
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessing inter lanthanide photophysical interactions in co-doped titanium dioxide nanoparticles for multiplex assays
Arijita Chakraborty ,
Gouranga H. Debnath and
Prasun Mukherjee*
,
Gouranga H. Debnath and
Prasun Mukherjee*
Centre for Research in Nanoscience and Nanotechnology, University of Calcutta, JD-2, Sector-III, Salt Lake, Kolkata-700106, West Bengal, India. E-mail: pmukherjee12@gmail.com
First published on 21st August 2017
Abstract
This study assesses the inter lanthanide photophysical interactions in trivalent lanthanide cations (Ln3+) co-doped titanium dioxide nanoparticles. As a case study, incorporation of neodymium (Nd3+) and samarium (Sm3+) to generate Ti(NdSm)O2 nanoparticles has been considered. The presence of co-doping offers a promising avenue for multiplex assays. The co-doped nanoparticles have characteristic visible emission at 584, 612, 664 and 726 nm respectively from Sm3+ and near infrared (NIR) emission at 912 and 1094 nm respectively from Nd3+, thus presenting composite doped nanoparticles with six distinct emission wavelengths spanning both the orange-red and NIR spectral window, using a single excitation wavelength. The photophysical properties of the Ti(NdSm)O2 nanoparticles have been compared with that observed in the singly doped Ti(Nd)O2 and Ti(Sm)O2 nanoparticles. Remarkable differences in the Ln3+ emission have been observed in the singly and doubly doped nanoparticles. Both the Nd3+ and Sm3+ emissions have been found to decrease in the Ti(NdSm)O2 nanoparticles, compared to those observed in the singly doped Ti(Nd)O2 and Ti(Sm)O2 nanoparticles. However, the extent of decrease in emission was found to be unequal for Nd3+ and Sm3+, with a decrease being marginally more prominent in Nd3+. The results have been rationalized by considering the Ln3+ as charge traps in the nanoparticles and associated relaxation pathways that are dictated by the spin selection rule. This photophysical rationalization was further tested and verified by performing experiments with the Ti(NdEr)O2 nanoparticles. The results presented provide important physical insight on the design criteria of co-doped semiconductor nanoparticles.
Introduction
The luminescence of trivalent lanthanide cations (Ln3+) is gaining increasing attention thanks to the unique characteristics of the core like nature of the 4f–4f transitions and finds use in biological imaging, bio-analytical applications, optoelectronics, telecommunications, lasers, sensing.1–16 The Ln3+ luminescence exhibits sharp emission bands spanning entire visible and near infrared (NIR) spectral range with minimum intra and inter Ln3+ spectral overlap, longer lifetime (typically in the range of microseconds to milliseconds), and resistance to photobleaching. These properties offer possibilities for multiplexing, time-gated measurements and longer data acquisition; hence opening up avenues for selective and sensitive detection with better signal to noise ratio.Development of Ln3+ containing luminophores for practical applications poses challenges because the molar extinction co-efficient of Ln3+ is extremely low (≤10 M−1 cm−1, as compared to 104 to 105 M−1 cm−1 for common organic fluorophores) and quenching of Ln3+ luminescence by the vibrational overtones of the common bonds present in nearby ligand and solvent molecules.5,17 These two factors, namely inefficient direct excitation and efficient environment induced luminescence quenching restrict easy realization of Ln3+ photoluminescence. Placing Ln3+ in an appropriate co-ordination environment with organic molecules as ligands,18–23 or incorporating these cations in suitable supramolecular assemblies including dendrimers, micelles, metal organic frameworks (MOF), semiconductor nanoparticles24–41 generate important avenues in which usable Ln3+ luminescence could be realized. A beneficial scenario would use placing the Ln3+ in a host matrix that absorbs the electromagnetic radiation efficiently and transfers the energy to the Ln3+ center, thereby realizing the Ln3+ photoluminescence from the composite host–guest system. In addition to the energy feeding process (optical antenna effect) the host matrix protects the Ln3+ luminescence from environmental quenching effects.
Towards the general goal to use Ln3+ photoluminescence for practical applications, we have been working on developing systems with semiconductor nanoparticles as the host, with relevant emphasis on understanding the underlying photophysical processes.38,39,42–47 Deciphering the light induced processes provide an opportunity to develop novel host (semiconductor nanoparticles)–guest (Ln3+) composite system with predictable photoluminescence properties, without necessarily approaching the problem on a combinatorial basis. Recently we have reported a systematic photoluminescence study with Ln3+ (Ln = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb) incorporated TiO2 [Ti(Ln)O2] nanoparticles and found Ti(Nd)O2 and Ti(Sm)O2 nanoparticles as the suitable candidates with significant host sensitized Ln3+ emission.46 TiO2 nanoparticles were considered as a model system to understand the host sensitized Ln3+ emission, as the ultraviolet-visible spectral region only has contribution from host sensitization, with direct excitation of Ln3+ being generally inefficient and 4f–5d transition along with the anion valence band to Ln3+ charge transfer (LMCT) energy lying out of the experimentally observed spectral window. The other Ti(Ln)O2 [besides Ti(Nd)O2 and Ti(Sm)O2] nanoparticles studied either showed moderate or no host sensitized Ln3+ emission. The luminescence sensitization process has been rationalized considering Ln3+ as the charge (electron and/or hole) traps in the semiconductor nanoparticles and the exciton recombination at these trap sites populating the Ln3+ luminescent energy level, thereby realizing the Ln3+ photoluminescence. It has been observed that in cases where Ln3+ ground and luminescent energy levels are optimally placed within the band gap of the host nanoparticles, the ground and luminescent energy levels of Ln3+ has the capability to trap the hole and electron respectively and such a co-localization of charge carriers in the Ln3+ trap site results in most efficient host sensitized dopant photoluminescence in the Ti(Ln)O2 nanoparticles.
Several researchers have investigated assemblies with multiple Ln3+ doping,26,48–58 towards the aim to develop multiplex assays which would generally reveal information from different locations in the context of complex diseases.59–65 DiMaio and co-workers48 have studied controlled energy transfer from Tb3+ to Eu3+ in LaF3 nanoparticles, with spatial restriction of the dopants in either the core or shell in a core–shell nanostructure assembly. Hanley and co-workers49 have reported Sm3+ and Eu3+ co-doped silica nanoparticles for multiplexed immunoassays, where inter lanthanide energy transfer was found to be absent. This work further compares a similar system where FITC and Cy3 were co-doped with significant energy transfer from FITC to Cy3, suggesting the usefulness of Ln3+ co-doping to develop multiplex assays. While this study investigates system having lanthanide moieties where inter Ln3+ energy transfer is absent, the study by DiMaio and co-workers48 clearly presents a case where such an electronic interaction is relevant; suggesting the importance of Ln3+ identity and their associated interactions in order to develop the co-doped nanoparticles for specific applications. Li and co-workers50 labelled silica nanoparticles with Eu3+ and Tb3+ chelates by covalent interaction and used the composite assembly to detect hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) by time-resolved immunofluorometric assays. The use of Ln3+ containing upconversion nanoparticles for multiplexing has been demonstrated by various researchers.51–54 Towards the development of near infrared (NIR) luminescent barcodes, Rosi, Petoud and co-workers26 reported a metal organic framework (MOF) containing Er3+ and Yb3+ where tunable NIR emission from both the cations are realizable as a function of Ln3+ concentration. Zheng and co-workers55 observed either luminescence enhancement or quenching of an Ln3+ in presence of another Ln3+ in co-doped fluoride nanocrystals. Lee and co-workers56 developed mixed Ln3+ (Dy/Eu, Ho/Eu, Ho/Tb) oxide nanoparticles for dual applications in magnetic resonance imaging (MRI) and photoluminescence imaging. Other researchers have also reported the use of lanthanide containing systems for multiplexing in biological assays.57,58 These studies collectively indicate that developing co-doped Ln3+ nanoparticles with simultaneous realization of distinct non-overlapping emission provide an avenue in the perspective of multiplex assays. Thus understanding and optimizing the inter lanthanide photophysical interactions is necessary.
While our previous works38,46,47 provide a foundation to rationalize the photophysical processes in singly Ln3+ incorporated semiconductor nanoparticles; these studies do not address the interactions in the cases of co-doping. Unravelling the photophysical interactions between Ln3+ of different identity is the primary objective of this study, where one Ln3+ may result in photoluminescence brightening or quenching of the other Ln3+ moiety. This study is organized as follows. The optimum extent of co-doping has been identified first by varying the Nd3+ and Sm3+ nominal doping extent in the Ti(NdSm)O2 nanoparticles. The choice of Nd3+ and Sm3+ as co-dopants is based on our observation of significant host sensitized Ln3+ emission in the Ti(Ln)O2 [Ln = Nd, Sm] nanoparticles, compared to the other Ti(Ln)O2 systems.46 This follows structural analysis of Ti(NdSm)O2 nanoparticles by X-ray diffraction (XRD) and Fourier transform infrared (FTIR) spectroscopy, corresponding comparisons with the singly doped Ti(Nd)O2 and Ti(Sm)O2 nanoparticles have been made. Photophysical properties have been studied in the appropriate doubly [Ti(NdSm)O2 and Ti(NdEr)O2 (vide infra)] doped nanoparticles, with relevant comparisons with the singly [Ti(Nd)O2, Ti(Sm)O2 and Ti(Er)O2] doped systems. Finally, an analysis is provided to rationalize the experimental observations.
Materials and methods
Chemicals
Tetra(n-butyl)titanate and lanthanide acetate hydrates (Ln = Nd, Sm, Er) (99.9%) were purchased from Alfa Aesar. Ethanol was purchased from Merck. IR grade potassium bromide (KBr) was purchased from Sigma-Aldrich. All chemicals were used as purchased without additional purification. Water used in all experiments was obtained from a Millipore system with a resistivity of 18.2 MΩ cm at 25 °C.Nanoparticle synthesis
The general synthetic protocol was adopted from the reports by Chen and co-workers66,67 and our previous work,46 which was further modified for the co-doping i.e. multiple distinct lanthanide incorporation within a single nanoparticle. For the synthesis of Ti(NdSm)O2 nanoparticles with nominal doping extent of 2% in each Ln3+, 58 μmol of each lanthanide (Ln: Nd and Sm)(III) acetate hydrate salt were dissolved in 200 μl water and 20 ml absolute ethanol with stirring at room temperature. 1 ml of tetra(n-butyl)titanate, dissolved in 20 ml ethanol was added to the previous mixture and stirring was continued with a magnetic stirrer for three hours. The resultant cloudy mixture was transferred to 50 ml Teflon lined autoclave to undergo the solvothermal treatment for 5 hours at 120 °C. The mixture was allowed to cool to room temperature. The as-synthesized materials were washed using absolute ethanol several times followed by centrifugation. The obtained precipitates were dried overnight at 60 °C. The sample was annealed at 500 °C for 2 hours. The syntheses of the Ti(NdSm)O2 nanoparticles with nominal doping extent of 1% and 4% in each Ln3+ were performed with 29 μmol and 0.116 mmol of each Ln3+ respectively, with the other conditions in the syntheses remaining same. The Ti(NdEr)O2 nanoparticles were synthesized with 2% nominal doping extent in each Ln3+ using the same synthetic procedure, with the incorporation of appropriate Ln3+ precursors.X-ray diffraction
X-ray powder diffraction pattern of the samples were collected by using a PANalytical X'pert PRO diffractometer, operated at a generator voltage of 40 kV and current of 30 mA with Cu Kα radiation (λ = 0.154 nm) within the 2θ scan range of 15° to 90°.Fourier transform infrared spectroscopy
Fourier transform infrared spectra were acquired using Jasco FTIR 6300 spectrometer. The spectra presented were collected from an average of 64 scans. During spectral acquisition, the resolution was maintained at 4 cm−1. The sample for the experiment was prepared using KBr pellet method. The spectra were recorded at room temperature. The data analysis was carried out using the software provided with the instrument.Electron microscopy measurements
The transmission electron microscopy (TEM) experiments were performed to identify the morphology of the sample by using TEM instrument from JEOL (model JEM-2100) operated with an acceleration potential of 200 kV. The sample was prepared by placing a drop of colloidal dispersion on a carbon coated copper grid. Extra sample was removed by drying the grid. The energy dispersive X-ray spectra (EDS) were acquired using the Zeiss model EVO 18 scanning electron microscopy instrument.Luminescence spectroscopy
The steady-state luminescence spectra were acquired in the Horiba Fluorolog 3-22 luminescence spectrometer. To obtain the emission spectra, samples were excited at 350 nm and the excitation spectra were acquired by monitoring the major emission bands. To collect the spectra in visible region, the excitation and emission slits were kept at 2 nm spectral resolution. In the case of NIR spectral region, excitation and emission slits were maintained at 8 nm and 4 nm spectral resolutions respectively. For the samples with low luminescence intensity in the visible region, the spectra were acquired with 4 nm spectral resolution for both the excitation and emission slits. In the NIR spectral window, the excitation and emission spectral resolutions were maintained at 14 nm and 40 nm respectively for the samples with low luminescence intensity. For photoluminescence lifetime measurements a delay time and detection window of 0.5 and 5 ms respectively were used for the nanoparticles and the corresponding values were kept at 0.0001 and 0.05 ms respectively for collecting the emission from Sm(III) acetate. For lifetime measurements, the nanoparticles were excited at 350 nm and the emission was collected at 612 nm. The excitation and emission wavelengths for collection of Sm(III) acetate lifetime were kept at 400 and 595 nm respectively. The lifetimes were fitted with sum of two decaying exponentials. All data analyses were performed using Origin 8.5 software. To collect the emission in the visible and NIR spectral region the R-928 photomultiplier tube (PMT) and a liquid nitrogen cooled indium gallium arsenide (InGaAs) detector [model DSSIGA(1-9)010L] were used, respectively. The nanoparticles were dispersed in water for the photoluminescence measurements. All measurements were performed at ambient conditions.The photoluminescence quantum yields for the Ln3+ luminescence in the visible spectral range were calculated based on a relative method with a comparison to the quantum yield of coumarin 153 (C153) dissolved in methanol (Φr = 0.42 (ref. 68)). Relative quantum yields Φx with all the experimentally observed Ln3+ emission bands were calculated using eqn (1);
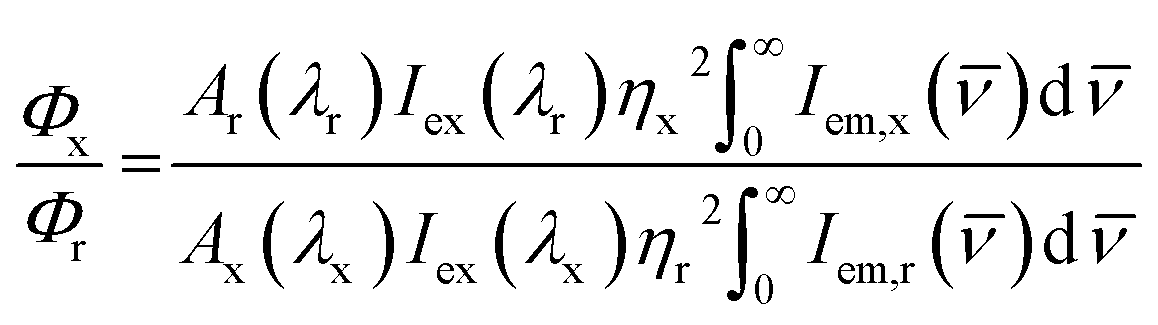 | (1) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ) is the luminescence intensity as a function of wavenumber . For the calculation of relative quantum yields in the NIR spectral region for the Nd3+ and Er3+ emission bands, the PMT and InGaAs detectors were calibrated with respect to the common band of Tm3+ emission in the Ti(Tm)O2 nanoparticles which is centered around 810 nm.46
) is the luminescence intensity as a function of wavenumber . For the calculation of relative quantum yields in the NIR spectral region for the Nd3+ and Er3+ emission bands, the PMT and InGaAs detectors were calibrated with respect to the common band of Tm3+ emission in the Ti(Tm)O2 nanoparticles which is centered around 810 nm.46
Results and discussion
X-ray diffraction
The X-ray diffraction patterns of the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown in Fig. 1. For the doubly doped system, a nominal doping extent of 2% in each Ln3+ has been considered (vide infra). The singly doped nanoparticles show characteristic diffraction peaks at 2θ = 25.4, 38.0, 48.3, 54.2, 55.3, 62.7, 69.1, 70.4 and 75.4 that has been correlated with the (101), (004), (200), (105), (211), (204), (116), (220) and (215) planes of anatase TiO2 crystal respectively. The nanoparticles with both Nd3+ and Sm3+ co-doped also exhibit similar diffraction pattern indicating that the crystal structure remains unchanged in the doubly doped TiO2 nanoparticles. Moreover, in all the nanoparticles studied, characteristic peak at 2θ = 27.4 from the (110) plane of the rutile phase was absent, suggesting that rutile phase does not have contribution in the systems investigated in the present work.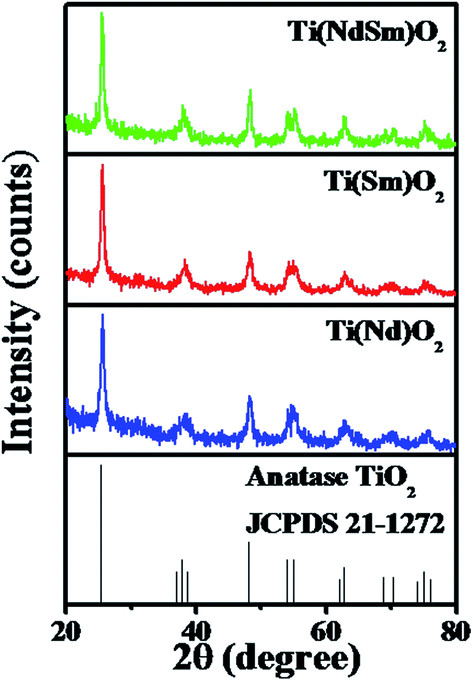 | ||
| Fig. 1 XRD profiles of the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown. | ||
To this end, we comment on the possible presence of the lanthanide impurity phases in the observed XRD patterns. For this exercise, a comparison of intensities has been made at 2θ = 65° (taken as a representative case), where no signal is present in the XRD patterns of either Ti(Ln)O2 [Ln = Nd, Sm], Ti(NdSm)O2 nanoparticles [as judged from the Joint Committee on Powder Diffraction Standards (JCPDS) card 21-1272] and the lanthanide impurity phases (as judged from the following JCPDS cards) with the corresponding intensity at the position of most intense lines of various lanthanide impurity phases, as mentioned in the parentheses, of neodymium oxide [JCPDS card numbers 65-6729 (Nd2O3, hexagonal, 2θ = 30.8°), 65-3184 (Nd2O3, cubic, 2θ = 28.0°), 46-1074 (NdO2, cubic, 2θ = 28.0°), 45-0087 (Nd6O11, cubic, 2θ = 28.0°)], NdxTiyOz [JCPDS cards 82-1095 (Nd2Ti3O9, tetragonal, 2θ = 29.6°), 70-2294 (NdTiO3, orthorhombic, 2θ = 32.4°), 70-1691 (Nd2Ti2O7, monoclinic, 2θ = 30.3°), 70-1544 (Nd2TiO5, orthorhombic, 2θ = 28.6°), 40-1051 (α-Nd2Ti4O11, 2θ = 51.2°)], samarium oxide [JCPDS cards 65-3183 (Sm2O3, cubic, 2θ = 28.3°), 43-1030 (Sm2O3, monoclinic, 2θ = 32.0°), 33-1146 (SmO, cubic, 2θ = 31.3°)] and SmxTiyOz [JCPDS cards 70-2295 (SmTiO3, orthorhombic, 2θ = 32.4°), 47-0283 (Sm2Ti2O7, orthorhombic, 2θ = 30.5°), 41-0497 (Sm4Ti3O12, monoclinic, 2θ = 27.5°), 22-1306 (Sm2TiO5, orthorhombic, 2θ = 29.0°), 35-0364 (β-Sm2TiO5, hexagonal, 2θ = 31.7°)]. These intensities are comparable in magnitude, necessarily reflecting noise in the observed XRD patterns where supposed lanthanide impurity phases should appear; suggesting that the observed signals from the nanoparticles studied do not have significant contribution from the lanthanide impurity phases.
Fourier transform infrared (FTIR) spectroscopy
The FTIR spectra of the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown in Fig. 2. For the doubly doped nanoparticles, a nominal doping extent of 2% in each Ln3+ has been considered (vide infra). All the nanoparticles investigated show characteristic infrared absorption bands at 470–490 and 660 cm−1 respectively, a signature of Ti–O–Ti bond vibrations. A comparison with the corresponding spectrum of Sm(III) acetate clearly reveals a different spectral signature, with characteristic bands at 1550 and 1460 cm−1 respectively originating from the carboxylate asymmetric and symmetric stretching vibrations. The absence of these bands in the nanoparticles investigated clearly indicates the absence of significant amount of free Ln(III) precursor salt in the nanoparticles studied and the Ln(III) related spectral signature being originated from the Ln(III) moieties that has interacted with the nanoparticles (vide infra).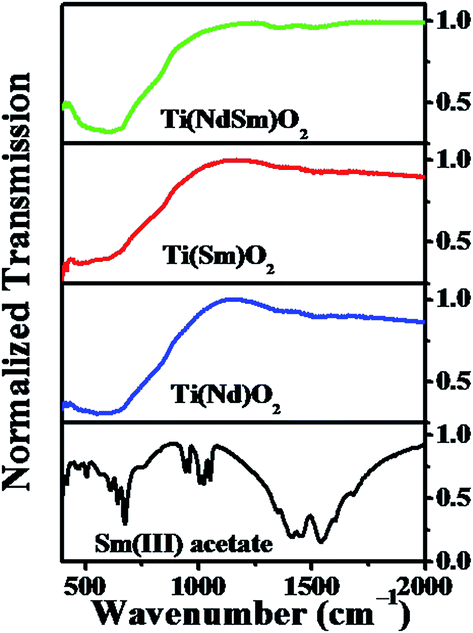 | ||
| Fig. 2 FTIR spectra of the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown. The corresponding spectra of Sm(III) acetate is also included. | ||
Electron microscopy
The transmission electron microscopy (TEM) image of the Ti(NdSm)O2 nanoparticles is shown in Fig. 3. The particles were found to be spherical in nature with the corresponding size distribution revealing a diameter of 2.6 ± 0.5 nm, reported as the average and standard deviation values. Our previous work46 reports a particle diameter of 3.5 ± 0.4 nm for the Ti(Sm)O2 nanoparticles. This suggests that co-doping Nd3+ and Sm3+ in the TiO2 nanoparticles do not affect the particle morphology to a significant extent. The high resolution transmission electron microscopy (HRTEM) image and the selected area electron diffraction (SAED) pattern identify the crystalline phases in the nanoparticles. The energy dispersive X-ray spectra (EDS) of the nanoparticles studied clearly identify the characteristic elemental peaks.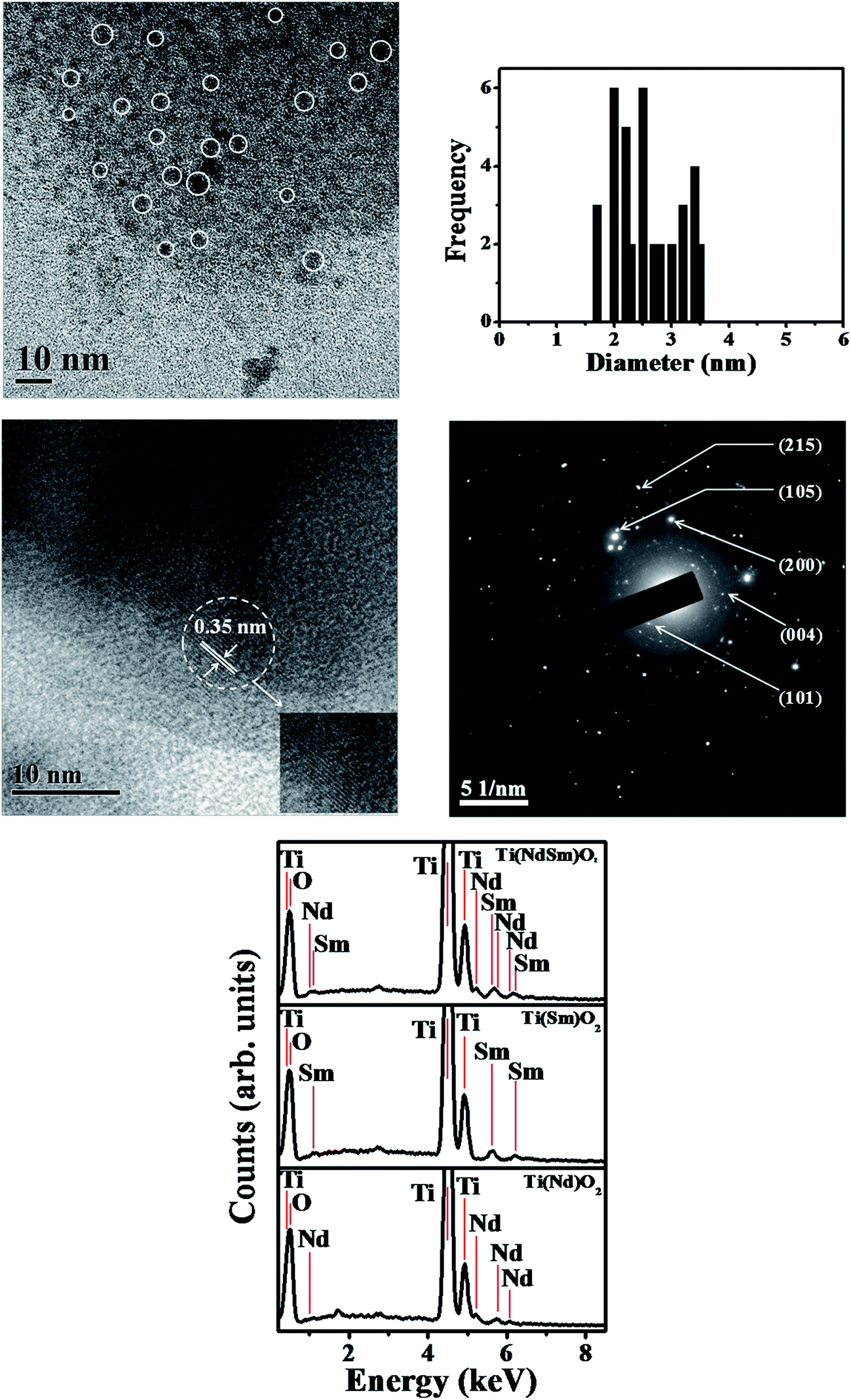 | ||
| Fig. 3 TEM image of the Ti(NdSm)O2 nanoparticles is shown in the top left panel, with the corresponding size distribution shown in the top right panel. The middle left and right panels show the HRTEM image and SAED pattern respectively. The bottom panel shows the EDS of various nanoparticles studied. | ||
Incorporation of Ln3+ in TiO2 nanoparticles generates lattice distortion and charge compensation, which is guided by the size and charge mismatch between the cationic ingredients. In a case study with europium doped titanium dioxide [Ti(Eu)O2] nanoparticles Chen and co-workers66 identified three distinct Eu3+ related sites in which for the two core sites the local site symmetry of Ti4+ deviates from D2d to D2 and C2v symmetry and one surface related site with C1 symmetry.
Photoluminescence spectroscopy
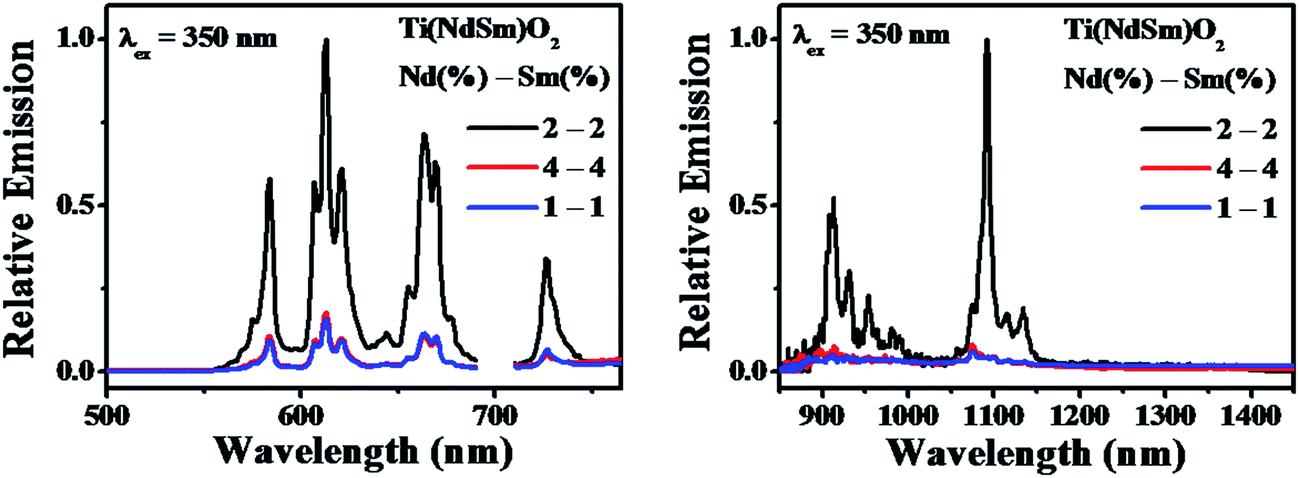 | ||
| Fig. 4 Photoluminescence emission spectra of the Ti(NdSm)O2 nanoparticles with varying nominal dopant extent are shown. The maximum intensity of the Sm3+ (left panel) and Nd3+ (right panel) emission of the sample with nominal doping extent of 2% in each Ln3+ has been normalized to unity in each panel, with the other spectra shown with respect to the intensity of this spectrum. | ||
It might be intuitively argued that the decreased luminescence intensity in the co-doped nanoparticles with nominal doping extent of 1% in each Ln3+ is associated with the lower amount of Ln3+ being present in the nanoparticles. The corresponding case with 4% in each Ln3+ poses an interesting case, where it demonstrates that merely increasing the doping extent does not make the dopant emission brighter. This could either originate from the inefficiency of dopant incorporation in the nanoparticles beyond a certain limit or due to the introduction of additional non-radiative decay pathways induced by high local dopant concentration. Important insight on this aspect comes from the elemental composition obtained from EDS measurements. The elemental composition for the nanoparticles studied is summarized in Table 1. These data reveal an increase in the amount of both Nd3+ and Sm3+ in the Ti(NdSm)O2 nanoparticles as a function of dopant concentration, suggesting the difficulty to explain the dopant luminescence quenching in the 4–4% co-doping case originating from inefficiency of dopant incorporation. Correspondingly, we propose that the luminescence quenching in this case most likely associate with the introduction of additional non-radiative decay paths that is correlated with high local concentration of the dopant cations.
| [Nd3+] (nominal)b | [Sm3+] (nominal)b | [Nd] (EDS)c | [Sm] (EDS)c | [Ti] (EDS)c | [O] (EDS)c |
|---|---|---|---|---|---|
| a The EDS values were obtained by elemental analysis from three different spatial locations and are presented as the average and the standard deviation values.b The nominal doping extent values were calculated with respect to the amount of tetra(n-butyl)titanate.c The atomic percent values from EDS were reported such that the sum of corresponding values of titanium, appropriate lanthanide and oxygen adds to 100. | |||||
| 2% | — | 0.40 ± 0.15 | — | 25.85 ± 2.63 | 73.75 ± 2.68 |
| — | 2% | — | 0.32 ± 0.14 | 25.71 ± 2.19 | 73.97 ± 2.24 |
| 1% | 1% | 0.12 ± 0.01 | 0.22 ± 0.01 | 23.45 ± 1.67 | 76.21 ± 1.67 |
| 2% | 2% | 0.36 ± 0.20 | 0.36 ± 0.08 | 24.03 ± 4.13 | 75.25 ± 4.20 |
| 4% | 4% | 0.75 ± 0.01 | 0.84 ± 0.02 | 26.27 ± 3.46 | 72.14 ± 3.46 |
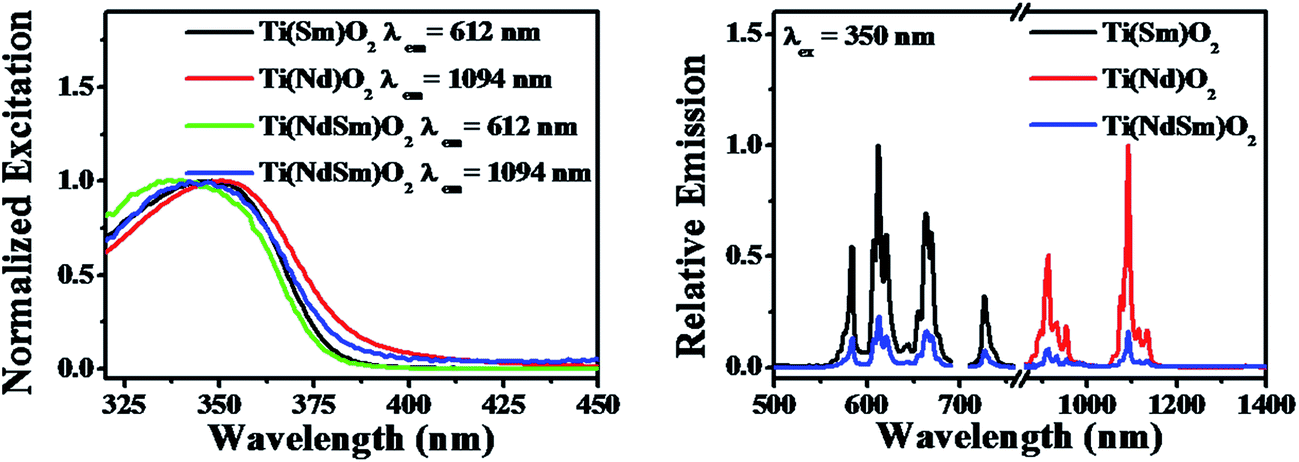 | ||
| Fig. 5 Photoluminescence excitation (left panel) and emission (right panel) spectra of the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown. In the right panel, the spectra for the singly doped nanoparticles have been normalized to unity, with the spectrum for the co-doped nanoparticles represented with respect to the singly doped spectra. | ||
The nanoparticles with both Nd3+ and Sm3+ co-doped [Ti(NdSm)O2] show characteristic emission bands from both the Nd3+ and Sm3+, giving access to both visible and near infrared (NIR) emission simultaneously with a single excitation source. Moreover, the emission lines are distinct due to core like feature of the 4f–4f transitions and do not have inter-band overlap. Thus the Ti(NdSm)O2 nanoparticles gives rise to six distinct emission bands, benefiting towards its usefulness as multiplex assays. Additionally, as the realization of Ln3+ photoluminescence has been achieved by host sensitization and not by direct excitation of Ln3+ moieties, the Stokes shift is large; thereby virtually eliminating the self quenching. Monitoring either the Nd3+ or Sm3+ emission at 1094 and 612 nm respectively in the Ti(NdSm)O2 nanoparticles, the excitation profiles appear very similar to that obtained in the two singly doped nanoparticles, demonstrating the operation of an optical antenna effect in the co-doped nanoparticles as well.
However, it is important to note that both the Nd3+ and Sm3+ emission decreases in the Ti(NdSm)O2 nanoparticles, compared to the corresponding cases in either the Ti(Nd)O2 or Ti(Sm)O2 nanoparticles. Moreover, this decrease was found to be uneven in the two cases. That is while the Nd3+ emission decreased by ∼6 times in the Ti(NdSm)O2 system compared to that in the Ti(Nd)O2 nanoparticles, the corresponding decrease for the Sm3+ emission was ∼4.5 times. The efficiency of the lanthanide emission has been compared by the emission quantum yield values and is summarized in Table 2. To shed light on the photophysical behavior, competitive mechanisms with regard to spectral overlap mediated energy transfer formulations (Förster and Dexter)69,70 and cation exchange71–76 has been considered. Important information regarding the cation exchange mechanism comes from the elemental composition (Table 1). The values related to Nd3+ and Sm3+ dopant incorporation were found to be very similar in the singly doped Ti(Ln)O2 [Ln = Nd, Sm] and doubly doped Ti(NdSm)O2 nanoparticles, suggesting the incorporation of both Nd3+ and Sm3+ is non-competitive during the formation of the Ti(NdSm)O2 nanoparticles. Hence, the inter-lanthanide cation exchange does not play significant contribution in the Ti(NdSm)O2 nanoparticles studied. A case where cation exchange being operative during the formation of the co-doped nanoparticles; the relative concentration of the displaced lanthanide cation would have been lesser. Accordingly, the observed difference in photoluminescence properties of the Ti(NdSm)O2 compared to that in the individually doped Ti(Ln)O2 [Ln = Nd, Sm] nanoparticles has predominant origin that is electronic in nature.
| System | ΦNd3+ | ΦSm3+ |
|---|---|---|
| a The values have been obtained from three independent measurements and are reported as the average and standard deviation values. | ||
| Ti(Nd)O2 | (9.1 ± 0.2) × 10−2 | — |
| Ti(Sm)O2 | — | (2.3 ± 0.5) × 10−2 |
| Ti(NdSm)O2 | (1.5 ± 0.2) × 10−2 | (0.53 ± 0.10) × 10−2 |
Successful incorporation of Ln3+ in core sites of the Ti(Ln)O2 nanoparticles studied in the present work comes from the excitation spectra upon monitoring the Ln3+ emission (Fig. 5 and 8), where the excitation profiles devoid of any direct sharp bands and photoluminescence lifetime measurements. The photoluminescence lifetime values for Sm3+ (taken as a representative Ln3+) in the Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles reveal a bi-exponential decay kinetics (Table 3), where the two lifetime components have been correlated to the lesser protected hence more quenching prone surface relates sites and more protected hence lesser quenching prone core related sites. Similar bi-exponential decay kinetics for Eu3+ emission in Ti(Eu)O2 nanoparticles has been reported by van Veggel and co-workers.33 It is important to note that Sm3+ has much shorter photoluminescence lifetime in either freely floating form47 or in protected molecular complex.19
| System | a1 | τ1 (ms) | a2 | τ2 (ms) | 〈τ〉a (ms) | Adjusted R2b |
|---|---|---|---|---|---|---|
| a 〈τ〉 = a1τ1 + a2τ2, with τ1 and τ2 being the two lifetime components having relative amplitudes of a1 and a2 respectively.b The adjusted R2 value considers the degrees of freedom during the fitting process and could be used as a gauge to determine the goodness of the fit.c From ref. 33.d From ref. 67.e From ref. 47.f From ref. 19, where H22IAM is the ligand. | ||||||
| Ti(Sm)O2 | 0.82 ± 0.02 | 0.42 ± 0.01 | 0.18 ± 0.02 | 3.4 ± 0.3 | 0.96 ± 0.11 | 0.98 |
| Ti(NdSm)O2 | 0.76 ± 0.02 | 0.53 ± 0.02 | 0.24 ± 0.02 | 3.6 ± 0.4 | 1.27 ± 0.19 | 0.97 |
| Ti(Eu)O2c | 0.23 | 0.40 | 0.77 | 1.2 | 1.0 | — |
| Ti(Sm)O2d | 0.40 | |||||
| Sm(III) Ace | 1.0 | 0.0048 | — | — | 0.0048 | |
| [SmR(+)BnMeH22IAM]f | 0.017 ± 0.002 | — | ||||
Our previous works on the photophysical processes in the Ti(Ln)O2 (ref. 46) [Ln = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb] and other semiconductor nanoparticles [Zn(Ln)S; Ln = Sm, Eu, Tb, Dy]38,44 Zn(Tb)S, Cd(Tb)S, Zn(Tb)Se and Cd(Tb)Se;38 Zn(Tb)S with varying size;43 near band gap matched Sn(Ln)O2 and Zn(Ln)S [Ln = Sm, Tb]47 have been rationalized considering the Ln3+ as the charge traps in the semiconductor nanoparticles and the exciton recombination at the Ln3+ related trap sites resulting in populating the Ln3+ luminescent energy levels, hence realizing the host sensitized Ln3+ luminescence from the doped nanoparticles. Construction of such relative energy level schematics where the Ln3+ ground and excited energy levels are placed with respect to the valence and conduction bands of the host material has been made following a method proposed by Dorenbos.77,78 This model relies on two fundamental assumptions, (i) the 4f binding energies of Ln3+ being universal and is system independent, by virtue of the core like nature of the 4f electrons and (ii) the energy of charge transfer from the anion valence band to the Eu3+ is equal to the difference in energy between the valence band and the Eu2+ ground energy level. An energy difference between Eu2+ and Eu3+ ground energy level has been considered as 5.7 eV for low band gap material (like the cases of TiO2 nanoparticles). The entire relative energy level schematics can be constructed using these inputs. The corresponding Jablonski diagrams for the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown in Fig. 6, with the associated photophysical processes discussed in the following text.
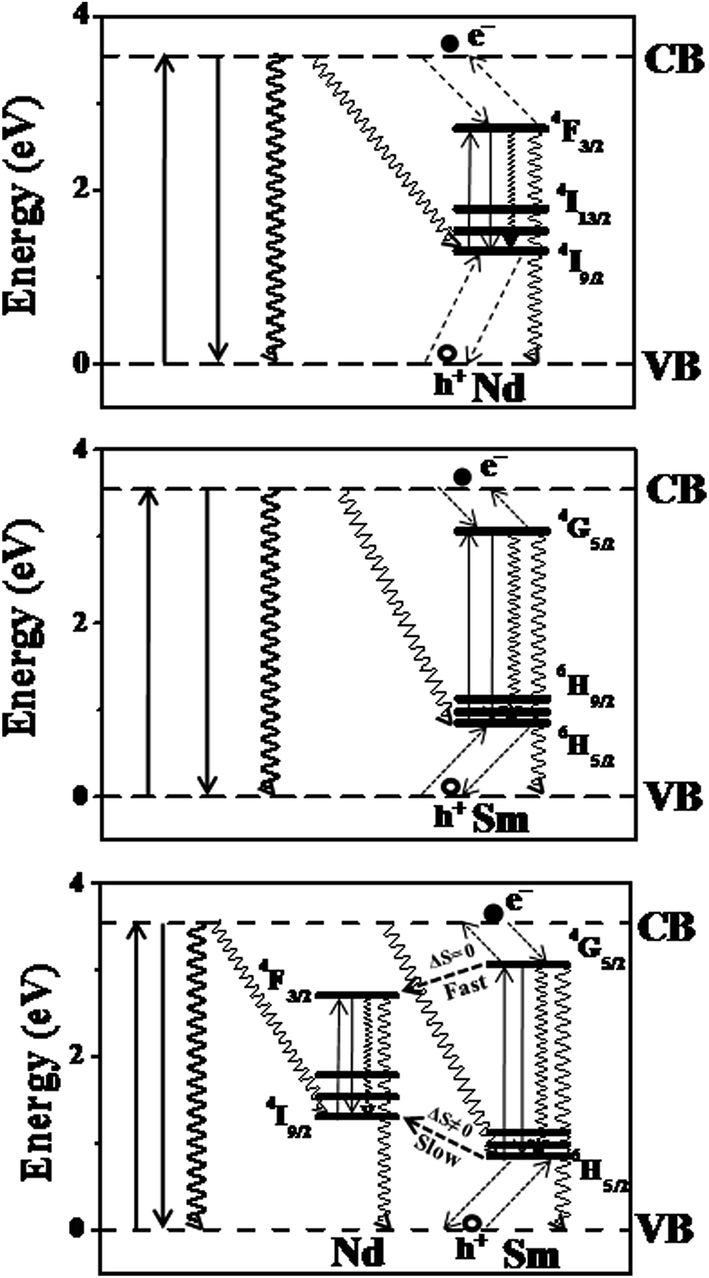 | ||
| Fig. 6 The Jablonski diagrams for the Ti(Nd)O2, Ti(Sm)O2 and Ti(NdSm)O2 nanoparticles are shown, with the identification of key photophysical processes. The dashed arrows represent the charge trapping and detrapping processes. The downward solid and squiggly arrows represent the radiative and non-radiative recombination processes respectively. | ||
In the Ti(Nd)O2 nanoparticles, absorption of light around 350 nm results in an excited electron in the conduction band while leaving a hole in the valence band. The 4I9/2 and 4F3/2 are being placed optimally above and below the valence and conduction band respectively resulting in potential hole and electron trapping in the Nd3+ ground and luminescent energy levels. Such a co-localization of charge carriers in the Nd3+ related trap site within a short time following the initial excitation competes with the time scale of other non-radiative decay mechanisms effectively and exciton recombination in the Nd3+ trap site resulting in the population of the luminescent energy level 4F3/2 in the Ti(Nd)O2 nanoparticles, thereby realizing the Nd3+ photoluminescence from the doped nanoparticles. Similarly, in the Ti(Sm)O2 nanoparticles, the corresponding energy levels 6H5/2 and 4G5/2 are responsible for the hole and electron trapping respectively, with the exciton recombination at the Sm3+ trap site resulting the population of 4G5/2 energy level, thereby generating Sm3+ photoluminescence from the Ti(Sm)O2 nanoparticles.
The case where both Nd3+ and Sm3+ are co-doped in the Ti(NdSm)O2 nanoparticles, the situation is complex as the interaction between the inter Ln3+ energy levels requires consideration. A visual inspection reveals that the Nd3+ ground energy level 4I9/2 lies above the corresponding level of Sm3+ 6H5/2. Similar consideration on the luminescent energy level shows that the Sm3+ 4G5/2 lies above the corresponding Nd3+ 4F3/2 energy level. These energy levels predict that the initially trapped hole and electron at the Sm3+ related trap should subsequently populate the Nd3+ energy levels. Accordingly, one might expect that the Nd3+ emission should enhance at the expense of Sm3+ emission in the Ti(NdSm)O2 nanoparticles, compared to the singly doped Ti(Nd)O2 and Ti(Sm)O2 nanoparticles respectively. Experimentally however this was not observed, where both the Nd3+ and Sm3+ emission decreases in the doubly doped nanoparticles compared to that in the singly doped counterparts, with the decrease being marginally more prominent in Nd3+ compared to that for the Sm3+ emission. This trend clearly indicate competitive role being operative.
We propose that the relaxation of the initially trapped charge carriers at the Sm3+ trap sites relax following the spin (S) selection rule, which states the transition is more favorable and fast when ΔS = 0. This means while the initially trapped electron from Sm3+ trap site is able to relax to the Nd3+ luminescent energy level within a short time that essentially competes with the depopulation of the Sm3+ luminescence, the situation is not same for the hole trapping case. As the spin quantum number is changing in the Sm3+ and Nd3+ ground energy levels, the hole trapping from the Sm3+ related site to the Nd3+ ground energy level is inefficient and a slow process and essentially is not complete within the excited state lifetime of Sm3+ in the Ti(NdSm)O2 nanoparticles. At this point, it is important to note that while the exciton recombination at the Ln3+ trap site with co-localization of hole and electron in the Ln3+ ground and luminescent energy levels would be the most efficient way to populate the luminescent energy level of the Ln3+ in host semiconductor nanoparticles, other related lesser efficient non-radiative recombination processes include the recombination of electron and hole at the valence and conduction band of the host lattice or electron and hole at the conduction band and the Ln3+ ground energy level or electron and hole at the Ln3+ luminescent energy level and valence band of the host lattice respectively. Such a photophysical rationalization with initial charge trapping in the Sm3+ trap site and subsequent fast relaxation of the excited electron from the Sm3+ to Nd3+ energy level and associated slow trapping of photo-generated hole from the Sm3+ to Nd3+ (that essentially competes with the microseconds to milliseconds lifetime of Sm3+ in the TiO2 based nanoparticles) accounts for the quenching of both Nd3+ and Sm3+ photoluminescence in the co-doped nanoparticles, compared to the corresponding nanoparticles with a single dopant moiety. Moreover, we anticipate that the slightly lesser quenching of the Sm3+ luminescence (compared to the case with Nd3+) in the Ti(NdSm)O2 nanoparticles, compared to that in the Ti(Sm)O2 nanoparticles, most likely associate with the initial trapping of the charge carriers at the Sm3+ sites and the initiation of subsequent exciton recombination, while such a process at the Nd3+ site experiences competitive relaxation processes and essentially would be lesser efficient.
To check this hypothesis that the experimental observations in the co-doped Ti(NdSm)O2 nanoparticles relate with the inter Ln3+ relaxation pathways that is dictated by the spin selection rule, we have undertaken experiments with Nd3+ and Er3+ co-doped TiO2 [Ti(NdEr)O2] nanoparticles. This combination of Ln3+ offers the access of ground and luminescent energy levels with ΔS = 0. The Jablonski diagram for the Ti(NdEr)O2 nanoparticles is shown in Fig. 7. However, Er3+ has more than one major luminescent energy levels and essentially the position of a particular luminescent energy level with respect to the conduction band of the host TiO2 nanoparticles matter while considering the Er3+ luminescence properties. The 4S3/2 and 4F9/2 energy levels of Er3+ lie above the Nd3+ 4F3/2 energy level, suggesting efficient electron relaxation from the Er3+ to Nd3+ in the Ti(NdEr)O2 nanoparticles. This however opens another possibility of relaxation from Nd3+ 4F3/2 to Er3+ 4I13/2 energy level. On the other hand, the initially trapped hole at the Er3+ 4I15/2 has capability to get trapped at the Nd3+ 4I9/2 energy level, with significant back hole transfer as these energy levels are nearly isoenergetic.
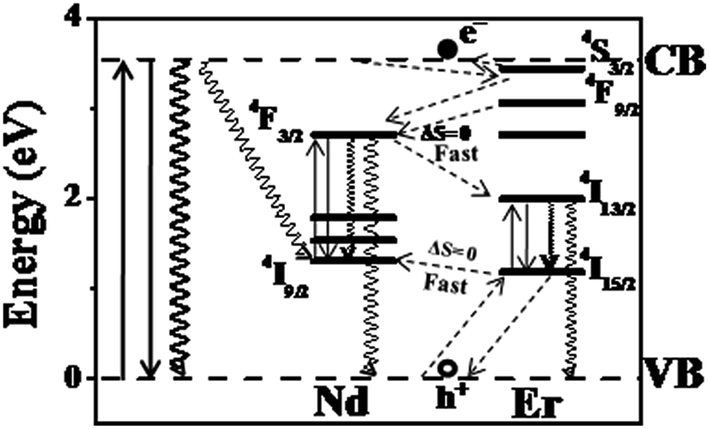 | ||
| Fig. 7 The Jablonski diagram for the Ti(NdEr)O2 nanoparticles is shown. | ||
The photoluminescence excitation and emission spectra of the Ti(NdEr)O2 nanoparticles are shown in Fig. 8. The relevant quantum yield values are summarized in Table 4. Clearly the emission at 565 and 665 nm originating from 4S3/2 → 4I15/2 and 4F9/2 → 4I15/2 respectively diminishes in the co-doped Ti(NdEr)O2 nanoparticles, compared to that in the Ti(Er)O2 nanoparticles. On the other hand, the emission intensity of 1550 nm band originating from 4I13/2 → 4I15/2 increases by ∼3 times in the doubly doped nanoparticles, compared to that in the singly Er3+ doped counterpart. The increase in Er3+ emission around 1550 nm in presence of Nd3+ is consistent with the electron relaxation from the 4F3/2 energy level of Nd3+ to the Er3+ 4I13/2 energy level. Moreover, the excitation spectrum monitoring the Er3+ 1550 nm emission in the Ti(NdEr)O2 nanoparticles also has contribution from host sensitized photoluminescence.
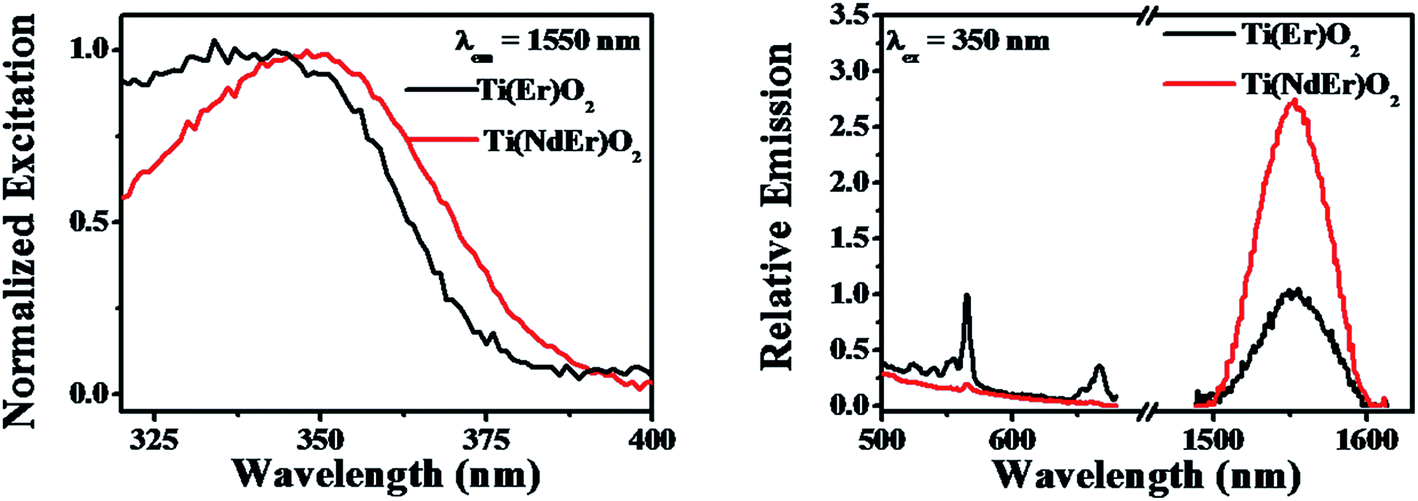 | ||
| Fig. 8 Photoluminescence excitation (left panel) and emission (right panel) spectra of the Ti(Nd)O2, Ti(Er)O2 and Ti(NdEr)O2 nanoparticles are shown. In the right panel, the emission intensity of the singly doped nanoparticles has been normalized to unity, with the intensity in the co-doped nanoparticles represented with respect to the singly doped system. | ||
| System | ΦNd3+ | ΦEr3+ [4S3/2 → 4I15/2] + ΦEr3+ [4F3/2 → 4I15/2] | ΦEr3+ [4I13/2 → 4I15/2] |
|---|---|---|---|
| a The values have been obtained from two independent measurements and are reported as the average and standard deviation values. | |||
| Ti(Nd)O2 | (9.1 ± 0.2) × 10−2 | — | — |
| Ti(Er)O2 | — | (1.9 ± 1.0) × 10−5 | (0.60 ± 0.14) × 10−6 |
| Ti(NdEr)O2 | (1.7 ± 0.1) × 10−2 | (0.13 ± 0.01) × 10−5 | (1.63 ± 0.17) × 10−6 |
The excitation spectra upon monitoring the Ln3+ emission in either the singly or doubly Ln3+ incorporated nanoparticles investigated in the present work only reveal a broad profile that is related to host sensitization, without noticeable contribution from the direct sharp intra-configurational excitation bands. The inability to observe such direct excitation bands most likely associate with the following points; (i) an estimation of the concentration of Ln3+ in the nanoparticles used in the photoluminescence spectroscopy measurements result in a value of ≤35 micromolar which is too low to observe extremely inefficient direct excitation, in presence of stronger contribution from host sensitization. It is noteworthy that van Veggel33 reported direct excitation bands in the Ti(Ln)O2 [Ln = Nd, Er] excitation profiles only when the nanoparticles concentration was high, whereas the same system with lower concentration of the nanoparticles did not produce the sharp lines in the excitation profile, (ii) in the framework of charge trapping mediated photoluminescence sensitization mechanism some of the Ln3+ energy levels lie closer or above the conduction band of the host lattice suggesting rapid autoionization of charge carriers. However, we note that in cases where sensitization involves an inter band gap Ln3+ energy level, a red shifted broad profile may result. Such scenario may arise in the Ti(NdEr)O2 nanoparticles, where the excitation spectrum upon monitoring the Er3+ emission at 1550 nm result in an enhanced lower energy contribution compared to that in the Ti(Er)O2 nanoparticles.
Finally, we note that while the photophysical rationalization presented in this work provides a qualitative basis to visualize the experimental observations, a complete picture requires the characterization of entire dynamics involving a wide range of time scales, with the charge trapping might be occurring in few picoseconds,79 nanoparticles and Ln3+ population decay occurring in the nanoseconds to microseconds–milliseconds range.5,38
Conclusions
Guided by the unique luminescence properties of the trivalent lanthanide cations (Ln3+) with its potential usefulness as multiplex assays and our recent work on singly Ln3+ doped titanium dioxide [Ti(Ln)O2] nanoparticles with the identification of Ti(Nd)O2 and Ti(Sm)O2 as the most promising systems having host sensitized dopant photoluminescence, this study develops a system where Nd3+ and Sm3+ have been co-doped in the TiO2 nanoparticles, to synthesize the Ti(NdSm)O2 nanoparticles. The co-doped nanoparticles benefits from simultaneous Sm3+ visible emission at 584, 612, 664 and 726 nm respectively and Nd3+ near infrared (NIR) emission at 912 and 1094 nm respectively. This provides an avenue to realize six distinct and non-overlapping emission bands spanning the orange-red and NIR spectral window using a single excitation wavelength, with a large Stokes shift. A comparison between the doubly doped and the corresponding singly doped nanoparticles reveals significant differences, with the Nd3+ and Sm3+ emission decreasing by ∼6 and ∼4.5 times in the co-doped system compared to that in the singly doped nanoparticles. The results have been rationalized considering the Ln3+ acting as charge traps in the semiconductor nanoparticles and associated inter Ln3+ (applicable in the co-doped nanoparticles) relaxation pathways that are governed by the spin selection rule. This proposed rationalization has been tested and verified by performing experiments with the Ti(NdEr)O2 nanoparticles, in which 1550 nm emission of Er3+ has been increased in intensity in the co-doped nanoparticles by ∼3 times, compared to that in the Ti(Er)O2 nanoparticles. To summarize, this work provides an avenue to develop a multiplex assay using Sm3+ and Nd3+ emission in the orange-red and NIR spectral range respectively. Future works may focus on developing composite doped nanoparticles having host sensitized dopant emission with distinct non-overlapping bands spanning the entire visible (blue, green and red) and NIR spectral domain simultaneously. The photophysical aspects discussed in the current work provides valuable foundation for developing such a composite system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial assistance from the Science and Engineering Research Board (SERB), Department of Science and Technology (DST) (SB/S1/PC-040/2013) is gratefully acknowledged. The authors thank Ms Urmila Goswami and Mr Prothyush Sengupta for help in the electron microscopy measurements. The authors also thank Prof. Dipankar Chattopadhyay for letting us use the XRD instrument in his laboratory and Mr Nayan Ranjan Saha for helping us with the XRD measurements.References
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS PubMed.
- N. Hildebrandt and H.-G. Löhmannsröben, Curr. Chem. Biol., 2007, 1, 167–186 CAS.
- A. Thibon and V. C. Pierre, Anal. Bioanal. Chem., 2009, 394, 107–120 CrossRef CAS PubMed.
- M. A. Alcala, S. Y. Kwan, C. M. Shade, M. Lang, H. Uh, M. Wang, S. G. Weber, D. L. Bartlett, S. Petoud and Y. J. Lee, Nanomedicine, 2011, 7, 249–258 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu and X. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- C. Bouzigues, T. Gacoin and A. Alexandrou, ACS Nano, 2011, 5, 8488–8505 CrossRef CAS PubMed.
- R. D. Teo, J. Termini and H. B. Gray, J. Med. Chem., 2016, 59, 6012–6024 CrossRef CAS PubMed.
- A. K. Hagan and T. Zuchner, Anal. Bioanal. Chem., 2011, 400, 2847–2864 CrossRef CAS PubMed.
- M. Sy, A. Nonat, N. Hildebrandt and L. J. Charbonnière, Chem. Commun., 2016, 52, 5080–5095 RSC.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–503 RSC.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- S. Petoud, G. Muller, E. G. Moore, J. Xu, J. Sokolnicki, J. P. Riehl, U. N. Le, S. M. Cohen and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 77–83 CrossRef CAS PubMed.
- H. Uh and S. Petoud, C. R. Chim., 2010, 13, 668–680 CrossRef CAS.
- J.-F. Lemonnier, L. Guénée, C. Beuchat, T. A. Wesolowski, P. Mukherjee, D. H. Waldeck, K. A. Gogick, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2011, 133, 16219–16234 CrossRef CAS PubMed.
- J.-F. Lemonnier, L. Babel, L. Guénée, P. Mukherjee, D. H. Waldeck, S. V. Eliseeva, S. Petoud and C. Piguet, Angew. Chem., Int. Ed., 2012, 51, 11302–11305 CrossRef CAS PubMed.
- J. Zhang, C. M. Shade, D. A. Chengelis and S. Petoud, J. Am. Chem. Soc., 2007, 129, 14834–14835 CrossRef CAS PubMed.
- J. P. Cross, M. Lauz, P. D. Badger and S. Petoud, J. Am. Chem. Soc., 2004, 126, 16278–16279 CrossRef CAS PubMed.
- C. S. Bonnet, L. Pellegatti, F. Buron, C. M. Shade, S. Villette, V. Kubiček, G. Guillaumet, F. Suzenet, S. Petoud and É. Tóth, Chem. Commun., 2010, 46, 124–126 RSC.
- K. A. White, D. A. Chengelis, K. A. Gogick, J. Stehman, N. L. Rosi and S. Petoud, J. Am. Chem. Soc., 2009, 131, 18069–18071 CrossRef CAS PubMed.
- K. A. White, D. A. Chengelis, M. Zeller, S. J. Geib, J. Szakos, S. Petoud and N. L. Rosi, Chem. Commun., 2009, 4506–4508 RSC.
- A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi and S. Petoud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199–17204 CrossRef CAS PubMed.
- D. A. Chengelis, A. M. Yingling, P. D. Badger, C. M. Shade and S. Petoud, J. Am. Chem. Soc., 2005, 127, 16752–16753 CrossRef CAS PubMed.
- X. Chen, W. Luo, Y. Liu and G. Liu, J. Rare Earths, 2007, 25, 515–525 CrossRef.
- Y. Liu, D. Tu, H. Zhu and X. Chen, Chem. Soc. Rev., 2013, 42, 6924–6958 RSC.
- W. Luo, Y. Liu and X. Chen, Sci. China Mater., 2015, 1–32 Search PubMed.
- J. W. Stouwdam and F. C. J. M. van Veggel, ChemPhysChem, 2004, 5, 743–746 CrossRef CAS PubMed.
- A. P. Jadhav, A. U. Pawar, U. Pal and Y. S. Kang, J. Mater. Chem. C, 2014, 2, 496–500 RSC.
- A. Pawar, A. Jadhav, C. W. Kim, H. G. Cha, U. Pal and Y. S. Kang, J. Lumin., 2015, 157, 131–136 CrossRef CAS.
- J. Planelles-Aragó, B. Julián-López, E. Cordoncillo, P. Escribano, F. Pellé, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 5193–5199 RSC.
- J. Planelles-Aragó, E. Cordoncillo, R. A. S. Ferreira, L. D. Carlos and P. Escribano, J. Mater. Chem., 2011, 21, 1162–1170 RSC.
- P. Mukherjee, C. M. Shade, A. M. Yingling, D. N. Lamont, D. H. Waldeck and S. Petoud, J. Phys. Chem. A, 2011, 115, 4031–4041 CrossRef CAS PubMed.
- P. Mukherjee, R. F. Sloan, C. M. Shade, D. H. Waldeck and S. Petoud, J. Phys. Chem. C, 2013, 117, 14451–14460 CAS.
- J. R. Dethlefsen, A. A. Mikhailovsky, P. T. Burks, A. Døssing and P. C. Ford, J. Phys. Chem. C, 2012, 116, 23713–23720 CAS.
- R. Martín-Rodríguez, R. Geitenbeek and A. Meijerink, J. Am. Chem. Soc., 2013, 135, 13668–13671 CrossRef PubMed.
- A. Ghatak, G. H. Debnath, M. Mandal and P. Mukherjee, RSC Adv., 2015, 5, 32920–32932 RSC.
- G. H. Debnath, A. Chakraborty, A. Ghatak, M. Mandal and P. Mukherjee, J. Phys. Chem. C, 2015, 119, 24132–24141 CAS.
- A. Chakraborty, G. H. Debnath, M. Ahir, S. Bhattacharya, P. Upadhyay, A. Adhikary and P. Mukherjee, RSC Adv., 2016, 6, 43304–43315 RSC.
- G. H. Debnath, A. Chakraborty and P. Mukherjee, RSC Adv., 2016, 6, 85230–85241 RSC.
- A. Chakraborty, G. H. Debnath, N. R. Saha, D. Chattopadhyay, D. H. Waldeck and P. Mukherjee, J. Phys. Chem. C, 2016, 120, 23870–23882 CAS.
- P. Manna, A. Chakraborty, G. H. Debnath and P. Mukherjee, J. Phys. Chem. Lett., 2017, 8, 2794–2798 CrossRef CAS PubMed.
- J. R. DiMaio, C. Sabatier, B. Kokuoz and J. Ballato, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1809–1813 CrossRef CAS PubMed.
- K. Murray, Y.-C. Cao, S. Ali and Q. Hanley, Analyst, 2010, 135, 2132–2138 RSC.
- Y. Xu and Q. Li, Clin. Chem., 2007, 53, 1503–1510 CAS.
- L. Cheng, K. Yang, S. Zhang, M. Shao, S. Lee and Z. Liu, Nano Res., 2010, 3, 722–732 CrossRef CAS.
- H. H. Gorris, R. Ali, S. M. Saleh and O. S. Wolfbeis, Adv. Mater., 2011, 23, 1652–1655 CrossRef CAS PubMed.
- H. Dong, L.-D. Sun, W. Feng, Y. Gu, F. Li and C.-H. Yan, ACS Nano, 2017, 11, 3289–3297 CrossRef CAS PubMed.
- R. Deng and X. Liu, Nat. Photonics, 2014, 8, 10–12 CrossRef CAS.
- D. Gao, H. Zheng, X. Zhang, W. Gao, Y. Tian, J. Li and M. Cui, Nanotechnology, 2011, 22, 175702 CrossRef PubMed.
- W. Xu, B. A. Bony, C. R. Kim, J. S. Baeck, Y. Chang, J. E. Bae, K. S. Chae, T. J. Kim and G. H. Lee, Sci. Rep., 2013, 3, 3210 CrossRef PubMed.
- S. H. Crayton, D. R. Elias, A. A. Zaki, Z. Cheng and A. Tsourkas, Biomaterials, 2012, 33, 1509–1519 CrossRef CAS PubMed.
- X. Yan, L. Yang and Q. Wang, Angew. Chem., Int. Ed., 2011, 50, 5130–5133 CrossRef CAS PubMed.
- X. Gao, L. Yang, J. A. Petros, F. F. Marshall, J. W. Simons and S. Nie, Curr. Opin. Biotechnol., 2005, 16, 63–72 CrossRef CAS PubMed.
- W. C. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS PubMed.
- X. Gao and S. Nie, Trends Biotechnol., 2003, 21, 371–373 CrossRef CAS PubMed.
- M. Hu, J. Yan, Y. He, H. Lu, L. Weng, S. Song, C. Fan and L. Wang, ACS Nano, 2010, 4, 488–494 CrossRef CAS PubMed.
- P. Ellmark, L. Belov, P. Huang, C. S. Lee, M. J. Solomon, D. K. Morgan and R. I. Christopherson, Proteomics, 2006, 6, 1791–1802 CrossRef CAS PubMed.
- H. Li, Z. Cao, Y. Zhang, C. Lau and J. Lu, Anal. Methods, 2010, 2, 1236–1242 RSC.
- M. V. Yezhelyev, X. Gao, Y. Xing, A. Al-Hajj, S. Nie and R. M. O'Regan, Lancet Oncol., 2006, 7, 657–667 CrossRef CAS PubMed.
- W. Luo, R. Li, G. Liu, M. R. Antonio and X. Chen, J. Phys. Chem. C, 2008, 112, 10370–10377 CAS.
- W. Luo, R. Li and X. Chen, J. Phys. Chem. C, 2009, 113, 8772–8777 CAS.
- J. E. Lewis and M. Maroncelli, Chem. Phys. Lett., 1998, 282, 197–203 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn, 2006 Search PubMed.
- S. Speiser, Chem. Rev., 1996, 96, 1953–1976 CrossRef CAS PubMed.
- D. H. Son, S. M. Hughes, Y. Yin and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- R. D. Robinson, B. Sadtler, D. O. Demchenko, C. K. Erdonmez, L.-W. Wang and A. P. Alivisatos, Science, 2007, 317, 355–358 CrossRef CAS PubMed.
- D. Mocatta, G. Cohen, J. Schattner, O. Millo, E. Rabani and U. Banin, Science, 2011, 332, 77–81 CrossRef CAS PubMed.
- C. Dong and F. C. J. M. van Veggel, ACS Nano, 2009, 3, 123–130 CrossRef CAS PubMed.
- B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759–19770 CAS.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- P. Dorenbos and E. van der Kolk, Appl. Phys. Lett., 2006, 89, 061122 CrossRef.
- P. Dorenbos, J. Alloys Compd., 2009, 488, 568–573 CrossRef CAS.
- P. Kambhampati, J. Phys. Chem. C, 2011, 115, 22089–22109 CAS.
| This journal is © The Royal Society of Chemistry 2017 |