhizome of L. chuanxiong,35,36 but most of them were isolated from the liposoluble fraction. In consideration of the bioavailability in vivo, the phthalide derivatives from the water-soluble extract of this plant deserve to be the primary focus of further studies. In the course of our study, eleven new phthalide derivatives (1–11) have been isolated and their structures were elucidated by various spectroscopic methods (Fig. 1). Subsequently, their neuroprotective activities on SH-SY5Y cell injury induced by H2O2 and OGD were evaluated at a concentration of 10 μM.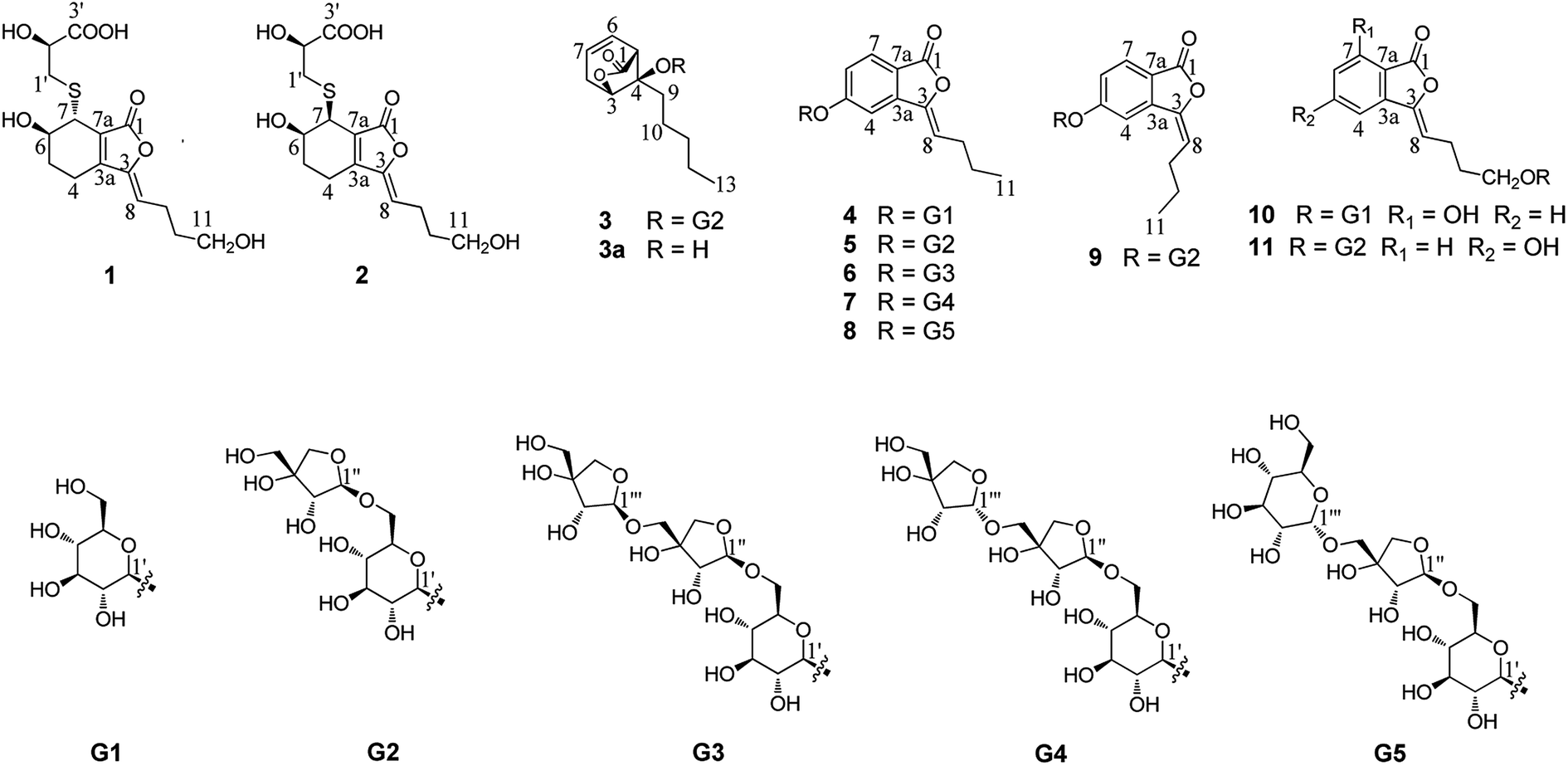 | ||
| Fig. 1 Chemical structures of 1–11. | ||
Results and discussion
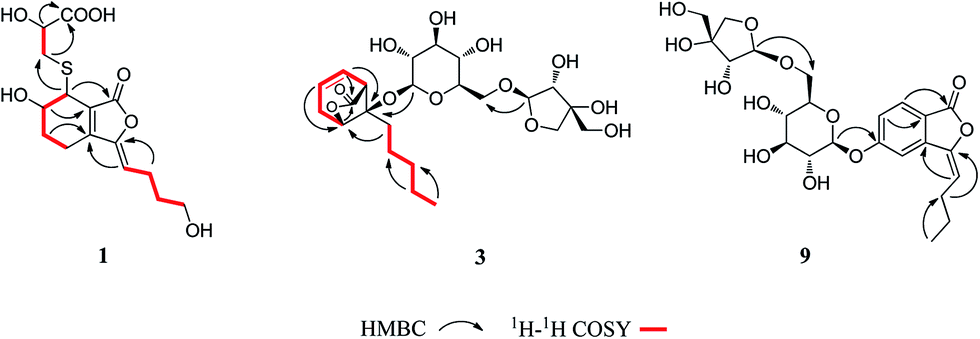
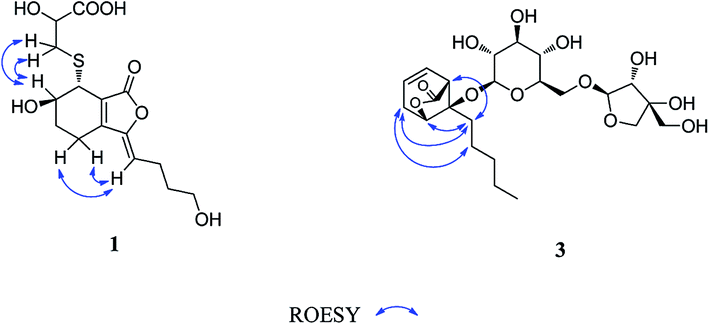
Compound 1 was obtained as a white amorphous powder, and its molecular formula was established as C15H20O7S by HRESIMS at m/z 345.0999 [M + H]+ (calcd 345.1003). The IR spectrum peaks at 3387 cm−1, 1741 cm−1 and 1682 cm−1 suggest the presence of hydroxy, γ-lactone and diene groups, respectively. In the 1H NMR spectra (Table 1), a double bond at δH 5.48 (1H, t, J = 7.5 Hz, H-8), six methylene resonances at δH 2.43 (2H, m, H-4), 1.81 (2H, m, H-5), 2.31 (2H, q, J = 7.5 Hz, H-9), 1.56 (2H, m, H-10), 3.41 (2H, t, J = 6.0 Hz, H-11), 2.89 (1H, dd, J = 7.0, 13.5 Hz, H-1′a), 3.02 (1H, dd, J = 4.5, 13.5 Hz, H-1′b), one methine resonance at δH 3.50 (1H, brs, H-7), and two oxygen-methine resonances at δH 4.10 (1H, brs, H-6) and 4.17 (1H, dd, J = 4.5, 7.0 Hz, H-2′) were presented. The 13C NMR spectrum (Table 1) shows 15 carbon resonances, including two carboxyl carbons at δC 173.9 (C-3′) and 168.1 (C-1), four olefinic carbons, five methylene carbons, a methine carbon resonance at δC 41.8 (C-7), an oxygen-methylene carbon resonance at δC 60.2 (C-11), and two oxygen-methine resonances at δC 67.5 (C-6) and 70.4 (C-2′). In the HMBC experiment (Fig. 2), the correlations from H-8 (δH 5.48) to C-3 (δC 147.7) and C-3a (δC 151.6) suggest the existence of a 1,3-butadiene moiety. Furthermore, it was confirmed to be connected to a 3-hydroxypropyl on the basis of the correlations of the 1H–1H COSY experiment (Fig. 2), including those between H-8 and H-9, H-9 and H-10, and H-10 and H-11. In addition, the 1H–1H COSY correlations from H-4 to H-5, from H-5 to H-6, and from H-6 to H-7 together with the HMBC correlations from H-6 (δH 4.10) to C-7a, from H-7 (δH 3.50) to C-7a and C-3a, from H-5 (δH 1.81) to C-3a, and from H-4 (δH 2.43) to C-3a and C-7a indicate the existence of a cyclohexene moiety. The 2-carboxyl-2-hydroxy-ethylthio moiety on position C-7 was confirmed by the 1H–1H COSY correlations from H-1′ (δH 2.89, 3.02) to H-2′ (δH 4.17) and the HMBC correlations from H-1′ and H-2′ to the carbonyl carbon at δC 173.9 and from H-7 to C-1′ at δC 36.9 in consideration of the changes of the chemical shifts of C-7 and C-1′. The HMBC correlation from H-7 to the carbonyl at δC 168.1 indicates that C-7a connected with the carbonyl. Thus, the planar structure was determined to be 6-hydroxy-7-(2-carboxyl-2-hydroxy-ethylthio)-3-(4-hydroxybutylidene)-4,5,6,7-tetrahydrophthalide. The absolute configuration of C-2′ in 1 was established by a dimolybdenum tetraacetate [Mo2(AcO)4]-induced circular dichroism procedure.37 The diagnostic Cotton effect approximately 344.5 nm was negative, so the absolute configuration of C-2′ in 1 was assigned as S. In the ROESY spectra (Fig. 3), the correlation between H-8 and H-4 proved the Z-configuration of the double bond on position C-3, and the correlation between H-6 and H-1′ suggested that their relative configuration was trans. Their absolute configuration was determined by comparing the experimental and calculated ECD data based on the time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31+G (d,p) level. Considering that the variations in the side chain resulted in too many conformations and had little effect on the CD spectrum of the tetrahydrophthalide core in 1, the selected conformers 1Ja (6S,7S) and 1Jb (6R,7R) were calculated after optimization (Fig. 4). A comparison of the theoretically calculated and experimental ECD curves (Fig. 5) permitted the assignment of the absolute configuration of 1 as 6R,7R. Finally, the structure of 1 was determined to be (3Z,3aE)-(6R,7R,2′S)-6-hydroxy-7-(2-carboxyl-2-hydroxyethylthio)-3-(4-hydroxybutylidene)-4,5,6,7-tetrahydrophthalide, named thiosenkyunolide A.| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 168.1 | 167.1 | 174.7 | |||
| 3 | 147.7 | 147.4 | 4.58, brs | 78.9 | ||
| 3a | 151.6 | 150.9 | ||||
| 4 | 2.43, m | 16.1 | 2.41, m | 19.6 | 84.2 | |
| 2.55, m | ||||||
| 5 | 1.81, m | 22.9 | 1.74, m | 25.8 | 3.21, dd (1.5, 7.5) | 47.7 |
| 6 | 4.10, brs | 67.5 | 3.95, dd (3.5, 10.5) | 68.2 | 5.87, m | 123.9 |
| 7 | 3.50, brs | 41.8 | 3.81, d (3.5) | 43.6 | 5.77, m | 127.9 |
| 7a | 124.2 | 126.1 | ||||
| 8 | 5.48, t (7.5) | 112.7 | 5.42, t (7.5) | 112.5 | 2.46, m | 31.3 |
| 9 | 2.31, q (7.5) | 22.4 | 2.29, q (7.5) | 22.4 | 1.67, m | 26.1 |
| 10 | 1.56, m | 31.8 | 1.55, m | 31.7 | 1.38, m | 21.7 |
| 11 | 3.41, t (6.0) | 60.2 | 3.40, t (6.5) | 60.2 | 1.19, m | 31.3 |
| 12 | 1.25, m | 21.7 | ||||
| 13 | 0.85, t (7.5) | 13.7 | ||||
| 1′ | 2.89, dd (7.0, 13.5) | 36.9 | 2.93, dd (7.0, 13.5) | 37.9 | 4.38, d (8.0) | 97.2 |
| 3.02, dd (4.5, 13.5) | 3.07, dd (4.5, 13.5) | |||||
| 2′ | 4.17, dd (4.5, 7.0) | 70.4 | 4.12, dd (4.5, 7.0) | 70.7 | 2.94, m | 73.2 |
| 3′ | 173.9 | 174.1 | 3.28, m | 75.2 | ||
| 4′ | 2.99, m | 70.1 | ||||
| 5′ | 3.14, m | 76.9 | ||||
| 6′ | 3.35, m | 67.9 | ||||
| 3.81, dd (1.5, 10.5) | ||||||
| 1′′ | 4.84, d (3.0) | 109.4 | ||||
| 2′′ | 3.74, brs | 75.9 | ||||
| 3′′ | 78.6 | |||||
| 4′′ | 3.58, d (9.5) | 73.4 | ||||
| 3.85, d (9.5) | ||||||
| 5′′ | 3.35, m | 63.4 | ||||
 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of 1, 3 and 9. | ||
 | ||
| Fig. 3 Key ROESY correlations of 1 and 3. | ||
 | ||
| Fig. 4 The structures of 1Ja, 1Jb, 2Ja and 2Jb. | ||
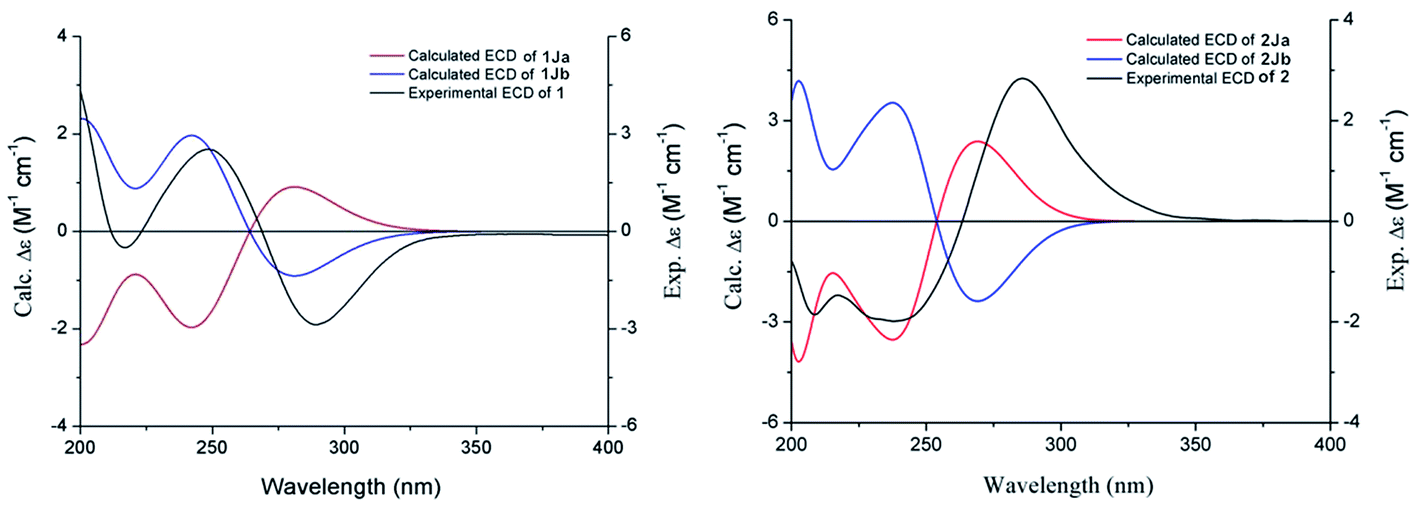 | ||
| Fig. 5 Experimental ECD and calculated ECD spectrum of 1 and 2 in MeOH. | ||
The HRESIMS of 2 gave the same molecular formula as 1, C15H20O7S, based on the [M + Na]+ ion peak at m/z 367.0827 (calcd 367.0822). The 1H and 13C NMR spectra of 2 (Table 1) were closely comparable to those of 1, with the minor differences occurring in the signals in the cyclohexene moiety, that is, the 13C NMR data were shifted downfield, except that of C-3a (Table 1). Further 2D NMR analysis, including HSQC, HMBC, and 1H–1H COSY experiments, confirmed that compound 2 was the optical isomer of compound 1. Using the same method, the absolute configuration of C-2′ in 2 was established as S by a dimolybdenum tetraacetate [Mo2(AcO)4]-induced circular dichroism procedure,37 whose diagnostic Cotton effect at approximately 345 nm was also negative. In the ROESY spectra, the correlation between H-8 and H-4 proved the Z-configuration of the double bond at position C-3, but the correlation between H-6 and H-1′ was not observed in 2 compared with 1, which suggested that the relative configuration between them was cis. The absolute configurations of 6R,7S were further confirmed by a comparison of the experimental ECD spectrum and calculated ECD data using the aforementioned methods (Fig. 5). Thus, the structure of 2 was determined to be (3Z,3aE)-(6R,7S,2′S)-6-hydroxy-7-(2-carboxyl-2-hydroxyethylthio)-3-(4-hydroxylbutylidene)-4,5,6,7-tetrahydrophthalide, named thiosenkyunolide B.
Compound 3 was obtained as a white amorphous powder. Its molecular formula was identified as C23H36O12 (m/z 503.2148 [M − H]−) by negative HRESIMS. The IR absorptions at 3336 cm−1, 1774 cm−1 and 1049 cm−1 represented a hydroxyl group, carbonyl group and alkenyl group, respectively. The 1H NMR and 13C NMR spectra (Table 1) of 3 displayed an amyl side chain that produced a methyl resonances [δH 0.85 (3H, t, J = 7.5 Hz, H-13); δC 13.7] and four methylene resonances [δH 1.25 (2H, m, H-12), 1.19 (2H, m, H-11), 1.38 (2H, m, H-10), 1.67 (2H, m, H-9); δC 21.7, 31.3, 21.7, 26.1, respectively]; two olefinic [δH 5.77 (1H, m, H-7), 5.87 (1H, m, H-6); δC 127.9, 123.9]; an oxymethine [δH 4.58 (1H, brs, H-3); δC 78.9] and a methine [δH 3.21 (1H, dd, J = 1.5, 7.5 Hz, H-5); δC 47.7]; and one methylene [2.46 (2H, m, H-8); δC 31.3]. Furthermore, a carbonyl carbon at δC 174.7 (C-1) and a quaternary carbon δC 84.2 (C-4) were observed. The presence of two anomeric protons at δH 4.38 (1H, d, J = 8.0 Hz, H-1′) and 4.84 (1H, d, J = 3.0 Hz, H-1′′) and eleven carbons proved that compound 3 contained two sugar moieties. Comparison of the NMR data of 3 with those of the known compound ligusticoside A16 suggested that these two compounds shared a similar skeleton except sugar moieties. In the HMBC spectrum, the correlations H-1′/C-4 and H-1′′/C-6′ proved that the glucose moiety was attached at C-4 and the pentose was attached at the C-6′ of glucose (Fig. 2). The configurations of the apiose and glucose were determined to be D-configurations by GC analysis after acidic hydrolysis and chiral derivatization (retention times at 18.26, 29.32 min). The β-anomeric configurations were deduced on the basis of their coupling constants (Glc: J = 8.0 Hz; Api: J = 3.0 Hz). In the ROESY spectrum of 3, the correlations of H-8/H-9, H-8/H-10, H-5/H-9, H-5/H-10 and H-3/H-9 indicated that H-3, H-5 and amyl chain were located on the same side (Fig. 3).
The absolute configuration of 3 was determined by comparing the experimental and calculated ECD data based on the TD-DFT method at the B3LYP/6-31+G (d,p) level. Considering the numerous conformations of sugar, we used the aglycone 3a which was obtained from the acidic hydrolysis for ECD calculations. Through all wavelengths, the absolute configuration of 3S,4R,5R was confirmed by matching the calculated spectrum with the experimental ECD data (Fig. 6). Thus, the structure of 3 was confirmed and named ligusticoside B.
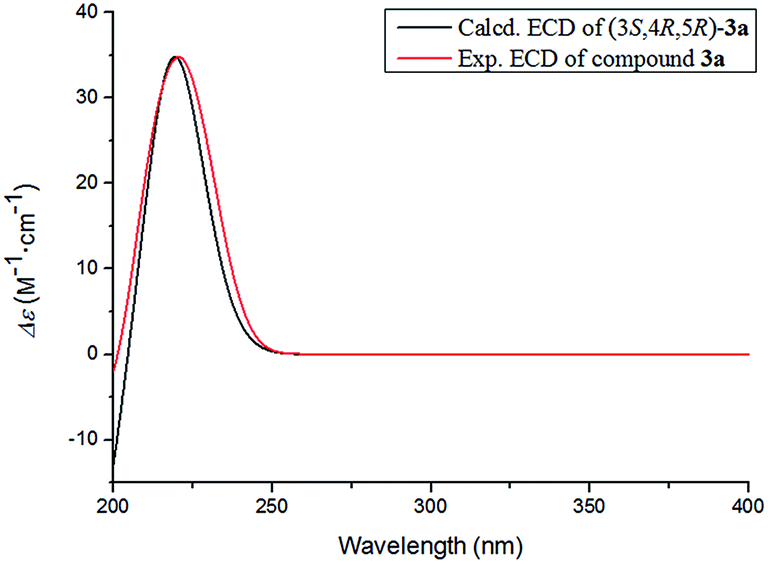 | ||
| Fig. 6 Experimental ECD and calculated ECD spectrum of 3a in MeOH. | ||
Compound 4 was obtained as a white amorphous powder. The positive HRESIMS gave the [M + H]+ ion peak at m/z 367.1399, in accordance with an empirical molecular formula of C18H22O8. The 1H NMR spectrum (Table 2) of 4 presented a methyl resonance at δH 0.95 (3H, t, J = 7.5 Hz, H-11), two methylene resonances at δH 1.51 (2H, m, H-10) and 2.36 (2H, q, J = 7.5 Hz, H-9) and an olefinic proton at δH 5.96 (1H, t, J = 8.0 Hz, H-8). An ABX system at δH 7.80 (1H, d, J = 8.5 Hz, H-7), 7.20 (1H, dd, J = 8.5, 2.0 Hz, H-6) and 7.59 (1H, d, J = 2.0 Hz, H-4) was presented. Additionally, the presence of multiple protons between δH 3.20 and 3.70 and the presence of a doublet at δH 5.12 (1H, d, J = 6.5 Hz, H-1′) suggested the occurrence of a glycose moiety. The 13C NMR spectrum (Table 2) of 4 showed 18 carbons, including a carbonyl carbon at δC 165.9 (C-1), a tertiary olefinic carbon at δC 109.5 (C-8), a quaternary olefinic carbon at δC 145.0 (C-3), six aromatic carbons at δC 141.4 (C-3a), 106.4 (C-4), 162.8 (C-5), 119.4 (C-6), 126.6 (C-7), and 116.9 (C-7a), and two methylene carbon resonances at δC 27.4 (C-9) and 22.0 (C-10). In addition, six oxygenated carbons that contributed to a glycose moiety were also observed at δC 99.9 (C-1′), 73.1 (C-2′), 76.6 (C-3′), 69.5 (C-4′), 77.2 (C-5′), and 60.5 (C-6′).
| Position | 4a | 5a | 6b | 7b | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| a 500 MHz for 1H NMR, 125 MHz for 13C NMR.b 600 MHz for 1H NMR, 150 MHz for 13C NMR. | ||||||||
| 1 | 165.9 | 165.9 | 165.8 | 165.8 | ||||
| 3 | 145.0 | 144.9 | 144.9 | 144.9 | ||||
| 3a | 141.4 | 141.3 | 141.2 | 141.2 | ||||
| 4 | 7.59, d (2.0) | 106.4 | 7.54, d (2.0) | 106.7 | 7.54, d (1.8) | 106.6 | 7.54, d (1.8) | 106.6 |
| 5 | 162.8 | 162.7 | 162.6 | 162.6 | ||||
| 6 | 7.20, dd (2.0, 8.5) | 119.4 | 7.23, dd (2.0, 8.5) | 119.1 | 7.23, dd (1.8, 9.0) | 119.0 | 7.23, dd (1.8, 8.4) | 119.0 |
| 7 | 7.80, d (8.5) | 126.6 | 7.83, d (8.5) | 126.7 | 7.82, d (9.0) | 126.6 | 7.82, d (8.4) | 126.6 |
| 7a | 116.9 | 117.0 | 117.0 | 117.0 | ||||
| 8 | 5.96, t (8.0) | 109.5 | 5.95, t (8.0) | 109.6 | 5.95, t (7.8) | 109.4 | 5.95, t (7.8) | 109.4 |
| 9 | 2.36, q (7.5) | 27.4 | 2.35, m | 27.4 | 2.36, m | 27.4 | 2.35, m | 27.3 |
| 10 | 1.51, m | 22.0 | 1.52, m | 22.0 | 1.52, m | 21.9 | 1.52, m | 21.9 |
| 11 | 0.95, t (7.5) | 13.8 | 0.95, t (7.5) | 13.8 | 0.95, t (7.8) | 13.7 | 0.95, t (7.2) | 13.7 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| Glc | ||||||||
| 1′ | 5.12, d (6.5) | 99.9 | 5.10, d (7.0) | 99.9 | 5.12, d (7.2) | 99.8 | 5.11, d (7.2) | 99.8 |
| 2′ | 3.20, m | 73.1 | 3.13, m | 73.1 | 3.29, m | 73.0 | 3.29, m | 73.0 |
| 3′ | 3.29, m | 76.6 | 3.30, m | 75.6 | 3.30, m | 75.6 | 3.28, m | 75.6 |
| 4′ | 3.29, m | 69.5 | 3.29, m | 69.8 | 3.16, m | 69.6 | 3.14, m | 69.6 |
| 5′ | 3.47, m | 77.2 | 3.63, m | 76.5 | 3.63, m | 76.4 | 3.62, m | 76.4 |
| 6′ | 3.47, m | 60.5 | 3.43, m | 67.4 | 3.45, dd (6.6, 10.8) | 67.3 | 3.45, dd (6.6, 10.8) | 67.2 |
| 3.70, m | 3.88, m | 3.88, d (10.8) | 3.88, m | |||||
|
||||||||
| Api-1 | ||||||||
| 1′′ | 4.80, d (3.0) | 109.2 | 4.80, d (3.0) | 108.7 | 4.80, d (3.0) | 108.9 | ||
| 2′′ | 3.73, dd (3.0, 6.5) | 75.9 | 3.73, dd (3.0, 6.6) | 76.1 | 3.69, dd (3.0, 6.6) | 76.8 | ||
| 3′′ | 78.8 | 78.7 | 77.6 | |||||
| 4′′ | 3.59, d (9.0) | 73.3 | 3.58, d (9.6) | 73.4 | 3.64, m | 73.6 | ||
| 3.86, m | 3.84, d (9.0) | 3.86, m | ||||||
| 5′′ | 3.33, m | 63.1 | 3.33, m | 69.1 | 3.36, d (9.6) | 69.8 | ||
| 3.50, d (9.6) | 3.62, m | |||||||
|
||||||||
| Api-2 | ||||||||
| 1′′′ | 4.81, d (3.6) | 109.0 | 4.85, d (4.8) | 102.1 | ||||
| 2′′′ | 3.76, dd (3.6, 6.0) | 75.6 | 3.76, dd (4.8, 9.6) | 71.4 | ||||
| 3′′′ | 77.4 | 76.1 | ||||||
| 4′′′ | 3.60, m | 73.3 | 3.65, m | 73.8 | ||||
| 3.84, d (9.0) | 3.90, d (9.6) | |||||||
| 5′′′ | 3.30, m | 63.1 | 3.23, dd (5.4, 9.6) | 63.0 | ||||
| 3.33, m | 3.28, m | |||||||
In the HMBC spectrum of 4, the correlations H-7/C-1, H-6/C-7a, H-4/C-3, H-8/C-3a, H-9/C-3, H-10/C-8 and H-11/C-9 confirmed the presence of a butenylphthalide moiety. The glucose moiety was attached at C-5 by the correlation of H-1′/C-5. In the NOE experiment, when irradiating H-4, H-8 generated a gain, and when irradiating the H-8, H-4 also generated gain signal (ESI†). Thus the geometric configuration of the double bond was confirmed as Z. The glucose was confirmed as having a D-configuration through the same method as used for 3. The relatively large coupling constant (J = 6.5 Hz) of the anomeric proton suggested that the glucose moiety was β-configured. Therefore, the structure of 4 was established as (Z)-3-butylidene-5-O-β-D-glucopyranosyl phthalide, named ligusticumside A.
The molecular formula of compound 5 was confirmed as C23H30O12 by the [M − H]− ion peak at m/z 497.1664 in HRESIMS. The 1H NMR spectrum (Table 2) of 5 presented a methyl resonance at δH 0.95 (3H, t, J = 7.5 Hz, H-11), two methylene resonances at δH 1.52 (2H, m, H-10) and 2.35 (2H, m, H-9) and an olefinic proton at δH 5.95 (1H, t, J = 8.0 Hz, H-8). An ABX system at δH 7.83 (1H, d, J = 8.5 Hz, H-7), 7.23 (1H, dd, J = 8.5, 2.0 Hz, H-6) and 7.54 (1H, d, J = 2.0 Hz, H-4) was presented. According to the above 1H NMR spectroscopic data, compound 5 had a similar skeleton to 4 except for the glycose moiety. The presence of multiple protons between δH 3.10 and δH 3.89 and the presence of two doublets at δH 5.10 (1H, d, J = 7.0 Hz, H-1′) and 4.80 (1H, d, J = 3.0 Hz, H-1′′) suggested the occurrence of two sugar moieties. Compared with the 13C NMR spectroscopic data of 4, compound 5 had an additional five carbons whose resonances at δC 109.2 (C-1′′), 75.9 (C-2′′), 78.8 (C-3′′), 73.3 (C-4′′), and 63.1 (C-5′′) were observed, and combined with the coupling constant (J = 3.0 Hz), the pentose was confirmed to be β-apiose. The apiose and glucose moieties were further determined to have D-configurations by the above method. The β-anomeric configuration was deduced on the basis of the coupling constant (Glc: J = 7.0 Hz). The connective position of apiose was identified by the correlation of H-6′/C-1′′, which confirmed that the C-1′′ of the apiofuranosyl was located at the C-6′ of the glucopyranosyl in the HMBC spectrum. The geometric configuration of the double bond was confirmed as Z by comparison with the data of 1H NMR and 13C NMR in 4. Thus, compound 5 was established as (Z)-3-butylidene-5-O-β-D-apiofuranosyl-(1 → 6)-β-D-glucopyranosyl phthalide, named ligusticumside B.
The molecular formula of 6 was assigned as C28H38O16 based on the [M + Na]+ ion peak at m/z 653.2046 (calcd 653.2052) in the HRESIMS. By carefully comparing the related data, 6 had similar structural characteristics to 4 and 5, with the main difference that 6 had a three-monosaccharide unit. In the 1H NMR spectrum (Table 2) of 6, the presence of multiple protons between δH 3.15 and δH 3.89 and the presence of three anomeric protons at δH 5.12 (1H, d, J = 7.2 Hz, H-1′), 4.80 (1H, d, J = 3.0 Hz, H-1′′) and 4.81 (1H, d, J = 3.6 Hz, H-1′′′) suggested the occurrence of three monosaccharide units. The 13C NMR resonances (Table 2) at δC 99.8, 108.7, and 109.0 indicated that 6 had a pyranose and two furanoses. The apiose and glucose were determined to have D-configurations through the same method as used above. The β-anomeric configurations were confirmed on the basis of their coupling constants (Glc: J = 7.2 Hz; Api-inner: J = 3.0 Hz; Api-outer: J = 3.6 Hz). In the HMBC spectrum, the glucose moiety was also attached at C-5 by the correlation of H-1′/C-5. The correlation H-6′/C-1′′ confirmed that the apiofuranosyl was located at the glucopyranosyl. The correlation H-5′′/C-1′′′ confirmed that the outer apiofuranosyl was located at the inner apiofuranosyl. Thus, compound 6 was established as (Z)-3-butylidene-5-O-β-D-apiofuranosyl-(1 → 5)-β-D-apiofuranosyl-(1 → 6)-β-D-glucopyranosyl phthalide, named ligusticumside C.
Compound 7 had the same molecular formula (C28H38O16) as 6 through the HRESIMS at m/z 653.2066 [M + Na]+. A careful comparison of the IR, UV and NMR data of 6 and 7 suggested that they had the same planar structure. The main difference was the chemical shifts of C-1′′′ and C-2′′′ of the outer apiose at δC 102.1 and 71.4 in 7, instead of δC 109.0 and 75.6 in 6, respectively. This was confirmed by the coupling constant of H-1′′′ (J = 4.8 Hz in 7 vs. J = 3.6 Hz in 6). Clearly, the outer sugar unit had an α configuration instead of the β configuration in 6. The sugar moieties were determined to be D-configurations of apiose and glucose through the same method as used above. Therefore, the structure of 7 was confirmed as (Z)-3-butylidene-5-O-α-D-apiofuranosyl-(1 → 5)-β-D-apiofuranosyl-(1 → 6)-β-D-glucopyranosyl phthalide, named ligusticumside D.
According to the [M + Na]+ ion peak at m/z 683.2164 in the HRESIMS, the molecular formula of 8 was assigned as C29H40O17. Comparing its NMR data with those of 7, both of them are trisaccharides of butylidenephthalide with the major difference between them being the type of the outer sugar. The presence of an anomeric proton at δH 4.64 (1H, d, J = 3.5 Hz, H-1′′′) and carbons at δC 99.2, 72.8, 72.1, 70.0, 73.3, and 60.8 suggested that the outer sugar of 8 was α-glucose instead of α-apiose in 7. The apiose and glucose were determined to have D-configurations by the same method as used above. Thus, the structure of 8 was confirmed as (Z)-3-butylidene-5-O-α-D-glucopyranosyl-(1 → 5)-β-D-apiofuranosyl-(1 → 6)-β-D-glucopyranosyl phthalide, named ligusticumside E.
The molecular formula of compound 9 was determined to be C18H22O8 through HRESIMS ([M + Na]+ 521.1629). The identical data of HRESIMS, IR, and UV suggested that 5 and 9 were isomers, with the difference being the geometric configuration of the double bond. In the 1H NMR spectrum of 9 (Table 3), an olefinic proton was presented at δH 5.90 (1H, t, J = 7.8 Hz, H-8). In the NOE experiment, when irradiating H-4, H-8 did not generate a gain, and when irradiating H-8, H-4 also did not generate a gain signal, which was entirely different from the results for 5 (ESI†). Additionally, the C-4 (δC 110.1) and C-8 (δC 114.2) were shifted significantly downfield in the 13C NMR spectrum when compared to those of 5 (C-4 at δC 106.7 and C-8 at δC 109.6). Thus, the double bond was proven to have an E configuration, and compound 9 was confirmed as (E)-3-butylidene-5-O-β-D-apiofuranosyl-(1 → 6)-β-D-glucopyranosyl phthalide, named ligusticumside F.
| Position | 8a | 9b | 10a | 11a | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| a 500 MHz for 1H NMR, 125 MHz for 13C NMR.b 600 MHz for 1H NMR, 150 MHz for 13C NMR. | ||||||||
| 1 | 165.9 | 165.6 | 164.4 | 166.1 | ||||
| 3 | 144.9 | 144.7 | 145.0 | 144.9 | ||||
| 3a | 141.3 | 139.4 | 141.0 | 141.8 | ||||
| 4 | 7.54, d (2.0) | 106.6 | 7.45, d (1.8) | 110.1 | 7.27, d (8.0) | 110.3 | 7.17, d (2.0) | 114.4 |
| 5 | 162.7 | 162.5 | 7.54, t (8.0) | 136.6 | 163.8 | |||
| 6 | 7.23, dd (2.0, 8.5) | 119.0 | 7.32, dd (1.8, 9.0) | 118.7 | 6.89, d (8.0) | 116.4 | 6.97, dd (2.0, 8.0) | 118.5 |
| 7 | 7.82, d (8.5) | 126.8 | 7.89, d (9.0) | 127.0 | 157.2 | 7.68, d (8.0) | 126.8 | |
| 7a | 117.0 | 118.7 | 109.4 | 105.6 | ||||
| 8 | 5.96, t (8.0) | 109.6 | 5.90, t (7.8) | 114.2 | 5.86, t (7.5) | 107.9 | 5.89, t (7.5) | 108.7 |
| 9 | 2.35, m | 27.4 | 2.52, m | 27.2 | 2.40, q (7.5) | 22.3 | 2.41, q (7.5) | 22.3 |
| 10 | 1.51, m | 22.0 | 1.57, m | 22.3 | 1.71, m | 28.9 | 1.72, m | 28.9 |
| 11 | 0.95, t (7.5) | 13.8 | 0.98, t (7.8) | 13.6 | 3.50, m | 68.1 | 3.51, m | 67.7 |
| 3.83, m | 3.80, m | |||||||
|
||||||||
| Glc-1 | ||||||||
| 1′ | 5.12, d (7.5) | 99.8 | 5.11, d (7.2) | 100.2 | 4.12, d (8.0) | 102.9 | 4.14, d (8.0) | 102.8 |
| 2′ | 3.29, m | 73.1 | 3.30, m | 73.0 | 2.94, t (8.0) | 73.4 | 2.95, overlap | 73.2 |
| 3′ | 3.29, m | 75.6 | 3.31, m | 75.7 | 3.06, m | 76.9 | 3.12, t (8.5) | 76.6 |
| 4′ | 3.16, m | 69.5 | 3.14, m | 69.8 | 3.03, q (9.0) | 70.1 | 2.98, overlap | 70.2 |
| 5′ | 3.64, m | 76.4 | 3.89, m | 76.2 | 3.11, m | 76.8 | 3.25, m | 75.5 |
| 6′ | 3.46, m | 67.4 | 3.44, dd (6.6, 10.8) | 67.5 | 3.42, dd (5.0, 11.0) | 61.1 | 3.39, dd (7.0, 11.0) | 68.2 |
| 3.89, m | 3.87, m | 4.12 d (8.0) | 3.82 m | |||||
|
||||||||
| Api | ||||||||
| 1′′ | 4.83, d (4.0) | 108.8 | 4.80, d (3.0) | 109.2 | 4.84, d (3.0) | 109.2 | ||
| 2′′ | 3.71, dd (4.0, 6.5) | 76.8 | 3.75, dd (3.0, 6.6) | 75.8 | 3.74, d (3.0) | 75.8 | ||
| 3′′ | 77.7 | 78.7 | 78.8 | |||||
| 4′′ | 3.65, m | 73.7 | 3.57, d (9.0) | 73.2 | 3.56, d (9.5) | 73.4 | ||
| 3.91, m | 3.86, d (9.0) | 3.84, d (9.5) | ||||||
| 5′′ | 3.25, m | 69.6 | 3.31, m | 62.9 | 3.30, t (11.0) | 63.1 | ||
| 3.64, m | 3.32, t (11.0) | |||||||
|
||||||||
| Glc-2 | ||||||||
| 1′′′ | 4.64, d (3.5) | 99.2 | ||||||
| 2′′′ | 3.29, m | 72.8 | ||||||
| 3′′′ | 3.16, m | 72.1 | ||||||
| 4′′′ | 3.07, m | 70.0 | ||||||
| 5′′′ | 3.40, m | 73.3 | ||||||
| 6′′′ | 3.45, m | 60.8 | ||||||
| 3.58, m | ||||||||
Compound 10 was obtained as a white amorphous powder. The HRESIMS showed a positive ion at m/z 405.1156 [M + Na]+ that matched the molecular formula C18H22O9. The IR spectrum presented hydroxyl (3395 cm−1) and γ-lactone (1757 cm−1) peaks. The 1H NMR spectrum of 10 (Table 3) showed three aromatic resonances at δH 7.27 (1H, d, J = 8.0 Hz, H-4), 7.54 (1H, t, J = 8.0 Hz, H-5), and 6.89 (1H, d, J = 8.0 Hz, H-6), which were assigned to a 1,2,3-trisubstituted benzene ring, an olefinic resonance at δH 5.86 (1H, t, J = 7.5 Hz, H-8), two methylene resonances at δH 2.40 (2H, q, J = 7.5 Hz, H-9) and 1.71 (2H, m, H-10), and a pair of oxygen-methylene resonances at δH 3.50 (1H, m, H-11a) and 3.83 (1H, m, H-11b), as well as the glucoside resonances. The 13C NMR experiment (Table 3) presented 18 carbon resonances, including one carbonyl carbon at δC 164.4 (C-1), six aromatic carbons, two olefinic carbons at δC 145.0 (C-3) and 107.9 (C-8), two methylene carbon resonances at δC 22.3 (C-9) and 28.9 (C-10), one oxygen-methylene resonance at δC 68.1 (C-11), and a set of glucose carbon resonances, implying that 10 was still a phthalide derivative with a glucose unit. The correlation of H-1′/C-11 in the HMBC spectrum suggested that the glucose moiety was located at C-11. The glucose was confirmed as having a D-configuration by the same method as used above. The β-configuration was deduced based on the coupling constant (J = 8.0 Hz) of H-1′. Thus, compound 10 was determined to be (Z)-3-(4-O-β-D-glucopyranosyl-butylidene)-7-hydroxyphthalide, named ligusticumside G.
Compound 11 had the molecular formula C23H30O13 by the analysis of its HRESIMS, which gave a positive ion at m/z 537.1587 [M + Na]+ (calcd 537.1579). An ABX system at δH 7.17 (1H, d, J = 2.0 Hz, H-4), 6.97 (1H, dd, J = 2.0, 8.0 Hz, H-6) and 7.68 (1H, d, J = 8.0 Hz, H-7) (Table 3) in the 1H NMR spectrum suggested the presence of the same trisubstituted aromatic ring as in 4 instead of that in 10. In the HMBC experiment, H-7 (δH 7.68) correlated with C-5 (δC 163.8) and C-3a (δC 141.8), H-6 (δH 6.97) correlated with C-4 (δC 114.4) and C-7a (δC 105.6), and H-4 (δH 7.17) correlated with C-6 (δC 118.5), C-7a (δC 105.6), and C-3 (δC 144.9), indicating that the aromatic hydroxyl was located at position C-5. The additional resonances at δH 4.84 (1H, d, J = 3.0 Hz, H-1′′) and from δH 3.84 to 3.30 were attributed to one pentose in 11. The 13C NMR spectrum showed 23 carbon resonances (Table 3), and the corresponding pentose resonances at δC 109.2 (C-1′′), 75.8 (C-2′′), 78.8 (C-3′′), 73.4 (C-4′′), and 63.1 (C-5′′) were observed. The apiofuranosyl and glucopyranosyl were determined to have D-configurations by the same method as used above (retention times at 18.26, 29.32 min). The coupling constants of the anomeric protons indicated the β-configuration (Glc: J = 8.0 Hz; Api: J = 3.0 Hz). Moreover, the correlation between H-1′′ (δH 4.84) and C-6′ (δC 68.2) confirmed that the C-1′′ of the apiofuranosyl was located at the C-6′ of the glucopyranosyl. Thus, compound 11 was confirmed as (Z)-3-[4-O-β-D-apiofuranosyl-(1 → 6)-β-D-glucopyranosyl-butylidene]-7-hydroxyphthalide, named ligusticumside H.
Compounds 1–11 were tested for their neuroprotective effects on SH-SY-5Y cell injury induced by H2O2 and OGD with L-NBP (L-3-n-butylphthalide) as a positive control. The results showed that compound 5 exhibited a weak neuroprotective effect with the increase cell viability rate of 15.60% and compound 4 exhibited a moderate neuroprotective effect with the increase cell viability rate of 24.35%, compared with the positive control L-NBP with increase cell viability rate of 18.26% on H2O2-induced neurotoxicity. And compound 4 exhibited a moderate neuroprotective effect with the increase cell viability rate of 13.72%, compared with the positive control L-NBP with increase cell viability rate of 4.03% on OGD-induced neurotoxicity.
Experimental
General experimental procedures
The optical rotations, UV spectra and ECD spectra were recorded with JASCO P-2000, V650 and J-815 spectrometer (JASCO, Easton, MD, USA), respectively. The Infrared spectra were measured on Nicolet 5700 spectrometer (Thermo Scientific, FL, USA). The NMR spectra were recorded with Varian 500 MHz (Bruker-Biospin, Billerica, MA, USA) and 600 MHz NMR spectrometers (Varian, Inc., Palo Alto, CA, USA). HRESIMS reports were obtained from Agilent 6520 HPLC-Q-TOF (Agilent Technologies, Waldbronn, Germany) and LCMS-IT-TOF system (Shimadzu Scientific Instruments Inc., Kyoto, Japan). Preparative HPLC was performed using a Shimadzu LC-10AT with a ODS-A column (250 mm × 20 mm, 5 μm; YMC Corp., Kyoto, Japan). The Agilent 1200 series system was used to carry on the HPLC-DAD analysis with an Apollo C18 column (250 mm × 4.6 mm, 5 μm; Alltech Corp., Lexington, KY, USA). The Agilent 7890A was used to carry on the GC analysis with a capillary column, HP-5 (60 m × 0.32 mm, with a 1 μm film; Agilent Technologies Inc., CA, USA). Macroporous resin Diaion HP-20 (Mitsubishi Chemical Corp., Tokyo, Japan), RP-C18 (50 μm, YMC Corp.), and Sephadex LH-20 (Pharmacia Fine Chemicals, Uppsala, Sweden) were used to column chromatograph.Plant material
The roots of Ligusticum chuanxiong Hort. were collected from Pengzhou Town, Sichuan Province in PRC, in Jun 2013 and identified by professor L. Ma. A voucher specimen (ID-S-2594) was deposited at the Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China.Extraction and isolation
The powdered rhizome of L. chuanxiong Hort. (100.0 kg) was exhaustively extracted with 80% EtOH under reflux condition. The solvent was evaporated by reduced pressure and then the residue (23.1 kg) was partitioned successively with EtOAc and n-BuOH. The n-BuOH-soluble portion (1300 g) was applied on a HP-20 column to give five fractions A–E through gradient elution with H2O, 15% ethanol, 30% ethanol, 50% ethanol, and 95% ethanol, respectively. Fraction C (103.0 g) was chromatographed over an RP-C18 column, eluting with H2O/MeOH (from 100:0 to 0:100) to give 16 fractions (C1–C16) on the basis of HPLC analyses. Fraction C5 was subjected to a Sephadex LH-20 with a gradient of increasing MeOH (0–100%) in H2O and then separated by preparative HPLC (MeOH/H2O, 30:70, V/V, HOAc, 0.01%) to give 2 (4 mg). Fraction C13 was separated by column chromatography over a Sephadex LH-20 using H2O as the eluent and was further purified by preparative HPLC (MeOH/H2O, 45:55, v/v, HOAc, 0.01%) to give 1 (64 mg). Compounds 10 (4 mg) and 11 (4 mg) were obtained from fraction C14 with the above method. Fraction D (48.0 g) was chromatographed over an RP-C18 column, eluting with H2O/MeOH (from 95:5 to 0:100) to give 24 fractions (D1–D24) on the basis of HPLC and TLC analyses. Fraction D9 was purified by a Sephadex LH-20 with a gradient of increasing MeOH (0–100%) in H2O and then separated by preparative HPLC (MeCN/H2O, 28:72, V/V, HOAc, 0.02%) to give 4 (70 mg), 5 (50 mg), 6 (3 mg), 7 (4 mg), 8 (3 mg), and 9 (10 mg). Fraction D8 was chromatographed over a Sephadex LH-20 with a gradient of increasing MeOH (0–100%) in H2O to give 19 fractions (D8-1–D8-19) on the basis of HPLC and TLC analyses. Fraction D8-2 was chromatographed over silica gel (EtOAC/MeOH/H2O, from 120:1:0.5 to 1:1:0.5) to give 3 (120 mg).
Structure characterization
ε): 201 (4.01), 277 (4.09) nm; [α]20D −49 (c 0.1 MeOH); HRESIMS m/z 345.0999 [M + H]+ (calcd 345.1003), m/z 367.0823 [M + Na]+ (calcd 367.0822); IR (KBr) νmax: 3387, 2938, 1741, 1682, 1633, 1514, 1414, 1230, 1184, 1093, 1043, 954 cm−1; 1H NMR and 13C NMR see Table 1.ε): 201 (3.84), 277 (4.06) nm; [α]20D +30 (c 0.1 MeOH); HRESIMS m/z 345.1008 [M + H]+ (calcd 345.1003), m/z 367.0827 [M + Na]+ (calcd 367.0822); IR (KBr) νmax: 3347, 2932, 2882, 1756, 1680, 1632, 1412, 1323, 1239, 1090, 1048, 1026, 997, 966 cm −1; 1H NMR and 13C NMR see Table 1.ε): 203 (4.18), 216 (3.90) nm; [α]20D +4 (c 0.1 MeOH); HRESIMS m/z 503.2148 [M − H]− (calcd 503.2134); IR (KBr) νmax: 3336, 2924, 1774, 1275, 1350, 1049 cm−1; 1H NMR and 13C NMR see Table 1.ε): 253 (4.42), 273 (4.16) nm; [α]20D −73 (c 0.1 MeOH); HRESIMS m/z 367.1399 [M + H]+ (calcd 367.1387); IR (KBr) νmax: 3377, 2929, 1759, 1689, 1613, 1484, 1297, 1078, 991 cm−1; 1H NMR and 13C NMR see Table 2.ε): 253 (4.35), 274 (4.07) nm; [α]20D −96 (c 0.1 MeOH); HRESIMS m/z 497.1664 [M − H]− (calcd 497.1665); IR (KBr) νmax: 3406, 2931, 1763, 1611, 1482, 1293, 1066, 1011 cm−1; 1H NMR and 13C NMR see Table 2.ε): 252 (4.19), 275 (3.92) nm; [α]20D −78 (c 0.1 MeOH); HRESIMS m/z 653.2046 [M + Na]+ (calcd 653.2052); IR (KBr) νmax: 3372, 2930, 1762, 1681, 1611, 1387, 1293, 1066, 930 cm−1; 1H NMR and 13C NMR see Table 2.ε): 253 (4.27), 272 (4.03) nm; [α]20D −30 (c 0.1 MeOH); HRESIMS m/z 653.2066 [M + Na]+ (calcd 653.2052); IR (KBr) νmax: 3398, 2932, 1765, 1687, 1613, 1295, 1072, 930 cm−1; 1H NMR and 13C NMR see Table 2.ε): 253 (4.38), 270 (4.16) nm; [α]20D −29 (c 0.1 MeOH); HRESIMS m/z 683.2164 [M + Na]+ (calcd 683.2158); IR (KBr) νmax: 3384, 2926, 1762, 1688, 1614, 1294, 1073, 928 cm−1; 1H NMR and 13C NMR see Table 3.ε): 254 (4.08), 272 (3.91) nm; [α]20D −21 (c 0.1 MeOH); HRESIMS m/z 521.1629 [M + Na]+ (calcd 521.1629); IR (KBr) νmax: 3399, 2931, 1765, 1689, 1612, 1483, 1294, 1072, 953 cm−1; 1H NMR and 13C NMR see Table 3.ε): 198 (3.96), 225 (4.14), 265 (3.95), 330 (3.66), 339 (3.70) nm; [α]20D −10 (c 0.1 MeOH); HRESIMS m/z 405.1156 [M + Na]+ (calcd 405.1156); IR (KBr) νmax: 3395, 2928, 2880, 1757, 1687, 1606, 1473, 1372, 1297, 1199, 1163, 1079, 1017, 895 cm−1; 1H NMR and 13C NMR see Table 3.ε): 254 (4.54) nm; [α]20D −30 (c 0.1 MeOH); HRESIMS m/z 537.1587 [M + Na]+ (calcd 537.1579); IR (KBr) νmax: 3375, 2932, 2884, 1753, 1689, 1610, 1481, 1467, 1381, 1295, 1165, 1057, 999, 932 cm−1; 1H NMR and 13C NMR see Table 3.Determination of the absolute configuration of sugar
Compounds 5 (2 mg) was dissolved in 1 mol L−1 CF3COOH (14 mL) and then the mixture was heated in 70 °C for 1 h. The mixture was then extracted three times with EtOAc, and the aqueous layer was freeze-dried to obtain residue. Using the same method with the literature,38 the residue was dissolved in anhydrous pyridine (2 mL), L-cysteine methyl ester hydrochloride (4 mg) was added, and then the mixture was heated in a water bath (60 °C) for 1 h. After the reaction solution was dried under vacuum, N-trimethylsilylimidazole (1 mL) was added, and the solution was heated in a water bath (60 °C) for 1 h and extracted three times with H2O/n-hexane. Then, the n-hexane layer was analyzed using GC under conditions as follows: injection temperature, 300 °C; detector temperature (FID), 300 °C; capillary column, HP-5 (30 m × 0.32 mm, Dikma); start temperature, 200 °C, raised to 260 °C at a rate of 10 °C min−1, and the final temperature maintained for 30 min; and N2 used as the carrier gas.Neuroprotective activities of compounds 1–11
The assay method of neuroprotective effects about compounds refer to the procedures of literature.39Conclusions
In the course of a search for neuroprotective compounds from Ligusticum chuanxiong Hort., eleven new phthalide derivatives were obtained. Among them, two are substituted with the rare mercaptopropionic acid. All compounds were tested for their neuroprotective effects, and it was observed that compound 4 had moderate effect against H2O2 and OGD-induced neurotoxicity in SH-SY5Y cells at 10 μM. These results could contribute to the better understanding of the therapeutic usage of Ligusticum chuanxiong Hort.Acknowledgements
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-010).References
- Editing Group for The Compilation of Chinese Herbal Medicines, The Compilation of Chinese Herbal Medicines, People′s Medical Publishing House, Beijing, 1996, pp. 133–135 Search PubMed.
- J. Huang, X. Q. Lu, C. Zhang, J. Lu, G. Y. Li, R. C. Lin and J. H. Wang, Fitoterapia, 2013, 91, 21–27 CrossRef CAS PubMed.
- C. P. Miao, S. H. Wu, B. Z. Luo, J. Wang and Y. W. Chen, Fitoterapia, 2010, 81, 1088–1090 CrossRef CAS PubMed.
- M. Kim, S. O. Kim, M. Lee, J. H. Lee, W. S. Jung, S. K. Moon, Y. S. Kim, K. H. Cho, C. N. Ko and E. H. Lee, Eur. J. Pharmacol., 2014, 740, 504–511 CrossRef CAS PubMed.
- S. L. Li, S. S. K. Chan, G. Lin, L. Ling, R. Yan, H. S. Chung and Y. K. Tam, Planta Med., 2003, 69, 445–451 CrossRef CAS PubMed.
- N. Y. Yang, D. C. Ren, J. A. Duan, X. H. Xu, N. Xie and L. J. Tian, Helv. Chim. Acta, 2009, 92, 291–297 CrossRef CAS.
- L. S. Lim, P. Shen, Y. H. Gong and E. L. Yong, Phytochemistry, 2006, 67, 728–734 CrossRef CAS PubMed.
- J. Huang, X. Q. Lu, J. Lu, G. Y. Li, H. Y. Wang, L. H. Li, R. C. Lin and J. H. Wang, J. Asian Nat. Prod. Res., 2013, 15, 1237–1242 CrossRef CAS PubMed.
- Y. H. Li, S. L. Peng, Y. Zhou, K. B. Yu and L. S. Ding, Planta Med., 2006, 72, 652–656 CAS.
- T. Naito, T. Katsuhara, K. Niitsu, Y. Ikeya, M. Okada and H. Mitsuhashi, Phytochemistry, 1992, 31, 639–642 CrossRef CAS.
- T. Naito, K. Niitsu, Y. Ikeya, M. Okada and H. Mitsuhashi, Phytochemistry, 1992, 31, 1787–1789 CrossRef CAS.
- T. Naito, Y. Ikeya, M. Okada, H. Mitsuhashi and M. Maruno, Phytochemistry, 1996, 41, 233–236 CrossRef CAS.
- J. Yang, X. L. Feng, Y. Yu, Q. Wang, J. Zou, C. X. Wang, Z. Q. Mu, X. S. Yao and H. Gao, China's Med., 2016, 11, 1–7 CrossRef PubMed.
- W. Wei, W. Xu and X. W. Yang, J. Asian Nat. Prod. Res., 2017, 19, 704–711 CrossRef CAS PubMed.
- P. O. Donkor, Y. Chen, L. Q. Ding and F. Qiu, J. Ethnopharmacol., 2016, 194, 530–548 CrossRef CAS PubMed.
- X. L. Chang, Z. Y. Jiang, Y. B. Ma, X. M. Zhang, K. W. K. Tsim and J. J. Chen, J. Asian Nat. Prod. Res., 2009, 11, 805–810 CrossRef CAS PubMed.
- M. J. Liang, L. C. He and G. D. Yang, Life Sci., 2005, 78, 128–133 CrossRef CAS PubMed.
- E. Y. Kim, J. H. Kim and M. R. Rhyu, Biol. Pharm. Bull., 2010, 33, 1360–1363 CAS.
- K. N. Nam, K. P. Kim, K. H. Cho, W. S. Jung, J. M. Park, S. Y. Cho, S. K. Park, T. H. Park, Y. S. Kim and E. H. Lee, Cell Biochem. Funct., 2013, 31, 707–712 CrossRef CAS PubMed.
- X. Yang, Y. C. Wang, L. L. Li, Y. C. Jin, L. Sironi, Y. Wang and Y. Wang, Fitoterapia, 2014, 95, 240–246 CrossRef PubMed.
- W. Li, S. Zhang, Q. H. Gao, J. W. Hou and T. T. Wei, Res. Chem. Intermed., 2004, 30, 605–613 CrossRef CAS.
- T. F. Lee, Y. L. Lin and Y. T. Huang, Planta Med., 2007, 73, 527–534 CrossRef CAS PubMed.
- H. Y. Qi, S. O. Siu, Y. Chen, Y. F. Han, I. K. Chu, Y. Tong, A. S. Y. Lau and J. H. Rong, Chem.-Biol. Interact., 2010, 183, 380–389 CrossRef CAS PubMed.
- L. Liu, Z. Q. Ning, S. Shan, K. Zhang, T. Deng, X. P. Lu and Y. Y. Cheng, Planta Med., 2005, 71, 808–813 CrossRef CAS PubMed.
- W. C. Ko, C. C. Liao, C. H. Shih, C. B. Lei and C. M. Chen, Planta Med., 2002, 68, 1004–1009 CrossRef CAS PubMed.
- J. W. Tian, F. H. Fu, W. L. Jiang, C. Y. Wang, F. Sun and T. P. Zhang, China J. Chin. Mater. Med., 2005, 30, 466–468 Search PubMed.
- W. X. Gong, Y. Z. Zhou, X. Li, X. X. Gao, J. S. Tian, X. M. Qin and G. H. Du, Molecules, 2016, 21, 549 CrossRef PubMed.
- T. F. Lee, Y. L. Lin and Y. T. Huang, Planta Med., 2007, 73, 527–534 CrossRef CAS PubMed.
- Y. L. Lin, T. F. Lee, Y. J. Huang and Y. T. Huang, J. Gastroenterol. Hepatol., 2006, 21, 1257–1265 CrossRef PubMed.
- Y. Sim and S. Shin, Arch. Pharmacal Res., 2008, 31, 497–502 CrossRef CAS PubMed.
- X. Kuang, J. R. Du, Y. X. Liu, G. Y. Zhang and H. Y. Peng, Pharmacol., Biochem. Behav., 2008, 88, 213–221 CrossRef CAS PubMed.
- J. Wang, J. R. Du, Y. Wang and C. Y. Wang, Acta Pharmacol. Sin., 2010, 31, 791–797 CrossRef CAS PubMed.
- J. R. Du, B. Bai, X. Kuang, Y. Yu, C. Y. Wang, Y. Ke, Y. J. Xu, A. H. C. Tzang and Z. M. Qian, J. Ethnopharmacol., 2006, 108, 54–58 CrossRef CAS PubMed.
- Q. Lu, T. Q. Qiu and H. Yang, Eur. J. Pharmacol., 2006, 542, 136–140 CrossRef CAS PubMed.
- X. Ran, L. Ma, C. Peng, H. Zhang and L. P. Qin, Pharm. Biol., 2011, 49, 1180–1189 CrossRef CAS PubMed.
- W. Wei, X. W. Wu and X. W. Yang, RSC Adv., 2016, 6, 61037–61046 RSC.
- G. Snatzke, U. Wagner and H. P. Wolff, Tetrahedron, 1981, 37, 349–361 CrossRef CAS.
- M. L. Gan, M. T. Liu, L. S. Gan, S. Lin, B. Liu, Y. L. Zhang, J. C. Zi, W. X. Song and J. G. Shi, J. Nat. Prod., 2012, 75, 1373–1382 CrossRef CAS PubMed.
- S. W. Huang, J. W. Qiao, X. Sun, P. Y. Gao, L. Z. Li, Q. B. Liu, B. Sun, D. L. Wu and S. J. Song, J. Funct. Foods, 2016, 24, 183–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1D NMR, 2D NMR HRMS, IR, and ECD spectra. See DOI: 10.1039/c7ra06813a |
| This journal is © The Royal Society of Chemistry 2017 |