
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manufacturing a super-active carbon using fast pyrolysis char from biomass and correlation study on structural features and phenol adsorption†
Hyewon Hwanga,
Olga Sahinb and
Joon Weon Choi *c
*c
aDepartment of Forest Sciences, Seoul National University, 599 Gwanak-ro, Gwanak-gu, Seoul, 08826, Korea
bInstitute of Catalytic Research (IKFT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1 76344 Eggenstein-Leopoldshafen, Germany
cGraduate School of International Agricultural Technology, Institute of Green-Bio Science and Technology, Seoul National University, Pyeongchang, Gangwon-do 25354, Korea. E-mail: cjw@snu.ac.kr; Fax: +82-33-339-5830; Tel: +82-33-339-5840
First published on 31st August 2017
Abstract
Fast pyrolysis char, obtained from two biomasses at 500 °C with a residence time of 1.3 s, was used as a precursor of nanoporous carbon material under different KOH loadings. In order to investigate a potential application as a biosorbent, a phenol removal test according to contact time and initial adsorbate concentration was also conducted in this study. During the process, a significant change in bonding structure of char focusing on weight loss and released volatiles was observed. In the presence of catalyst, aromatic hydrocarbons including BTEX were significantly released along with intermolecular bonding cleavage. After KOH activation at 700 °C for 2 h, the produced carbon material was characterized by van Krevelen diagrams, XRD, Raman spectroscopy, and XPS analysis. The produced activated carbon has a highly aromatic structure compared with commercial activated carbon (AC). In addition, a super-active carbon with a large pore volume (1.58 cm3 g−1) and high specific surface area (2711 m2 g−1) was successfully prepared, and the pore size largely depended on catalyst loading. Furthermore, the Langmuir adsorption capacity of the produced activated carbon for phenol was 625 mg g−1, which is superior to that of AC (500 mg g−1). Interestingly, a correlation study revealed that mesopore volume has a strong positive correlation with adsorption capacity, probably due to an increase in mass transfer and active site accessibility. This work suggests the possibility of developing green carbon materials using a co-product of the biorefinery process as a natural adsorbent that exceeds the performance of AC and also expands insights into their thermal conversion behavior during the process.
1. Introduction
Fast pyrolysis is one of the most promising thermal conversion processes to produce liquid alternative fuel from dry biomass sources in high yield (∼70 wt%).1 Although the process has been optimized for liquid oil production, it is also accompanied by a valuable co-product, char, a solid-state material mainly composed of carbon.2,3Because of the low specific surface area of fast pyrolysis char, very little is known about its application and it has commonly been used for combustion for energy production. However, it has great potential to be utilized as a precursor of porous carbonaceous material due to its high carbon content.4,5 The fast pyrolysis char can be converted to high-active carbon through post thermochemical treatment such as activation using chemical agents which provides better availability. Improved quality carbon material is mainly used as a bio-adsorbent,6,7 and can be used in the field of catalyst supports8 and electrode materials.9
The world activated carbon market exceeded $3 billion in 2015 and is anticipated to gradually rise at a compound annual growth rate (CAGR) of 11.9% by 2019.10 In addition, as concerns related to environmental problems grow, there is an increasing demand for renewable resources to replace coal, specifically bituminous coal, the main raw material of activated carbon. Therefore, if pyrolysis char could be used as a precursor of activated carbon, it would be expected to have a positive effect on the environment as well as provide an economic benefit.
However, there have been few studies conducted on the production of activated carbon using fast pyrolysis char, and in particular, studies on the application as biosorbent have been limited. Instead, a great deal of research has focused on activation processes using coal11 or biomass such as wood,12 nut shells,13 and wastes,14 requiring an additional carbonization step for long times.
Although activation is a well-known process for the commonly used precursors mentioned above, the mechanism has not been fully elucidated due to the complexity of its various parameters. Indeed, most research has mainly represented chemical and structural characteristics of the produced activated carbon and did not deal with the mechanism of structure formation. For the catalytic activation process, a few studies15–17 have suggested chemical reactions with KOH, widely used activating agent due to its high efficiency in the production of activated carbon with large pore volumes and specific surface areas, mostly focused on the interactions between KOH and carbon solids and changes in the state of the catalyst, not carbon material itself. Thus, study on the degradation procedure of raw material as catalytic activation proceeds is still lacking.
In this study, it was hypothesized that the observation of weight loss tendency and kinds of released volatile compounds during thermal conversion process could provide key information to understand the cleavage process of structural bonding. In addition, KOH activation could result in production of highly-porous activated carbon from fast pyrolysis char. Last but not least, evaluating pollutant removal efficiency by use of the produced carbon material could be strongly associated with its potential to replace commercial one.
In this context, utilization of fast pyrolysis char, co-product from biofuel technology, to a precursor for activated carbon by KOH activation was carried out in this study. Before a practical activation, modification of the char structure was investigated by determining changes in weight loss point of structural degradation and volatile compounds released during the catalytic conversion of pyrolysis char with KOH. For chemical activation, overall experiments were designed by varying biomass type (softwood and hardwood) and catalyst loading, which are significant factors in the process. Finally, all the samples were subjected to pollutant adsorption test. Phenol, a versatile precursor for plastics, drugs, and herbicide industries, was selected in this study because it is the representative one of priority pollutants determined by the U.S. Environmental Protection Agency (EPA) and it can cause serious problems such as protein degeneration, tissue erosion, paralysis of the central nervous system, and damage to brain functions at low concentrations when entering drinking water systems.18,19 Because adsorption capacity is significantly affected by properties of activated carbon, a correlation study was also intensely carried out to determine the most important factor. Furthermore, adsorption capacity as well as chemical, structural, morphological, and textural properties of each carbon product were compared with commercial activated carbon. The present work will help to enhance both value of fast pyrolysis char and efficiency of water remediation by conversion of co-product into a high-performance biosorbent, which is an approach to total biorefinery.
2. Materials and methods
2.1 Feedstock analysis and fast pyrolysis char preparation
Two kinds of wood, yellow poplar (YP) and Japanese red pine (JP), were kindly provided by the Korea Forest Research Institute (Seoul, Republic of Korea) and were used as the biomass feedstock in this study. Samples were ground and sieved through 0.5 mm mesh, followed by drying in an oven overnight before use. The moisture content of each sample was measured to be less than 2 wt%. Biomass samples were analyzed by elemental analysis (C, H, and N) using a US/CHNS-932 (LECO, USA) and the ratio of oxygen was calculated by difference. Compositional analysis for holocellulose, lignin, and ash content was conducted according to Wise's method,20 TAPPI method (T222 om-88), and National Renewable Energy Laboratory (NREL) standard procedures, respectively. In order to determine the inorganic content of biomass, inductively coupled plasma-emission spectroscopy (ICP-ES) analysis was performed using an ICPS-1000IV instrument (Shimadzu, Japan). Before the analysis, 0.5 g of samples were digested with an addition of 10 mL of acid mixture (HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl:H2O2 (8:1:1, v/v)) by microwave (Multiwave 3000, Anton Paar). The heating and pressure program were set as follows. Temperature is ramped to 550 °C by 5°C min−1 and held for 5 min, then ramped to 1200 °C with the same heating rate, and held 10 min at final temperature. The reaction pressure was maintained at 55 bar with a 0.3 bar s−1 during the process. After the reaction, liquefied sample was diluted to 50 mL and analyzed. Table S1† shows data for the fundamental analysis of the feedstocks.
:HCl:H2O2 (8:1:1, v/v)) by microwave (Multiwave 3000, Anton Paar). The heating and pressure program were set as follows. Temperature is ramped to 550 °C by 5°C min−1 and held for 5 min, then ramped to 1200 °C with the same heating rate, and held 10 min at final temperature. The reaction pressure was maintained at 55 bar with a 0.3 bar s−1 during the process. After the reaction, liquefied sample was diluted to 50 mL and analyzed. Table S1† shows data for the fundamental analysis of the feedstocks.
The char, a carbonaceous precursor for activated carbon, was obtained from fast pyrolysis of the two biomass samples under conditions of 500 °C and 1.3 s. The schematic of an experimental apparatus, fluidized bed reactor, for fast pyrolysis have been provided in previous studies2,3 and the detailed experimental procedure is as follow. Each biomass was prepared dry and injected into the heated reactor by a screw-type feeder at a rate of about 150 g h−1 under 10 L min−1 of N2 flow rate. Biomass then was rapidly decomposed, some were recovered as liquid oil via an ethanol cooler and an electrostatic precipitator, and some were collected as solid char by a cyclone connected to the reactor. After the experiment, enough gas was allowed to flow for 30 min to react the residual volatiles, and then the temperature of the reactor was lowered. Finally, the collected solid char was weighed and stored in a sealed vial. The char yield was 8.4 wt% for YP and 12.9 wt% for JP based on the wet basis of biomass samples. The produced char from YP and JP was labeled YPC and JPC, respectively.
2.2 Structural degradation analysis of fast pyrolysis char during catalytic thermal conversion
As for analytical approach, in order to investigate the structural degradation behavior of fast pyrolysis char in the presence of catalyst, thermogravimetric (TG) and differential thermogravimetric (DTG) analyses and pyrolysis-gas chromatography-mass spectrometry (Py-GC-MS) were conducted. The samples for both experiments were prepared as a mixture of ground char and KOH catalyst (Sigma Aldrich), with catalyst ratios of 0, 2:1, and 4:1 (catalyst:char, w/w).
For TG-DTG analysis, approximately 10 mg of each mixture sample was placed in a crucible in the instrument chamber (TGA/DSC 3 + (LF-1100) (Mettler-Toledo AG, Switzerland)) and heated at 10 °C min−1 in the temperature range of 40 °C to 700 °C under a nitrogen atmosphere.
Analytical pyrolysis to identify released volatile compounds from fast pyrolysis char samples during catalytic pyrolysis was conducted using Py-GC-MS equipped with a coil-type CDS Pyroprobe 5000 (CDS Analytical Inc., Oxford, PA, USA) and GC-MS/FID (Agilent Technologies 7890A/Agilent Technologies 5975A, USA) with a DB-5 capillary column (30 m × 0.25 mm ID × 0.25 μm film thickness). The prepared mixture sample including 3 mg of char was introduced into a quartz tube, which was then sealed by glass wool at both ends. For quantitative analysis, 1 μL of internal standard (IS; 1.3 mg of fluoranthene per mL of methanol) was also added to each sample. Finally, the sample was pyrolyzed to 700 °C at a heating rate of 10 °C ms−1 and was maintained for 20 s. Subsequently, the released volatile products were on-line transferred to a GC-MS/FID, and the program was set as follows. The GC injector and detector temperatures were set at 250 °C and 300 °C, respectively. The oven temperature program began with a 50 °C hold for 5 min, and the temperature was increased at a heating rate of 3 °C min−1 to 280 °C and maintained for 10 min. The separated compounds were transferred into the ionization source of a quadrupole MS, and ionization was conducted in electron impact (EI) mode at 70 eV. The volatile compounds were identified based on NIST MS Search 2.0 (NIST/EPA/NIH Mass Spectral Library; NIST 02), and quantitative analysis was achieved with consideration of response factors (Rf) of each compound, as suggested in a previous study.21 The amount of volatiles released from char with different catalyst ratios was calculated with the following equation.
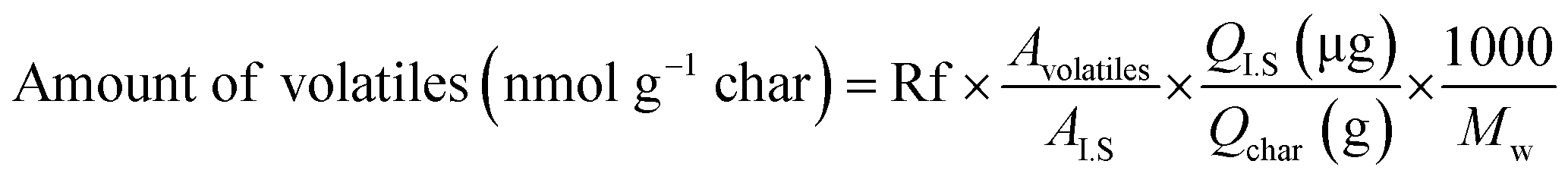 | (1) |
2.3 Manufacturing of nanoporous carbon from fast pyrolysis char by catalytic activation
Chemical activation of fast pyrolysis char was performed to produce nanoporous activated carbon using batch-type reactor placed at the Institute of Catalysis Research and Technology (IKFT) of Karlsruhe Institute of Technology (KIT). For each experiment, a mixture of pyrolysis char (1.5 g) and KOH (0, 3, or 6 g for ratio conditions of 0, 2:1, or 4:1, respectively, w/w) was prepared and purged with nitrogen (250 mL min−1) for 10 min to retain the inert condition of the reactor before activation. The reactor was then introduced into a furnace that had previously reached the reaction temperature of 700 °C, and this temperature was maintained for 2 h with a constant flow of nitrogen gas. After activation, the reactor was removed and allowed to cool to room temperature. The mixture was washed with 250 mL of HCl solution (1 M), followed by 2 L of D.I. water to remove the KOH catalyst and residual HCl, respectively. No precipitation was observed in any experiment after adding a droplet of AgNO3 (0.1 M) to the filtrate, which confirmed that HCl had been thoroughly removed. The product was dried in an oven overnight, and the yield was calculated. All experiments were carried out in triplicate, and the produced activated carbon was referred to as YP0, YP2, YP4, JP0, JP2, and JP4 according to the type of precursor and ratio of catalyst.
2.4 Characterization of activated carbon
Elemental analysis for carbon product was carried out by the same method as referred in Section 2.1. The surface morphologies of the samples were observed by field-emission scanning electron microscopy (FESEM), using a SIGMA (Carl Zeiss, UK) at various magnifications. For surface structure analysis, X-ray diffraction (XRD) and Raman spectroscopy were carried out using D8 ADVANCE with DAVINCI (BRUKER, Germany) at the National Instrumentation Center for Environmental Management (NICEM) and T64000 (HORIBA Scientific, Japan) at the National Center for Inter-university Research Facilities (NCIRF) at Seoul National University, respectively. X-ray photoelectron spectroscopy (XPS) measurements were conducted to determine surface chemistry of the samples using AXIS-HSi (KRATOS, UK). Binding energies for high-resolution scanning of C 1s spectra were fitted at 284.5 eV for the lowest binding energy peak (C 1s (1)), and peak deconvolution was accomplished using casaXPS software. Zeta potential was measured using Zetasizer-nano series (Malvern, UK). The suspension sample was prepared by adding fine char powder to 0.05 N NaCl solution, and adjusting the pH from 3 to 11 with HCl and NaOH solution. The mixture was agitated for better wettability and dispersion over 24 h, and sonicated before the measurement. The texture features were characterized by Brunauer–Emmett–Teller (BET) method for specific surface area from N2 adsorption, the Barrett–Joyner–Halenda (BJH) method for pore size and average diameter using adsorption branch, and the t-plot method for micropore structure using an ASAP 2010 (Micrometrics, Canada) with a N2 adsorption–desorption isotherm at 77 K.2.5 Phenol adsorption experiment
Phenol (Sigma Aldrich) adsorption tests for fast pyrolysis char and produced activated carbon were performed at 28 °C under two different parameters; contact times and concentrations of phenol solution. For the adsorption, 0.05 g of char sample was placed into flasks and 100 mL of phenol solution was added. The each flask was sealed by parafilm and then shaken at 150 rpm. The effect of contact times on adsorption capability was determined under different time intervals (0.5, 1, 2, 3, 5, 7, and 20 h) using 200 mg L−1 of phenol solution. The effect of initial adsorbate concentrations was investigated using various phenol solutions (25, 50, 100, 150, and 200 mg L−1) with fixed contact times for 20 h to reach equilibrium state of the solution. After the adsorption, absorbance of the filtrate was measured using UV-vis spectrophotometer (EVOLUTION 201, USA) at the wavelength of 270 nm and phenol concentration was calibrated from the absorbance data. The amount of adsorbed phenol at the equilibrium qe (mg g−1) was calculated as follows:
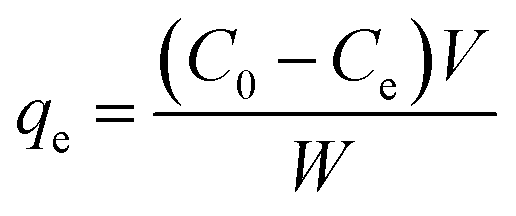 | (2) |
Overall scheme of this study, including three main parts of analytical and practical approach, was provided in Fig. 1. Furthermore, commercial activated carbon (AC; purchased from Sigma-Aldrich) was characterized and applied to phenol adsorption by the same methods as above in order to compare with the resultant activated carbon samples in this study. All experiments were carried out in duplicate, and the average values are presented.
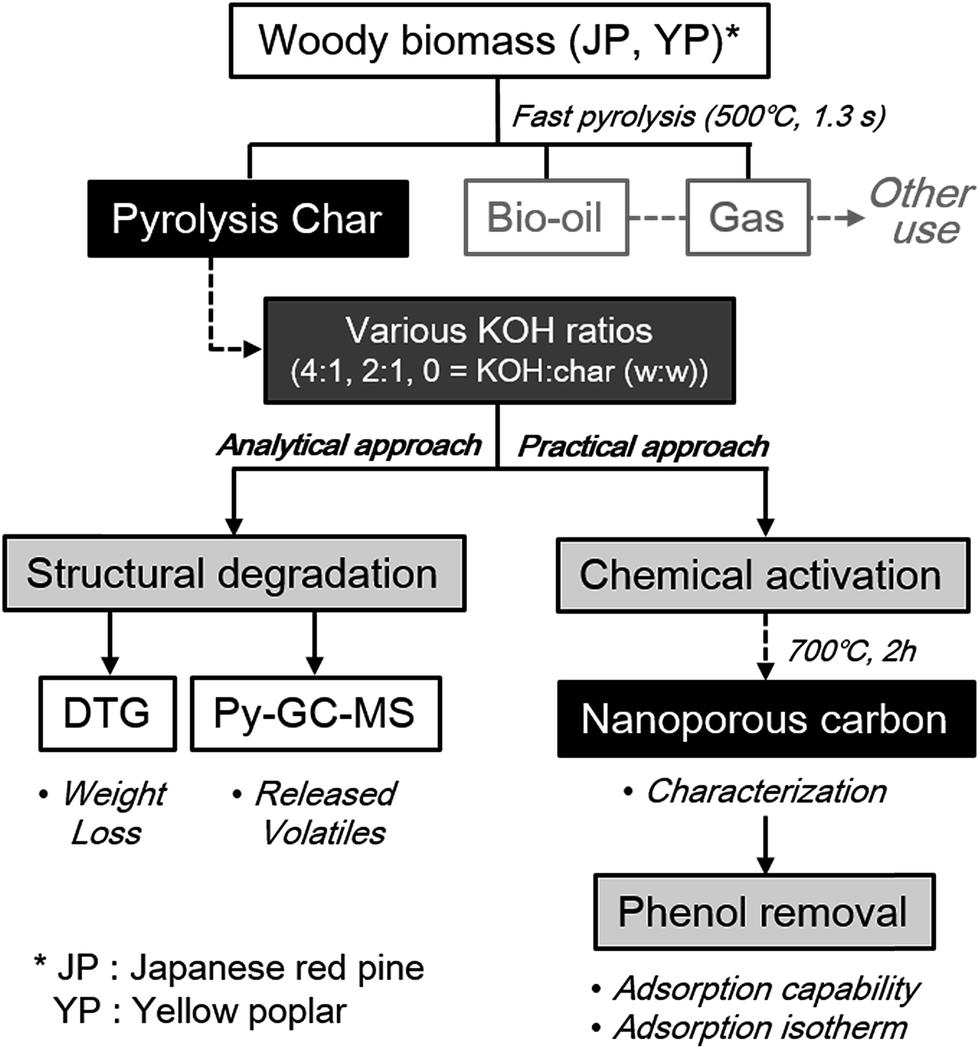 | ||
| Fig. 1 Overall scheme of analytical and practical approach to utilize co-product solid from fast pyrolysis to nanoporous material under different catalyst loading and feedstock type. | ||
3. Results and discussion
3.1 Structural degradation of char during catalytic thermal conversion by DTG and Py-GC-MS
Before conducting a practical activation process to produce activated carbon, a study of structural degradation behavior of fast pyrolysis char in the presence of catalyst was investigated. Changes in sample weight as temperature increased and volatile compounds released from char during catalytic thermal decomposition were analyzed by DTG and Py-GC-MS analysis, respectively.Fig. 2 shows the DTG curves of pyrolysis char samples with or without KOH catalyst under a nitrogen atmosphere. The curves for pyrolysis char in the absence of KOH (JPC and YPC) appeared almost linear due to gradual decomposition over the temperature range up to 700 °C. In the presence of KOH, however, the curves had two main broad peaks, which indicates that rapid thermal degradation occurred in the temperature range around 200–300 °C.
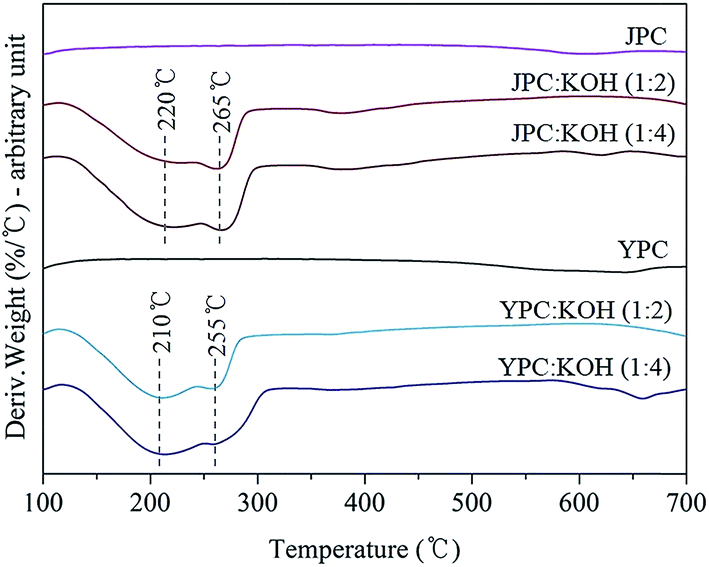 | ||
| Fig. 2 DTG curves of pyrolysis char with an addition of different KOH ratios. | ||
The thermal decomposition behavior of pyrolysis char samples in this study, produced at 500 °C in a fast pyrolysis process, likely corresponds to that of lignin or a condensed structure, which remained over the temperature range, rather than whole biomass commonly known for considerable decomposition at 200–300 °C due to cleavage of glycosidic bonds in cellulose and degradation of hemicellulose. In that temperature range, remaining propanoid side chains, β–β, C–C, and aryl ether linkages can be cleaved22–24 to form highly reactive and unstable intermediates that lead to further rearrangement reactions that increase stability. Therefore, peaks could appear by cleavage of the K–C structure complex derived from interaction between KOH and char as the temperature increased. Then, scission of chemical bonds in the structure of pyrolysis char, that were scarcely reacted or broken without catalyst, was facilitated, resulting in lower thermal stability.
As a result of volatile compounds released from pyrolysis char under different KOH ratios (Table 1), a total of 10 aromatic hydrocarbon (AH) and poly AH (PAH) compounds were detected as the main substances, instead of the oxygenated chemicals that are commonly produced from biomass pyrolysis. This result suggests that pyrolysis char already consists of an aromatic backbone structure, and these volatile compounds could derive from the macromolecular lattice of carbonized char by chemical reactions such as dehydrogenation, decarbonylation, and deoxygenation.
| Type | Compound | YPC:KOH |
JPC:KOH |
||||
|---|---|---|---|---|---|---|---|
| No. catalyst | 1:2 |
1:4 |
No. catalyst | 1:2 |
1:4 |
||
| a Unit: nmol g−1 char.b Aromatic hydrocarbon.c Polycyclic aromatic hydrocarbon. | |||||||
| AHb | Benzene | 2.08 | 6.37 | 8.88 | 10.01 | 14.81 | 14.53 |
| AH | Toluene | 0.79 | 3.18 | 5.30 | 2.69 | 7.35 | 8.84 |
| AH | Ethylbenzene | 0.00 | 0.00 | 0.07 | 0.00 | 0.21 | 0.30 |
| AH | p-Xylene | 0.00 | 0.89 | 1.80 | 0.14 | 1.86 | 2.53 |
| AH | m-Xylene | 0.09 | 0.37 | 0.45 | 0.09 | 0.52 | 0.80 |
| AH | 1,3,5-Trimethylbenzene | 0.04 | 0.21 | 0.80 | 0.20 | 0.19 | 0.41 |
| Total AH | 2.99 | 11.02 | 17.31 | 13.13 | 24.95 | 27.41 | |
| PAHc | Indene | 0.00 | 0.08 | 0.39 | 0.00 | 0.28 | 0.60 |
| PAH | Naphthalene | 0.10 | 0.12 | 0.39 | 0.82 | 0.76 | 0.81 |
| PAH | 1-Methyl-naphthalene | 0.00 | 0.00 | 0.19 | 0.07 | 0.32 | 0.46 |
| PAH | 9H-Fluorene | 0.00 | 0.04 | 0.19 | 0.03 | 0.21 | 0.30 |
| Total PAH | 0.10 | 0.24 | 1.15 | 0.92 | 1.56 | 2.16 | |
| Total | 3.09 | 11.26 | 18.46 | 14.05 | 26.51 | 29.56 | |
As the KOH ratio increased, the total amounts of volatiles increased to 18.46 nmol g−1 for JPC and 29.56 nmol g−1 for YPC, which are approximately 6 times and 2 times higher than the amounts without catalyst, respectively. This result corresponds to the DTG results that KOH enhanced degradation of the crystalline char structure, which also leads to pore creation by release of monomeric aromatics during catalytic activation.
Furthermore, the ratio of AH was much higher than that of PAH, and more than 85.1% of the total amount of volatiles consisted of benzene, toluene, ethylbenzene, and xylenes, generally referred to as BTEX, which are valuable compounds in chemical manufacturing. Among these, benzene was the most abundant, accounting for 65.0% and 61.5% of the total volatiles from YPC and JPC, respectively. Although the absolute quantity of benzene increased, its relative proportion gradually decreased with an increase in KOH ratio, whereas the quantities of other compounds increased, specifically toluene and xylenes. There seem to be several possible explanations for this result. It could be assumed that inter-molecular cleavage along with degradation of the char structure occurs more frequently than demethylation as catalyst ratio increased. On the other hand, benzene could be an intermediate that can be converted into other products under a high ratio of KOH. For instance, gaseous products or alkylated aromatics could be formed by further cracking or an electrophilic substitution reaction by metallic K derived from KOH acting as a catalyst.
For feedstock types, it appears that JPC has a more rigid structure than YPC based on the following results. First, the residue amount for JPC was 83.8%, higher than the 76.8% for YPC, at the final DTG analysis temperature. Next, the main degradation peaks appeared at 220 °C and 265 °C for JPC, which were 10 °C higher than those of YPC. In addition, the amount of volatiles detected from Py-GC-MS analysis of JPC was lower, indicating that its structure is more difficult to decompose. These observations could be attributed to the difference in structural characteristics of the original feedstock. It is generally accepted that there are covalent bonds inside the biomass called lignin-carbohydrate complex (LCC) that differ in softwood and hardwood. According to Jin et al., more than half of cellulose in softwood is combined with lignin, while about one-sixth of the cellulose occurs as lignin complexes in hardwood.25 In addition, coniferous lignin is thermally more stable and prone to form a more condensed structure than deciduous lignin due to highly reactive carbonium cations of guaiacyl nuclei as electron-rich aromatic carbons.26,27 Therefore, these differences in inherent structure of feedstock could cause structural rigidity of JPC derived from softwood. During the subsequent catalytic activation, breakdown and rearrangement of the aromatic backbone structure of pyrolysis char could be facilitated by KOH through cleavage of the thermally labile portion of C–C and ether linkages in char, leading to an increase in volatilization of aromatics.
3.2 Yields of activated carbon produced from fast pyrolysis char under different KOH ratios
After the practical activation process of fast pyrolysis char using a lab-scale furnace reactor, the yield of activated carbon product was shown in Table 2.| Product | Yield (wt%) |
|---|---|
| YP0 | 78.8 ± 0.1 |
| YP2 | 59.2 ± 0.4 |
| YP4 | 40.5 ± 1.0 |
| JP0 | 91.9 ± 0.1 |
| JP2 | 65.5 ± 2.3 |
| JP4 | 44.5 ± 0.4 |
Under the same activation process conditions, the activated carbon yield from JPC was higher than that of YPC, also supporting results of the structural degradation studies in Section 3.1 that the condensed and rigid structure of JPC could cause difficulties in thermal degradation.
Without KOH, 78.8 wt% and 91.9 wt% of carbon products were obtained for YP0 and JP0, respectively, and decomposition would occur mainly by the high temperature of 700 °C. As the ratio of KOH increased, the residue yield significantly decreased, and even at a catalyst ratio of 4, the yield fell to about half that of the non-catalytic conditions. Therefore, the presence of KOH had a great impact on the production of activated carbon through volatilization of large portion of pyrolysis char. This result could be explained by the change in reactivity and thermal lability of the bonding structure of char by KOH, leading to significant weight loss, as shown in the results of Fig. 2. In addition, the difference in yield between YPC and JPC was highest without catalyst and gradually decreased with increasing catalyst ratio. This result suggests that the catalytic influence of KOH enhanced degradation of the rigid structure of char that is scarcely decomposed solely by high temperature.
3.3 Graphitization and aromatization of pyrolysis char during the KOH activation process
Chemical and structural characteristics of activated carbon products were studied by elemental composition, XRD, Raman spectroscopy, and XPS analysis. From elemental analysis, the carbon ratio of pyrolysis char was 77.6% and 81.9% for YPC and JPC, respectively. As the KOH ratio increased, the product carbon proportion gradually increased to 90.1–90.4%, which was higher than that of AC (86.0%), whereas the hydrogen and oxygen proportions significantly decreased.Changes in elemental composition in terms of H/C and O/C ratios for biomass feedstock, fast pyrolysis char, and activated carbon products were studied by Van Krevelen plot, as shown in Fig. 3. Compared to the original feedstock, pyrolysis char had much lower H/C and O/C ratios, which demonstrates that significant structural change of biomass as well as dehydration reactions occurred during fast pyrolysis at 500 °C. For JP0 and YP0 carbon product treated at 700 °C under non-catalyst conditions, both atomic H/C and O/C ratios decreased compared to those of JPC and YPC. Specifically, the decrease in H/C ratio was more prominent, suggesting that dehydrogenation reactions proceeded at high temperature.
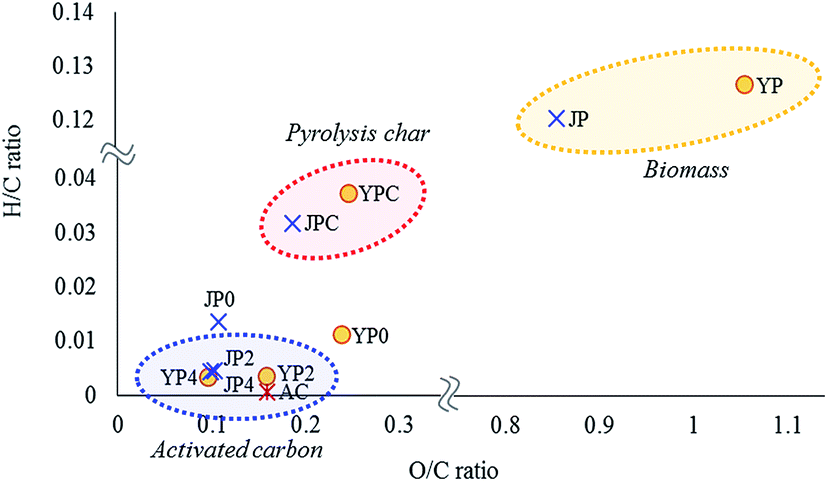 | ||
| Fig. 3 Van Krevelen diagram of biomass, pyrolysis char, and activated carbon products. | ||
In the presence of KOH, further deoxygenation and dehydrogenation reactions proceeded, and the atomic H/C and O/C ratios of carbon products decreased as much as that of AC, indicating that carbonized material can be obtained by the catalytic activation process using the co-product of fast pyrolysis. In addition, the decrease in these atomic ratios is indicative of growth of aromatic structures in the carbon material;28 thus, aromaticity of the product gradually increased with KOH activation. This result is related to the observation in Section 3.1 of an increase in release of aromatic volatiles, which could result from development of a high-degree aromatic structure due to dehydrogenation and deoxygenation enhanced by the catalyst.
JP seemed to be a more suitable precursor for activated carbon product due to its lower H/C and O/C ratios compared with those of YP. Indeed, the H/C and O/C ratios of JP-derived carbon products were lower until the KOH ratio was 2. At a KOH ratio of 4, however, the decreases in these ratios were similar regardless of biomass type, demonstrating that a high catalyst loading was effective in promoting degradation of the rigid char structure, as also shown by the yield data in Table 2.
Fig. 4 provides representative data on the structural characteristics of carbon products including XRD analysis and Raman spectroscopy. The XRD pattern in Fig. 4(a) shows two broad peaks (15.9° and 20–24°) of JP and YP for the (110) and (200) crystallographic planes with abundant hydroxyl groups involved in a number of intra- and intermolecular bonds in cellulose.29,30 These peaks disappeared after the fast pyrolysis process, and new peaks corresponding to sp3-hybridized lattice reflections (around 43°) and graphite fraction (at 21°, 23.5°, and 26.6°) were observed for pyrolysis char and non-catalytic activated carbon products, which can be attributed to degradation of the original biomass structure and creation of crystalline regions. The intensities around 21°, 22°, and 26.6° were also observed for AC, associated with a graphitic structure. In the presence of KOH, the sharp peaks lost intensity, and a broad peak with weak intensity appeared around 43.5°, indicating that intragraphitic layers were decomposed and amorphous carbon material with high porosity formed in the catalytic activation process.31
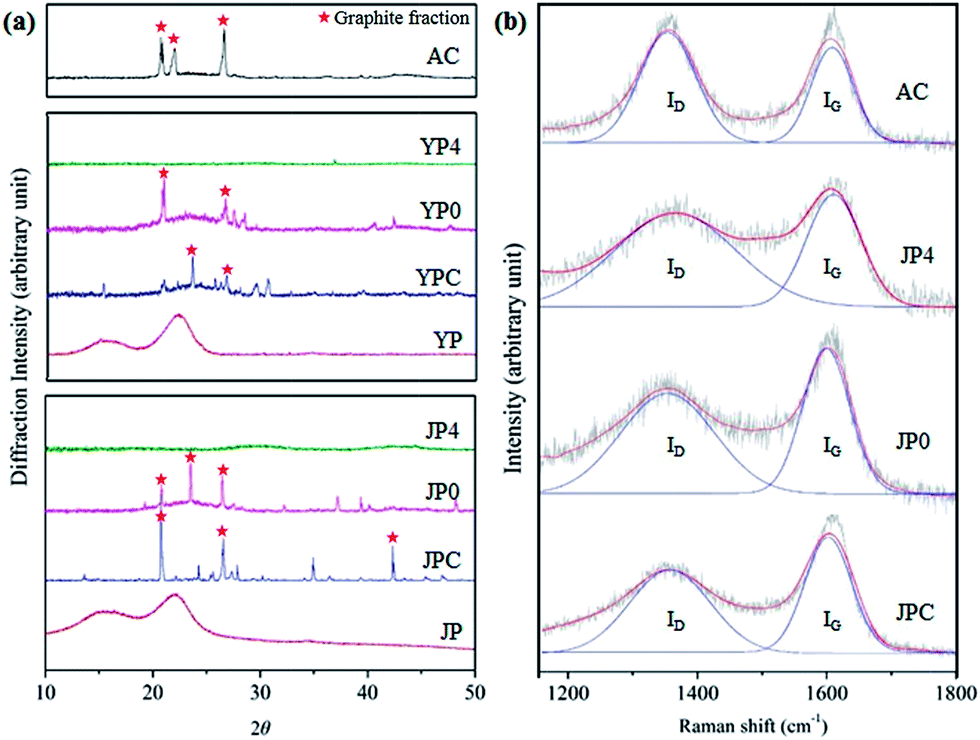 | ||
| Fig. 4 Structural analysis by (a) XRD and (b) Raman spectroscopy for comparison of the carbon product in this study and commercial activated carbon. | ||
From Raman spectra of carbon materials (Fig. 4(b)), two main peaks were deconvoluted and represent the presence of a disordered graphitic lattice, C–C bonds between aromatic rings and aromatics, and a medium to large aromatic ring system around 1350 cm−1 for the ID peak and assignment of graphite region and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds around 1580 cm−1 for the IG peak.32–34 After catalytic activation, the main peaks of JP4 were broader and showed a reduction of the IG peak compared to those of JPC. This could be attributed to development of amorphous structure rather than graphitization by reaction of KOH and fast pyrolysis char surface.35,36
C bonds around 1580 cm−1 for the IG peak.32–34 After catalytic activation, the main peaks of JP4 were broader and showed a reduction of the IG peak compared to those of JPC. This could be attributed to development of amorphous structure rather than graphitization by reaction of KOH and fast pyrolysis char surface.35,36
A numerical comparison of disorder and crystallinity of char samples was determined based on the ratio of the integrated intensity of ID/IG peaks, as shown in Table 3. The ratios of YPC and JPC were 1.09 and 1.24, which increased to 1.51 and 1.85, respectively, when the KOH ratio was 4. The increase in ID/IG ratio is attributed to the growth of large aromatic ring systems and the presence of dangling bonds in plane terminations of disordered graphite region in the solid sample.32–34 Thus, significant aromatization of pyrolysis char samples occurred under catalytic activation. These results are in close accord with other observations in this study, enhancement of dehydrogenation and deoxygenation reactions, as suggested in Py-GC-MS and Van Krevelen diagram analyses.
| Chemical composition (%) | C 1s (1) | C 1s (2) | C 1s (3) | C 1s (4) | C 1s (5) | ID/IG |
|---|---|---|---|---|---|---|
| sp2-graphitic or C–C, C–H | C–O | CO |
OC–O |
CO32− | ||
| YPC | 85.0 | 15.0 | — | — | — | 1.09 |
| YP0 | 83.6 | 13.2 | — | 3.2 | — | 1.18 |
| YP4 | 78.3 | 14.7 | 7.0 | — | — | 1.51 |
| JPC | 96.2 | 3.8 | — | — | — | 1.24 |
| JP0 | 93.6 | 6.4 | — | — | — | 1.37 |
| JP4 | 90.7 | 8.2 | 1.1 | — | — | 1.85 |
| AC | 88.9 | 11.1 | — | — | — | 1.55 |
In addition, surface functional characteristics were examined by XPS analysis, and the data are provided in Fig. S1† and Table 3. The results from deconvolution of C 1s were classified according to functional groups presented by Zielke et al.37 as follows: C 1s (1) sp2-graphitic bond, C–C, or C–H (284.5 eV); C 1s (2) hydroxyl or ether, C–O (286.0 eV); C 1s (3) carbonyl or quinone, CO (287.0 eV); C 1s (4) carboxyl or ester, OC–O (288.6 eV); and C 1s (5) carbonate, CO32− (289.5 eV). As seen in Fig. S1,† development of aromatic or conjugated structures after KOH activation was also confirmed by observing the π–π shake-up satellite tail at a few eV higher than the binding energy of the main peak38 in XPS spectra of JP4. The produced carbon products mainly consisted of the C 1s (1) peak, which could result from a deoxygenated and carbonized structure by high temperature, and they had comparable content to that of AC. Among carbon and oxygen bonds, the C 1s (2) peak for C–O bonds was the dominant peak for all carbon products, in agreement with other studies.4,39,40 When inorganic oxide was added to carbon material and the temperature increased, the oxide would transfer to the surface of the char, and bonds between metal and oxygen atoms would dissociate, followed by conversion of –O– free radicals to stable CO bonds.41 This would result in the appearance of a C 1s (3) peak for both YP4 and JP4.
Zeta potential of the activated carbon product and AC was also measured at different pH values as presented in Fig. S2.† As solution pH increased, zeta potential decreased and all the samples have negative zeta potentials over the entire pH ranges, being indicative of negatively charged surface due to the dissociation of acidic functional groups. This accordance with other researches as shown for typical activated carbon.42,43 Catalytic activated carbon exhibited a smaller level of negative charge than non-catalytic product and pyrolysis char. This suggests that reduction degree increased, which could enhance the π–π interaction in the subsequent aromatic organic material adsorption.44
From the results of structural analysis of the produced activated carbon, it could be concluded that both graphitization and aromatization proceeded, with the latter being the more prominent reaction during catalytic activation, along with development of amorphous structure, which could create more edges and corners to react with other substances.
3.4 Changes in morphological and textural features of carbon product
For examination of surface morphological features, SEM analysis was conducted at different magnifications, and the images are shown in Fig. S3.† Before activation, deformation of the biomass structure was observed for YPC and JPC due to pyrolysis at 500 °C. For YP4 and JP4, irregularity of the rupturing surfaces was more developed by catalytic cracking; thus, the edge portion could be extended instead of a smooth plane. Specifically, JP4 showed a more fragmented structure than YP4, suggesting there might be more edges that possess reactive sites.The N2 adsorption and desorption isotherms are given in Fig. S4.† According to International Union of Pure and Applied Chemistry (IUPAC) classification, catalyzed activated carbon is of type I and IV, representing micropore and mesopore structure. Also, hysteresis loop is shown for catalytic activated carbon and AC, which indicates that bimodal pore structure therein. The features for BET surface area and porosity of the raw carbon material and activated carbon products are presented in Table 4 and Fig. 5. The surface area of fast pyrolysis char from YPC and JPC was low at 9 m2 g−1 and 27 m2 g−1, respectively. As the KOH ratio increased, the surface area dramatically increased to 1908 m2 g−1 (YP4) and 2711 m2 g−1 (JP4), which was about three times higher than that of AC. The total pore volume of carbon products generated from catalytic activation also increased to 1.58 cm3 g−1 for JP4, and most of the volume was composed of mesopores. Interestingly, the proportion and amount of micropores were highest at a KOH ratio of 2, and approximately 68% of the total volume consisted of micropores. Thus, micropores could be transformed into large size pores with an increase in catalyst ratio, while the BET surface area and total pore volume increased. This indicates that it might be possible to selectively produce a micro- or meso-porous material for an intended purpose by controlling the catalyst ratio.
| Stotala (m2 g−1) | Sexternalb (m2 g−1) | Vtotalc (cm3 g−1) | Vmicrod (cm3 g−1) | Vmesoe (cm3 g−1) | Davgf (Å) | |
|---|---|---|---|---|---|---|
| a BET specific surface area measured by N2 adsorption data in the P/P0 range from 0.06 to 0.20.b External surface area calculated using t-plot method.c Total pore volume determined from the adsorption isotherm at P/P0 = 0.99.d Micropore volume calculated using t-plot method.e Mesopore volume calculated by the difference between Vtotal and Vmicro.f Average pore diameter determined by the adsorption data using BJH method. | ||||||
| YPC | 9 | — | 0.14 | — | 0.14 | 56.3 |
| YP0 | 71 | 47 | 0.05 | — | 0.05 | 43.0 |
| YP2 | 1825 | 504 | 0.90 | 0.61 | 0.29 | 25.4 |
| YP4 | 1908 | 1670 | 0.97 | 0.08 | 0.89 | 23.2 |
| JPC | 27 | — | 0.03 | — | 0.03 | 51.4 |
| JP0 | 116 | 75 | 0.08 | 0.02 | 0.06 | 42.9 |
| JP2 | 1812 | 716 | 0.88 | 0.50 | 0.38 | 21.4 |
| JP4 | 2711 | 1782 | 1.58 | 0.41 | 1.17 | 28.0 |
| AC | 900 | 446 | 0.82 | 0.21 | 0.61 | 49.8 |
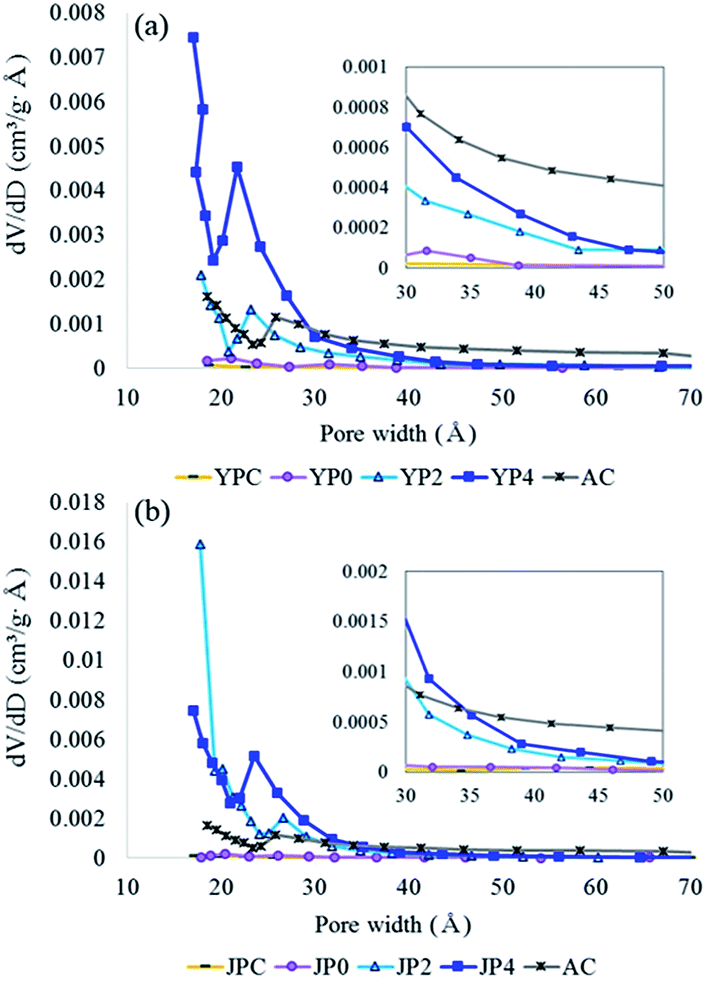 | ||
| Fig. 5 Pore size distribution for activated carbon produced under different KOH ratios products ((a): YP-derived carbon products, (b): JP-derived carbon products; data was determined by BJH method from the adsorption branch). | ||
Fig. 5 compares the pore size distribution (PSD) of carbon products before and after the activation process with different KOH ratios. From the PSD graph of fast pyrolysis char and non-catalytic activated carbon, a less developed porous structure was observed for both biomass types. During KOH activation, pores with width less than 30 Å were mainly generated, while the pore size of AC was relatively evenly distributed. The decrease in the pore distribution around 20–25 Å of catalytic activated carbon and AC is probably due to tensile strength effect generated by the structural properties as observed in the hysteresis loop in Fig. S4,† which has been reported in other studies.45–47 The number of supra-nanometer pores significantly increased with increasing KOH ratio, specifically for JP-derived activated carbon rather than YP-derived product. Consequently, the results of this study suggest that fast pyrolysis char can be utilized to prepare highly active carbon materials with comparable properties to commercial activated carbon, and these materials are expected to show high performance in a variety of application fields.
3.5 Evaluation of phenol adsorption capacity comparing to commercial activated carbon
In order to investigate application potential of the produced activated carbon derived from fast pyrolysis char as natural adsorbent for water remediation, phenol removal test was conducted.Fig. 6 shows the phenol adsorption capacity of the carbon products and AC using 200 mg L−1 of phenol standard solution according to different contact times. The adsorption process proceeded rapidly and it reached equilibrium in the initial 30 min, remaining constant for following times. For fast pyrolysis char and non-catalytic activated carbon samples, adsorption uptake was low and the values were fluctuated probably due to weak-binding interaction caused by a lack of active sites therein. On the other hand, KOH-activated carbon products showed largely improved phenol adsorption capacity, which was higher than that of AC.
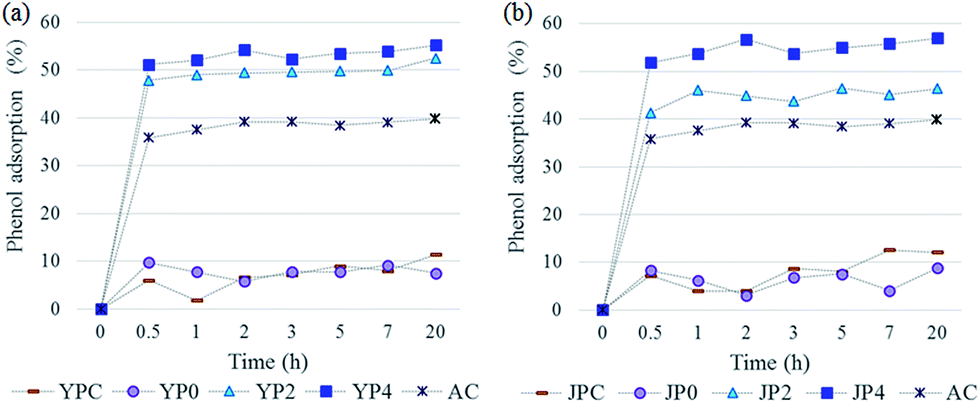 | ||
| Fig. 6 Evaluation of phenol adsorption capacity on carbon products according to contact time ((a): activated carbon from fast pyrolysis char of yellow poplar, (b): activated carbon from fast pyrolysis char of Japanese red pine). | ||
As for different solution concentration condition (25–200 mg L−1), the amount of phenol uptake increased with an increase in the solution concentration, but a relative adsorption rate based on initial solution gradually decreased for all samples. Thus, it can be assumed that adsorption could be no longer carried out after a certain amount of adsorption had proceeded and not all reactive sites in carbon products could be bound to adsorbate.
In order to assess the applicability of carbon products in this study for phenol adsorption process, isotherm model study was carried out. Two adsorption isotherms, Langmuir (eqn (3)) and Freundlich model (eqn (4)) which are most frequently used,48–51 were applied to determine the correlation between the adsorbed amount of phenol and the equilibrium concentration of solutions under different solution concentrations. The models equations are represented as follows:
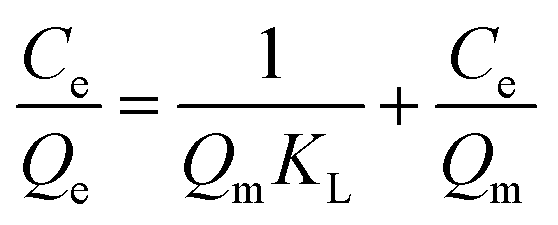 | (3) |
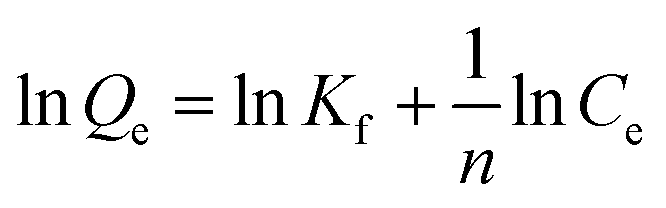 | (4) |
| Langmuir model | Freundlich model | |||||
|---|---|---|---|---|---|---|
| Qm (mg g−1) | KL | R2 | n | Kf | R2 | |
| YPC | 6.2 | 0.040 | 0.9867 | 0.74 | 1.285 | 0.4387 |
| YP0 | 5.0 | 0.004 | 0.9310 | 0.39 | 2.389 | 0.8750 |
| YP2 | 208.3 | 0.119 | 0.9994 | 2.82 | 46.647 | 0.9989 |
| YP4 | 454.5 | 0.186 | 0.9999 | 3.82 | 148.295 | 0.9770 |
| JPC | 7.4 | 0.005 | 0.4789 | 0.74 | 1.740 | 0.5320 |
| JP0 | 36.9 | 0.005 | 0.9077 | 0.37 | 2.418 | 0.7521 |
| JP2 | 192.3 | 0.111 | 0.9992 | 1.82 | 41.758 | 0.9940 |
| JP4 | 625.0 | 0.038 | 0.9996 | 1.96 | 54.533 | 0.9989 |
| AC | 500.0 | 0.075 | 0.9798 | 2.55 | 84.513 | 0.9391 |
Fast pyrolysis char and non-catalytic activated carbon products did not matched isotherm models due to their unreliable adsorption behavior. For KOH-activated carbon products, however, it can be observed that Langmuir isotherm model described adequately with high R2 value. Thus, adsorption of phenol could occur through monolayer coverage with identical affinity for the adsorbate on a homogeneous surface of activated carbon products in this study. This result also corresponds to other previous studies.52–54
In addition, JP4 showed the highest adsorption capacity (Qm), 625.0 mg g−1, which was higher than that of AC (500.0 mg g−1). This result was also superior to that of other researches, the adsorption capacity of 243.47 mg g−1 (ref. 55) and 149.25 mg g−1 (ref. 52) for phenol obtained by using wood- and rattan-based activated carbon, respectively. Thus, changes in structural features of char precursor during KOH activation significantly influenced on the adsorption capacity. Specifically, highly improved specific surface area and aromaticity could lead to an increase in π–π dispersion interaction as well as reaction between active site and adsorbate, major mechanisms related to phenol adsorption.
To further study on the correlation between features of carbon product and adsorption capacity (Qm), plots for Qm versus each property factors (carbon ratio, oxygen ratio, ID/IG ratio, Stotal, Sexternal, Vtotal, Vmicro, and Vmeso) were drawn. As a result, several factors including BET surface area, pore volume, and aromaticity determined by ID/IG ratio indicated a positive correlation with Qm value. Interestingly, among those factors, a high degree of correlation (R2 = 0.99) between mesopore volume and Qm was observed. As the mesopore volume of activated carbon product increased, Qm value also increases linearly (in Fig. 7(a)).
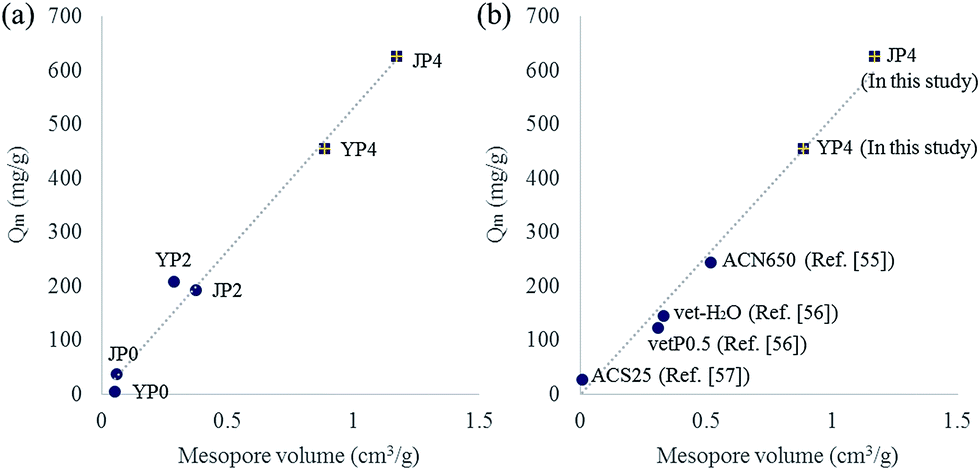 | ||
| Fig. 7 Correlation between Langmuir isotherm Q and mesopore volume of activated carbon ((a): plot for activated carbon produced in this study, (b): plot for activated carbon referred from other study). | ||
Fig. 7(b) shows the plot including data of the most developed activated carbon from this study and other studies.55–57 JP4 and YP4 in this study has higher Qm value than that from other studies, and the value has a strong positive correlation with the increase of mesopore volume. Generally, micropore has been considered as a dominant factor impacting the adsorption behavior because it could provide adsorptive sites rather than mesopore. However, mesopore can play an important role in enhancing adsorption capacity by expanding diffusion path for adsorbate to reach active sites.54 Therefore, a creation of mesopore in carbon product during the process could make the phenol adsorption reaction more favorable by enhancing rapid mass transfer as well as pore accessibility.
4. Conclusions
An approach to structural assessment of nanoporous activated carbon derived from fast pyrolysis char under various catalyst loadings and its application to natural adsorbent for phenol removal was carried out. As a result of Py-GC-MS analyses, an increase in release of aromatic hydrocarbons including a considerable proportion of BTEX were determined, which could be attributed that carbon–carbon bonds in solid char is more likely to be cleaved by KOH catalyst. After the activation process, a high-degree aromatic structure compared to AC was developed with a KOH ratio of 4. Moreover, successful production of microporous or mesoporous carbon material was accomplished with the maximum BET surface area of 2711 m2 g−1 (JP4), and the created pore size was largely influenced by catalyst loading. As for phenol removal, JP4 showed higher adsorption capability than that of AC and Langmuir model was more suitable to describe adsorption process in this study. In addition, a strong positive correlation between Vmeso and Qm value from Langmuir model was observed, indicating that enhanced mass transfer and pore accessibility by mesopore. The results of this study suggest that fast pyrolysis char, co-product of the process, can be utilized to prepare a superior natural adsorbent for phenol as well as highly active carbon materials comparing to AC, and the carbon materials are expected to be applied low-cost and high performance in a variety of application fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through Agri-Bio Industry Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (No. 115092022SB020), and by Korea Forest Service (Korea Forestry Promotion Institute) through ‘R&D Program for Forest Science Technology (Project No. 2017052A00-1720-BB02)’.References
- A. V. Bridgewater, Therm. Sci., 2004, 8, 21–50 CrossRef
.
- H. Hwang, S. Oh, I.-G. Choi and J. W. Choi, J. Anal. Appl. Pyrolysis, 2015, 113, 27–34 CrossRef CAS
- H. Hwang, S. Oh, T.-S. Cho, I.-G. Choi and J. W. Choi, Bioresour. Technol., 2013, 150, 359–366 CrossRef CAS PubMed
- R. Azargohar, S. Nanda, J. A. Kozinski, A. K. Dalai and R. Sutarto, Fuel, 2014, 125, 90–100 CrossRef CAS
- K. H. Kim, J.-Y. Kim, T.-S. Cho and J. W. Choi, Bioresour. Technol., 2012, 118, 158–162 CrossRef CAS PubMed
- I. M. Lima, A. A. Boateng and K. T. Klasson, J. Chem. Technol. Biotechnol., 2010, 85, 1515–1521 CAS
- A. Dąbrowski, P. Podkościelny, Z. Hubicki and M. Barczak, Chemosphere, 2005, 58, 1049–1070 CrossRef PubMed
- J. R. Kastner, J. Miller, D. P. Geller, J. Locklin, L. H. Keith and T. Johnson, Catal. Today, 2012, 190, 122–132 CrossRef CAS
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC
- G. V. Research, Activated Carbon Market Analysis By Product (Powdered Activated Carbon (PAC), Granular Activated Carbon (GAC)), By Application (Liquid Phase, Gas Phase), By End-Use (Water Treatment, Food & Beverages, Pharmaceutical & Medical, Automotive, Air Purification) And Segment Forecasts To 2024, 2016.
- G.-z. Gong, X. Qiang, Y.-f. Zheng, S.-f. Ye and Y.-f. Chen, New Carbon Mater., 2009, 24, 141–146 CrossRef CAS
- P. González-García, T. Centeno, E. Urones-Garrote, D. Ávila-Brande and L. Otero-Díaz, Appl. Surf. Sci., 2013, 265, 731–737 CrossRef
- E. Schröder, K. Thomauske, C. Weber, A. Hornung and V. Tumiatti, J. Anal. Appl. Pyrolysis, 2007, 79, 106–111 CrossRef
- J. Romanos, M. Beckner, T. Rash, L. Firlej, B. Kuchta, P. Yu, G. Suppes, C. Wexler and P. Pfeifer, Nanotechnology, 2011, 23, 015401 CrossRef PubMed
- T. Otowa, R. Tanibata and M. Itoh, Gas Sep. Purif., 1993, 7, 241–245 CrossRef CAS
- D. Lozano-Castello, J. Calo, D. Cazorla-Amoros and A. Linares-Solano, Carbon, 2007, 45, 2529–2536 CrossRef CAS
- E. Raymundo-Pinero, P. Azais, T. Cacciaguerra, D. Cazorla-Amorós, A. Linares-Solano and F. Béguin, Carbon, 2005, 43, 786–795 CrossRef CAS
- A. Knop and L. A. Pilato, Phenolic resins: chemistry, applications and performance, Springer Science & Business Media, 2013 Search PubMed
- R. Naseem and S. Tahir, Water Res., 2001, 35, 3982–3986 CrossRef CAS PubMed
- L. E. Wise, Paper Trade J., 1946, 122, 35–43 CAS
- J.-Y. Kim, J. H. Lee, J. Park, J. K. Kim, D. An, I. K. Song and J. W. Choi, J. Anal. Appl. Pyrolysis, 2015, 114, 273–280 CrossRef CAS
- W. Fiddler, W. E. Parker, A. E. Wasserman and R. C. Doerr, J. Agric. Food Chem., 1967, 15, 757–761 CrossRef CAS
- C. H. Ludwig and K. Sarkanen, Lignins: occurrence, formation, structure and reactions, Wiley-Interscience, 1971 Search PubMed
- R. Brežný, I. Šurina and M. Košík, Holzforschung, 1984, 38, 19–24 CrossRef
- Z. Jin, K. S. Katsumata, T. B. T. Lam and K. Iiyama, Biopolymers, 2006, 83, 103–110 CrossRef CAS PubMed
- M. Müller-Hagedorn, H. Bockhorn, L. Krebs and U. Müller, J. Anal. Appl. Pyrolysis, 2003, 68, 231–249 CrossRef
- K. Shimada, S. Hosoya and T. Ikeda, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef CAS
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46 CrossRef CAS PubMed
- M. Wada and T. Okano, Cellulose, 2001, 8, 183–188 CrossRef CAS
- M.-C. Popescu, C.-M. Popescu, G. Lisa and Y. Sakata, J. Mol. Struct., 2011, 988, 65–72 CrossRef CAS
- A. Dandekar, R. Baker and M. Vannice, Carbon, 1998, 36, 1821–1831 CrossRef CAS
- X. Li, J.-i. Hayashi and C.-Z. Li, Fuel, 2006, 85, 1700–1707 CrossRef CAS
- N. Shimodaira and A. Masui, J. Appl. Phys., 2002, 92, 902–909 CrossRef CAS
- Y. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 557–563 CrossRef CAS
- W. Li, L.-S. Zhang, Q. Wang, Y. Yu, Z. Chen, C.-Y. Cao and W.-G. Song, J. Mater. Chem., 2012, 22, 15342–15347 RSC
- L.-S. Zhang, W. Li, Z.-M. Cui and W.-G. Song, J. Phys. Chem. C, 2009, 113, 20594–20598 CAS
- U. Zielke, K. Hüttinger and W. Hoffman, Carbon, 1996, 34, 983–998 CrossRef CAS
- J. A. Gardella Jr, S. A. Ferguson and R. L. Chin, Appl. Spectrosc., 1986, 40, 224–232 CrossRef
- Y. Zhao, D. Feng, Y. Zhang, Y. Huang and S. Sun, Fuel Process. Technol., 2016, 141, 54–60 CrossRef CAS
- J. Dıaz-Terán, D. Nevskaia, J. Fierro, A. López-Peinado and A. Jerez, Microporous Mesoporous Mater., 2003, 60, 173–181 CrossRef
- J. Xiang, S. Hu, L. Sun, M. Xu, P. Li and X. Sun, J. Chem. Ind. Eng., 2006, 57, 2180 CAS
- G. Annadurai, R.-S. Juang and D.-J. Lee, J. Hazard. Mater., 2002, 92, 263–274 CrossRef CAS PubMed
- L. Chen, T. Ji, L. Mu, Y. Shi, L. Brisbin, Z. Guo, M. A. Khan, D. P. Young and J. Zhu, RSC Adv., 2016, 6, 2259–2269 RSC
- X. Wang, S. Huang, L. Zhu, X. Tian, S. Li and H. Tang, Carbon, 2014, 69, 101–112 CrossRef CAS
- J. C. Groen, L. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS
- W. Li, D. Chen, F. Xia, J. Z. Tan, P.-P. Huang, W.-G. Song, N. M. Nursam and R. A. Caruso, Environ. Sci.: Nano, 2016, 3, 94–106 RSC
- L. Cao, D. Chen, W. Li and R. A. Caruso, ACS Appl. Mater. Interfaces, 2014, 6, 13129–13137 CAS
- W. Li, C.-Y. Cao, L.-Y. Wu, M.-F. Ge and W.-G. Song, J. Hazard. Mater., 2011, 198, 143–150 CrossRef CAS PubMed
- H. Li, W. Li, Y. Zhang, T. Wang, B. Wang, W. Xu, L. Jiang, W. Song, C. Shu and C. Wang, J. Mater. Chem., 2011, 21, 7878–7881 RSC
- N. M. Nursam, J. Z. Tan, X. Wang, W. Li, F. Xia and R. A. Caruso, ChemistrySelect, 2016, 1, 4868–4878 CrossRef CAS
- J. Z. Tan, N. M. Nursam, F. Xia, M.-A. Sani, W. Li, X. Wang and R. A. Caruso, ACS Appl. Mater. Interfaces, 2017, 9, 4540–4547 CAS
- B. Hameed and A. Rahman, J. Hazard. Mater., 2008, 160, 576–581 CrossRef CAS PubMed
- B. Abussaud, H. A. Asmaly, T. A. Saleh, V. K. Gupta and M. A. Atieh, J. Mol. Liq., 2016, 213, 351–359 CrossRef CAS
- C.-T. Hsieh and H. Teng, Carbon, 2000, 38, 863–869 CrossRef CAS
- G. Yang, H. Chen, H. Qin and Y. Feng, Appl. Surf. Sci., 2014, 293, 299–305 CrossRef CAS
- S. Altenor, B. Carene, E. Emmanuel, J. Lambert, J.-J. Ehrhardt and S. Gaspard, J. Hazard. Mater., 2009, 165, 1029–1039 CrossRef CAS PubMed
- N. G. Rincón-Silva, J. C. Moreno-Piraján and L. Giraldo, Adsorption, 2016, 22, 33–48 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06910c |
| This journal is © The Royal Society of Chemistry 2017 |