
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the inhomogeneity and intermediates in the photosensitized degradation of rhodamine B by Ag3PO4 nanoparticles from an ensemble to a single molecule approach†
Beibei Xu,
Xiaojuan Wang,
Chaofeng Zhu,
Xia Ran,
Tianfeng Li* and
Lijun Guo *
*
School of Physics and Electronics, Henan University, Kaifeng 475004, P. R. China. E-mail: litianfeng@henu.edu.cn; juneguo@henu.edu.cn
First published on 22nd August 2017
Abstract
The photoinduced dynamics related to the degradation of a surface adsorbate by a semiconductive catalyst is critical for understanding the photocatalytic mechanism and improving the catalytic property of nanoscale materials. Herein, we report the investigation of the inhomogeneous interactions between Ag3PO4 nanoparticles and rhodamine B (RhB), and the direct observation of the intermediates generated in the photodegradation of RhB, using ensemble-averaged as well as single-molecule time-resolved fluorescence spectroscopies. The results demonstrate the existence of electron injection from RhB into the conduction band of Ag3PO4 and the formation of a deethylation intermediate before the subsequent degradation process. The fluorescence diversity both in lifetime and intensity fluctuation indicates an inhomogeneous interfacial interaction between the RhB molecule and Ag3PO4 nanoparticle with surface heterogeneity. Based on the lifetime distribution, the duration time of single-molecule events and the related dynamical analysis, it was revealed that the RhB molecule adsorbed on the active site of the Ag3PO4 nanoparticle has a higher injection efficiency and better photocatalytic activity. Moreover, the lifetime evolution derived from a subsection of the single-molecule emission trajectories proved that the electron injection occurred prior to the degradation through the attack of free radical O2˙−. These findings provide new insights into the heterogeneous interactions and dynamical information of the photosensitized degradation in an adsorbate/semiconductor catalyst.
Introduction
Photocatalytic and solar energy conversion nanoscale materials and their applications, such as in the degradation of organic compounds,1–3 hydrogen production by water photolysis,4 and dye-sensitized solar cells (DSSCs),5 have attracted much attention in the past decades. The efficiency of photocatalysis and optical energy conversion in semiconductor nanoscale photocatalysts is often affected by various parameters, including the particle size, crystalline phase, lattice or surface defects, surface area, and morphology of the particles.6 The complex interactions between nanostructures and their surface adsorbates play critical roles both in photocatalytic and photosensitizing processes, since these interactions greatly affect the separation efficiency of electron–hole pairs, the generation rate of free radicals, and the injection path of electrons.7–9 Further understanding of such complex dynamical interactions between nanoparticles and absorbates would be of great significance for designing novel materials, improving material properties, and elucidating the fundamental mechanisms of photocatalysis and pollutant degradation.To date, considerable research efforts have been devoted to examining the pathway and mechanism of photocatalytically degrading organics by various nanoscale semiconductors to promote the separation of electron–hole pairs and therefore to enhance the photocatalytic efficiency. Rhodamine B (RhB) has often been employed as a model molecule in characterizing the degradation effect. However, the degradation process of RhB and the generated intermediate products have been investigated mostly based on ensemble-averaged methods. It has been proposed that the active sites on a catalyst surface could serve as strong electron-trapping sites to enhance the photocatalytic efficiency. Specifically, the defect sites at surfaces or interfaces of TiO2 films have been found to promote the separation of photogenerated electron–hole pairs and therefore affect the photoactivity of defective TiO2 films.10,11 In another case where complexes were formed by a nanoparticle and adsorbate, it was found that the inhomogeneous interaction between a dye molecule and semiconductor surface also strongly affects the interfacial electron transfer (IET) and thus the sensitized efficiency in dye-sensitized solar cell (DSSC) systems, which have been investigated using ultrafast spectroscopy and ensemble-averaged approaches.12,13 Nevertheless, there remains an urgent need for a deeper understanding of the detailed interactions occurring on a nanoparticle surface. However, because these chemical reactions take place on heterogeneous surfaces, the spatial and temporal inhomogeneities are hard to identify and analyze using ensemble experiments. Single-molecule, single-particle fluorescence and single molecule fluorescence lifetime imaging microscopy (FLIM) have been proved to be the very powerful tools for investigating heterogeneous processes because of their high sensitivity and selectivity, simplicity of data collection, and high spatial and temporal resolution.14–18 Indeed, Majima and coworkers used single-molecule dynamical observations and proposed that the reaction sites in the effective reduction of probe molecules are located on the facets with a higher surface energy.19 The IET processes between a single dye molecule and TiO2 nanoparticle surface demonstrate an inhomogeneity and sensitizing efficiency.20,21 Compared with intensity imaging, fluorescence lifetime imaging is less susceptible to artifacts arising from scattered light, photobleaching, or non-uniform illumination of the sample, and it can thus provide an accurate approach to probing the molecular interaction changes, molecular and IET dynamics, and reaction intermediate states22–25 in photocatalysis at a single molecule level.
Ag3PO4 nanoparticles represent a novel photocatalyst with a band gap of 2.45 eV, which makes them a promising photocatalyst for degrading pollutants under visible light irradiation. Meanwhile, the energy of the conduction band (ECB) in Ag3PO4 is fairly low to be able to accept the photoexcited electrons of adsorbed molecules, when the lowest unoccupied molecular orbital (LUMO) of the adsorbate is higher than the ECB.26 Ge and coworker reported that the photodegradation of methyl orange over Ag3PO4 catalyst was greatly enhanced in the presence of RhB because of the injected electrons from the RhB molecule to the catalysts.27 In this work, we choose the RhB–Ag3PO4 nanoparticle system as a model to systematically investigate the heterogeneous catalysis and dynamical interaction between RhB and Ag3PO4 nanoparticles, and particularly to probe the generation of intermediate products in the photodegradation by using ensemble-averaged spectroscopy and single-molecule fluorescence lifetime microscopy.
Experimental
Materials and sample preparation
Silver acetate (CH3COOAg, >99%) was purchased from Aladdin, and disodium hydrogen phosphate (Na2HPO4·12H2O, >99%) was purchased from Kermel. All of the other reagents were commercially obtained and used without further purification. Deionized water from a Milli-Q system was used in the preparation of materials. All the cover glasses (Fisher, 18 mm × 18 mm, thickness ∼ 170 μm) were thoroughly cleaned by sonication in deionized water, ethanol, acetone, and deionized water, respectively for 20 min, and then dried using nitrogen gas before use. The Ag3PO4 crystals were synthesized by a simple precipitation process. Briefly, CH3COOAg (200 mg) was dissolved in 100 mL aqueous solution. Na2HPO4 aqueous solution (0.15 M) was added drop by drop to the above solution with a magnetic stirrer, which was then continuously stirred for 4 h to form a golden yellow precipitation. The obtained products were repeatedly washed with deionized water to remove the remaining CH3COO− and then dried in an oven at 50 °C for 6 h.28 The samples for the single-molecule experiments were prepared according to the previous literature.20 For a control experiment, 25 μL of 0.05 nM solution of RhB was spin-coated onto a coverslip at 3000 rpm. The sample of RhB on Ag3PO4 particles was prepared by first spin-coating 25 μL Ag3PO4 dispersed solution on a clean coverslip at 2000 rpm, followed by overlaying 25 μL of 0.05 nM RhB solution onto the above coverslip.Characterization
The morphology of the as-prepared photocatalysts was characterized by field emission scanning electron microscopy (SEM JSM-6700F, Hitachi). The UV-Vis absorption spectra of the solid materials were recorded using a Cary 5000 UV-Vis-NIR spectrophotometer equipped with an integrating sphere attachment. The absorption spectrum of the resulting solution after photocatalysis was obtained using a PerkinElmer Lambda 35 spectrometer. The fluorescence emission spectrum was recorded at room temperature with excitation at 488 nm on a PerkinElmer fluorescence spectrometer LS55.Photocatalytic activity measurements
The photocatalytic activity of Ag3PO4 nanoparticles was characterized by the de-colorization of RhB solution. The optical system for the photocatalytic reaction comprised a 250 W Xe lamp and a cutoff filter (λ > 400 nm, NBeT Beijing). In the photocatalytic experiments, 15 mg of Ag3PO4 nanoparticles was dispersed in RhB (100 mL, 10−5 M) aqueous solution and then stirred in the dark for 30 min to reach an absorption–desorption equilibrium of RhB on the surface of Ag3PO4 nanoparticles. With visible light irradiation, 2.5 mL of reaction solution was taken out at a given time. After centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 5 min), the supernatants were analyzed by monitoring the absorption maximum with a Lambda 35 UV-Vis spectrophotometer.
000 rpm, 5 min), the supernatants were analyzed by monitoring the absorption maximum with a Lambda 35 UV-Vis spectrophotometer.
Ensemble-averaged florescence lifetime measurements
Ensemble fluorescence decay traces were recorded using the time-correlated single photon counting (TCSPC) method with a home-built fluorescence lifetime setup (Harp300, Picoquant). A broadband tunable pulse femtosecond Ti:sapphire laser (Chameleon Ultra II, Coherent Inc.) equipped with a pulse selection system (Conoptics, Model 305) was used to obtain the excitation repetition frequency of 40 MHz. The 488 nm femtosecond excitation source was generated from the second harmonic generation with a BBO nonlinear crystal. The lens-focused fluorescence emission was separated into two channels by a dichroic mirror (ZT532rdc) and passed through a band-pass filter (ET540/30 m, Chroma) and a long-pass filter (AT575lp, Chroma). The photons from different channels were detected by two micro photon devices (MPD, Picoquant) and delivered into a TCSPC system for analysis.Single-molecule fluorescence lifetime measurements and imaging
Single-molecule fluorescence images were recorded on an inverted scanning confocal microscope (IX-73, Olympus) equipped with a 100× oil immersion objective (1.4 NA, Olympus) and a close-loop nanoscale-precision piezoelectric scanning stage (Physik Instrument). A pulsed excitation source of 488 nm was generated with a BBO crystal as described previously, and the excitation pulse energy at the sample was set below 0.25 pJ. The laser beam was reflected by a dichroic beam splitter (ZT488rdc) and was focused to 100× objective on the sample. The fluorescence was collected with the same objective, passed through a long-pass filter (ET525lp, Chroma), and focused on an MPD detector (100 × 100 μm2). Synchronization of the data acquisition with the scanner movement was controlled by SymPhoTime software using the time-tagged time-resolved (TTTR) measurement mode, in which each photon image was recorded by a raster scanning an area of 20 × 20 μm2 with a resolution of 128 × 128 pixels.Results and discussion
Ensemble-averaged observations of photodegradation
Fig. 1 shows the SEM image and absorption spectrum of the as-prepared Ag3PO4 nanoparticles with an average diameter of about 300 nm. The crystal structure and purity of these nanoscale catalysts were further characterized by their XRD and XPS spectra (Fig. S1 and S2†), which are consistent with the reported results in the literature.27 The UV-Vis absorption spectrum indicates the particles can absorb solar light with a wavelength shorter than 520 nm (Fig. 1b). The band gap Eg of the Ag3PO4 nanoparticle was determined to be 2.30 eV according to the equation: αhν = A(hν − Eg)n/2, where α, ν, and A are the absorption coefficient, light frequency, and proportionality constant, respectively.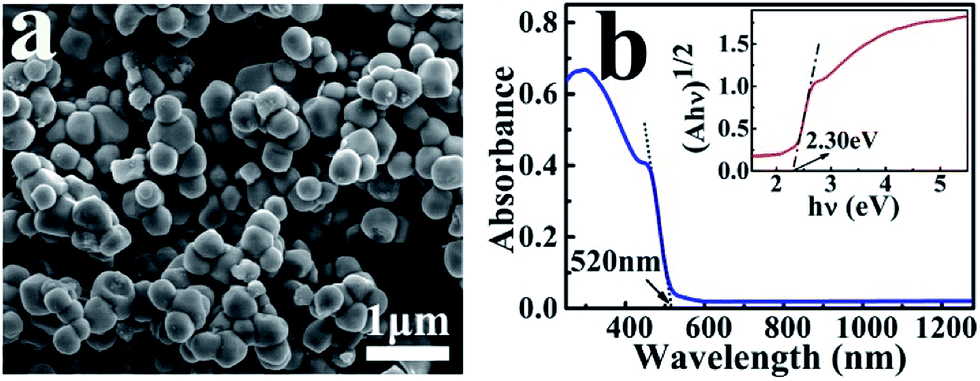 | ||
| Fig. 1 SEM image (a) and UV-Vis absorption spectrum (b) of the as-prepared Ag3PO4 nanoparticles. The inset is the plot of (αhν)1/2 versus (hν) for the Ag3PO4 to derive the band gap. | ||
The photocatalytic characterization of Ag3PO4 nanoparticles was monitored by observing the temporal changes of the maximum absorption of RhB under visible light irradiation (Fig. 2a). After 45 min, the absorption band at 554 nm was rapidly decreased and simultaneously broadened, which is similar to observations reported in the TiO2/RhB system.10,29–31 Concomitantly, the absorption maximum of the degraded solution exhibited a slight blue-shift from 554 nm to 530 nm. This blue-shift of the absorption band could be attributed to the typical process of the photochemical deethylation of RhB from the attack of active oxygen species on the N-ethyl group. In the first 35 min of visible irradiation, the absorption maximum continuously decreased with the ongoing photocatalysis of Ag3PO4 nanoparticles, due to the generated deethylated intermediates and degraded RhB. In the subsequent irradiation duration, the absorbance decreased continuously but the position of the absorption maximum no long shifted, manifesting that the intermediates were further degraded afterwards (inset of Fig. 2a). Correspondingly, the generation of intermediates in the photocatalytic process was also observed from the evolution of the fluorescence spectra of RhB degraded by Ag3PO4 nanoparticles under visible light irradiation (Fig. 2b). We found that the emission maximum of RhB at 575 nm decreased and a new band around 535 nm appeared as the reaction proceeded, confirming the generation of the deethylated intermediate. Consistently, the fluorescence intensity of the produced intermediates increased in the first 35 min and then decreased with further irradiation, similar to the above observations in the absorption spectroscopy.29,30
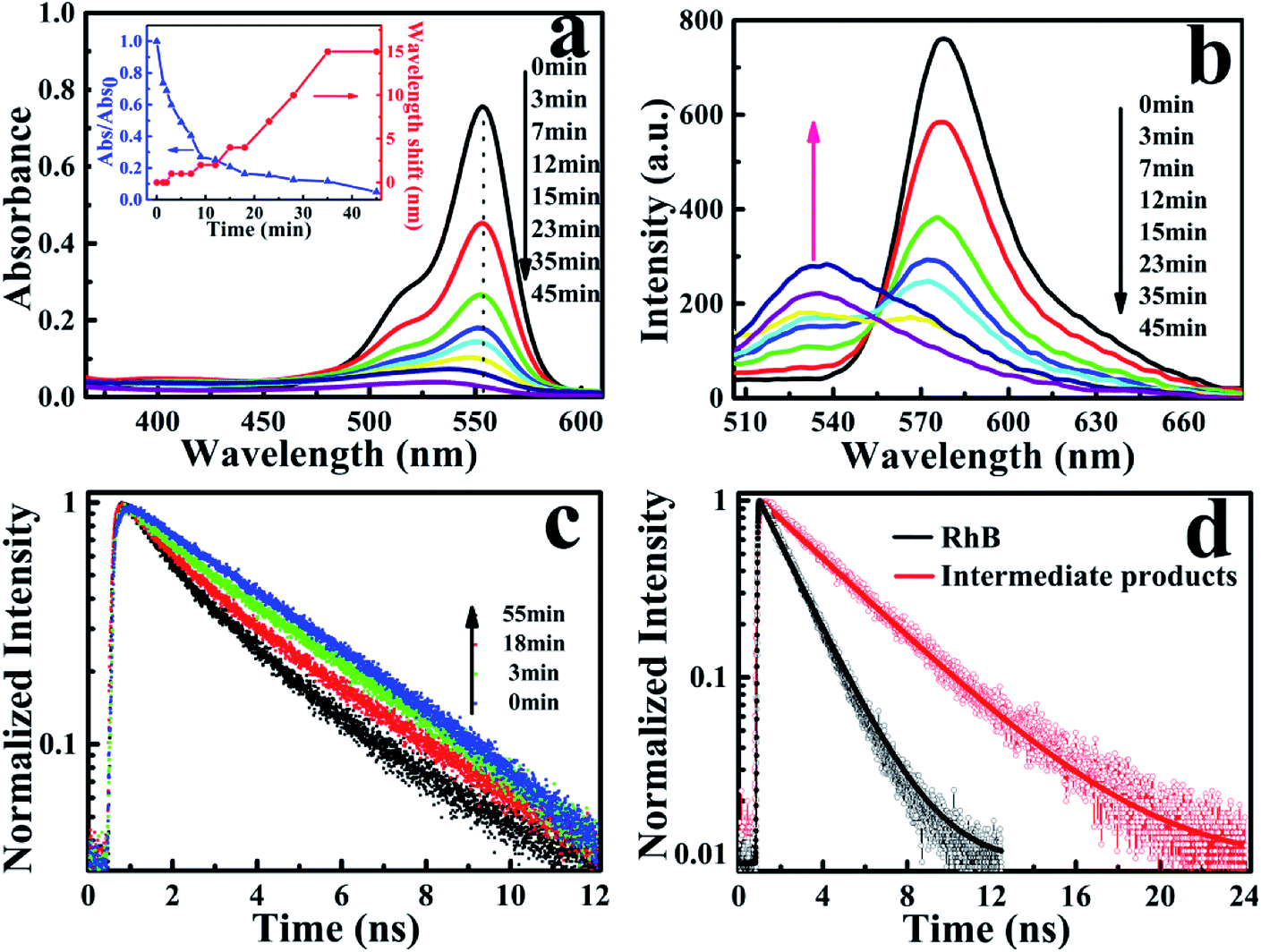 | ||
| Fig. 2 (a) UV-Vis spectral changes with visible irradiation time in the photodegradation of RhB solution (1 × 10−6 M) by Ag3PO4 nanoparticles. Inset: correlation between the absorbance changes at absorption maximum (blue line) and the corresponding wavelength shifts (red line); (b) fluorescence spectral evolution of the RhB/Ag3PO4 system under visible light irradiation. (c) Fluorescence decay curves (525–555 nm) of the RhB/Ag3PO4 system in aqueous solution under different visible irradiation times. (d) Typical fluorescence lifetime of RhB (black line) and the intermediate deethylation products (red line) in aqueous solution. | ||
To further identify the deethylation intermediates produced in the photocatalysis, we performed fluorescence lifetime measurements by monitoring the emissions of RhB and the intermediates, using a resemble-averaged two-channel TCSPC setup with tens of picosecond resolution. Briefly, the collected fluorescence signals from the photocatalytic solution were separated into two paths by a 532 nm dichroic mirror (ZT532rdc) and passed through a long-pass filter (AT575lp) for RhB and a band-pass filter (ET540/30 nm) for the deethylation intermediate, then detected by two MPDs, respectively. As shown in Fig. 2c, the decay curves of RhB (λem > 575 nm) demonstrate little change before and after the photocatalytic reaction (not shown). However, the emission transient traces of the deethylation intermediates (525 nm < λem < 555 nm) exhibit an obvious evolution with the photocatalysis because the intermediate has a longer fluorescence lifetime. In fact, the photons emitted from RhB in the shorter wavelength region can be also detected by the channel of 525 nm < λem < 555 nm, so the decay curves in this shorter wavelength region demonstrate a two-component exponential feature, where the ratio of these two components depends on the ongoing photocatalysis process. Consequently, the fluorescence lifetime (τ1) of RhB in aqueous solution was determined to be 1.75 ns, while a 3.85 ns lifetime (τ2) of the deethylation intermediate was obtained by fitting the trace after 55 min irradiation32,33 (Fig. 2d). According to I(t) = A1exp(−t/τ1) + A2exp(−t/τ2), we obtained the ratio of the two lifetime components varying with the irradiation time, and summarized the results in Table 1. These parameters clearly demonstrate that the amount of reactant RhB decreases while the produced intermediate with a longer lifetime increases as the irradiation time goes on. Therefore, this shows that the fluorescence quenching behavior with visible light irradiation originates from the degradation of RhB and the formation of deethylation intermediate.
| Time (min) | 1 | 5 | 9 | 15 | 28 | 45 |
| (τ1 = 1.75 ns) | 80% | 55.4% | 41.1% | 33.1% | 28.7% | 19.5% |
| (τ2 = 3.85 ns) | 20% | 44.6% | 58.9% | 66.9% | 71.3% | 80.5% |
In general, the RhB photodegradation consists of competitive or concomitant processes, namely the deethylation by photosensitization degradation and the destruction of the conjugated structure by photocatalytic processes.34 For the latter case, the aromatic chromophore is attacked by the photogenerated active holes at the catalyst surface, leading to a direct decomposition of RhB without producing fluorescent intermediates. It has been proposed that electron transfer from the singlet excited state of the adsorbed dye to the CB of the semiconductor is the principal pathway to induce the initial step for producing deethylation from the RhB molecule.10 In this situation, the strong interaction between the adsorbed dye and semiconductor surface is an important criterion for efficient electron or charge transfer. In the RhB/Ag3PO4 system, we observed the deethylation of RhB from the above ensemble experiments, indicating an electron-injection process or a photosensitization process taking place from the singlet excited state of RhB to the CB of Ag3PO4. In the next section, we focus on the investigation to gain new insights into the photosensitized degradation and the generation dynamics of the intermediates, especially at a single-molecule level.
Photosensitization from ensemble to a single molecule
To observe the electron-injection process from RhB to Ag3PO4 nanoparticles, we performed steady-state and time-revolved fluorescence measurements without visible light irradiation. As shown in Fig. 3a, the original RhB solution shows a strong fluorescence at 580 nm with the excitation of 488 nm, while the fluorescence intensity is remarkably decreased with the addition of Ag3PO4 nanoparticles suspension into the RhB aqueous solution. This quenching effect is attributed to the electron transfer from the excited singlet state of RhB to the conduction band of the Ag3PO4 nanoparticles,35 constituting an electron-injection process before the degradation of RhB molecule. This electron transfer was further confirmed by fluorescence lifetime measurements in the atmosphere. Two typical fluorescence decay traces of RhB molecules with and without Ag3PO4 are shown in Fig. 3b. The decay curve of RhB molecules without Ag3PO4 demonstrates a longer fluorescence lifetime. In contrast, the fluorescence lifetime of RhB molecules on the Ag3PO4 surface is dramatically shortened, indicating the IET between RhB and Ag3PO4 nanoparticles and confirming the rationality of the above-mentioned photosensitizing degradation. Usually, unconstrained bi-exponential and even multi-exponential fittings have to be considered to extract the decay components of adsorbed molecules on a catalyst surface with different active site distributions.36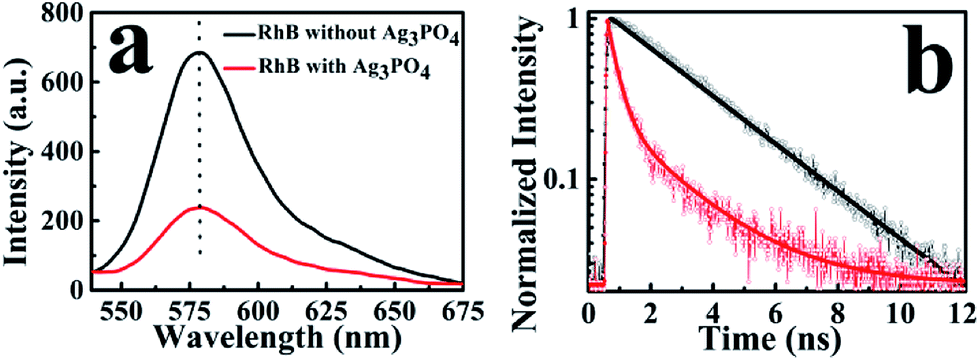 | ||
| Fig. 3 (a) Emission spectra of RhB molecules with and without Ag3PO4 nanoparticles in aqueous solution, (b) typical fluorescence decay curves of RhB molecules (black line) and RhB molecules on the Ag3PO4 surface without water (red line). | ||
To gain a new insight into this inhomogeneity, we performed single-molecule fluorescence lifetime measurements combined with confocal fluorescence microscopy to observe the behavior of a single RhB molecule on the Ag3PO4 surface. Fig. 4a and b show the single-molecule fluorescence lifetime images of RhB on bare cover glass and on Ag3PO4 nanoparticles-coated cover glass (20 × 20 μm2, 128 × 128 pixels matrix, 2 ms dwell time) under the same ambient conditions. The brightness and color of the light spot represent the fluorescence intensity and lifetime of a single molecule, respectively. Evidently, the fluorescence lifetime of some RhB molecules on Ag3PO4 particles-coated cover glass was shorter than that on bare cover glass, suggesting the occurrence of IET between these RhB molecules and the Ag3PO4 nanoparticles. It should be mentioned that a brighter molecule can have a shorter lifetime or a long lifetime, and there is no direct relation between the fluorescence intensity and fluorescence lifetime.37 Fig. 4c and d show the single-molecule fluorescence emission trajectories of RhB on the bare cover glass and on Ag3PO4 particles surface (binning time, 10 ms), respectively. Compared to a nearly constant emission on the glass surface, the fluorescence intensity of RhB on Ag3PO4 nanoparticle surface displays a strong fluctuation and blinking with the “dark” time ranging from sub-second to seconds, similar to the reports on PF/TiO2 (ref. 38) and CdTe/PI-CA.39 With photoexcitation, the excited state of an individual molecule either undergoes radiative emission to yield a photon that contributes to a bright state or undergoes a non-radiative electron transfer process that contributes to the dark state.21 In the fluorescence trajectory of RhB on Ag3PO4 particles surface, the “bright” state mostly reflects a low IET activity associated with the radiative relaxation from an excited state to ground state. Correspondingly, the “dark” state with the intensity close to background level indicates a high IET efficiency, demonstrating a quenching effect of the fluorescence emission.
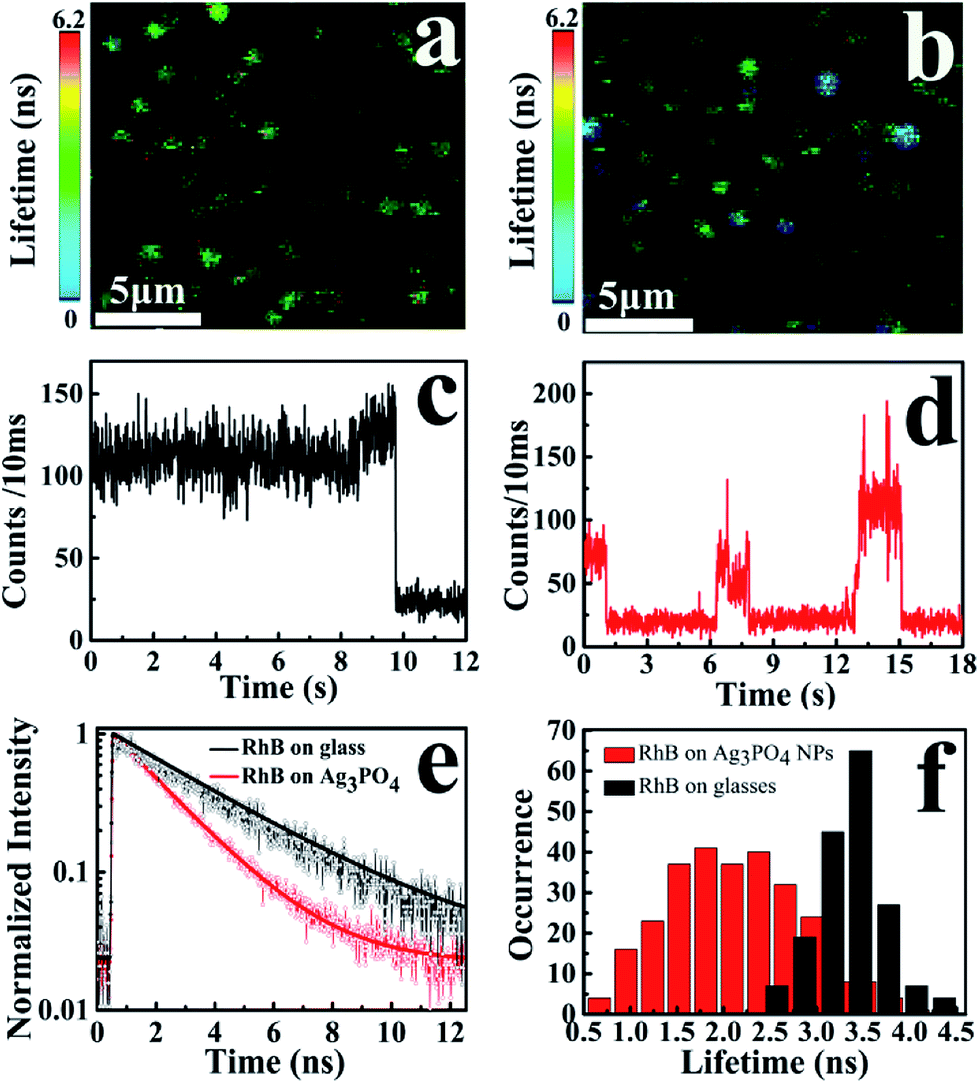 | ||
| Fig. 4 Single-molecule fluorescence images of RhB on (a) bare cover glass and (b) Ag3PO4 particles-coated cover glass (image size, 20 × 20 μm2) under the same fluorescence lifetime imaging conditions. (c, d) Typical fluorescence intensity trajectories of single-molecule RhB on (c) bare cover glass and (d) Ag3PO4 particles-coated cover glass, with the binning time of 10 ms. (e) Typical fluorescence lifetime decay curves of RhB on bare cover glass (black line) and on Ag3PO4 particles-coated cover (red line). (f) Single-molecule fluorescence lifetime distributions of RhB molecules on the bare cover glass (black) and on Ag3PO4 particle surface (red). | ||
The interaction and surrounding microenvironment of each molecule on the Ag3PO4 surface differs from time to time and from site to site, thus the lifetime distribution of RhB is supposed to be inhomogeneous as well. Fig. 4e displays two typical fluorescence decay traces of RhB molecules on bare cover glass and on Ag3PO4 nanoparticles-coated cover glass. Both traces can be fitted with a single exponential decay with the average fluorescence lifetime of 3.45 ns and 1.85 ns for RhB and RhB/Ag3PO4, confirming the photosensitization through electron transfer from excited RhB to Ag3PO4. Fig. 4f shows the statistical analysis of the fluorescence lifetime distribution of single RhB molecule on bare cover glass and on Ag3PO4 particles surface. We found a narrower distribution of fluorescence lifetime without Ag3PO4 nanoparticles, ranging from 2.4 ns to 4.4 ns with a 0.6 ns full width at half maximum (FWHM). On the contrary, a wider distribution of fluorescence lifetimes ranging from 0.6 to 4.2 ns with a 1.4 ns FWHM was observed for RhB on the Ag3PO4 nanoparticle surface. The average fluorescence lifetime (∼2.0 ns) for RhB on the Ag3PO4 particles surface was obviously smaller than that (∼3.5 ns) for RhB on the glass surface, indicating a dye-sensitizing feature and electron injection from excited RhB to Ag3PO4, which is consistent with the observations in the ensemble experiments.
The broad lifetime distribution reflects different IET reactivity dynamics and the complexity of interactions and electronic coupling between RhB and Ag3PO4 nanoparticles. To further confirm the heterogeneous photosensitizing in the RhB/Ag3PO4 system, we recorded hundreds of emission trajectories of RhB molecules on Ag3PO4 NPs-coated cover glass. Fig. 5 shows two typical single-molecule trajectories and their corresponding fluorescence decays. In the weak interaction case, the “bright” states of RhB emission dominate the intensity trajectory, even though a certain amount of “dark” states can be observed (Fig. 5a). These molecules emit relatively more photons and exhibit a longer fluorescence lifetime before photobleaching or being degraded (Fig. 5b and c), and thus can be attributed to the physically adsorbed RhB molecules on the Ag3PO4 surface. In contrast, the RhB molecule adsorbed on the active site of Ag3PO4 nanoparticles has a higher IET efficiency and a shorter fluorescence lifetime (Fig. 5d and f), and emits a small amount of photons before being photobleached or photodegraded (Fig. 5e). Correspondingly, the intensity trajectory demonstrates a drastic fluctuation, where the “dark” states and “bright” states appear alternatively for this strong interaction case. The ratio between the radiative and non-radiative rates is primarily modulated by the interaction mode or strength with the catalyst surface. Therefore, we can infer that the photosensitizing activity is strongly related to the adsorption sites on the particle surface and the energetic coupling between the excited singlet state of RhB and the conduction band of a Ag3PO4 nanoparticle. Similar single-molecule fluorescence behavior has been observed in other organic dye/semiconductor nanoparticle systems.20,21,38 On the other hand, we can also speculate that the subsequent photodegradation of RhB by Ag3PO4 nanoparticles is associated with the distribution of active sites on the Ag3PO4 surface.
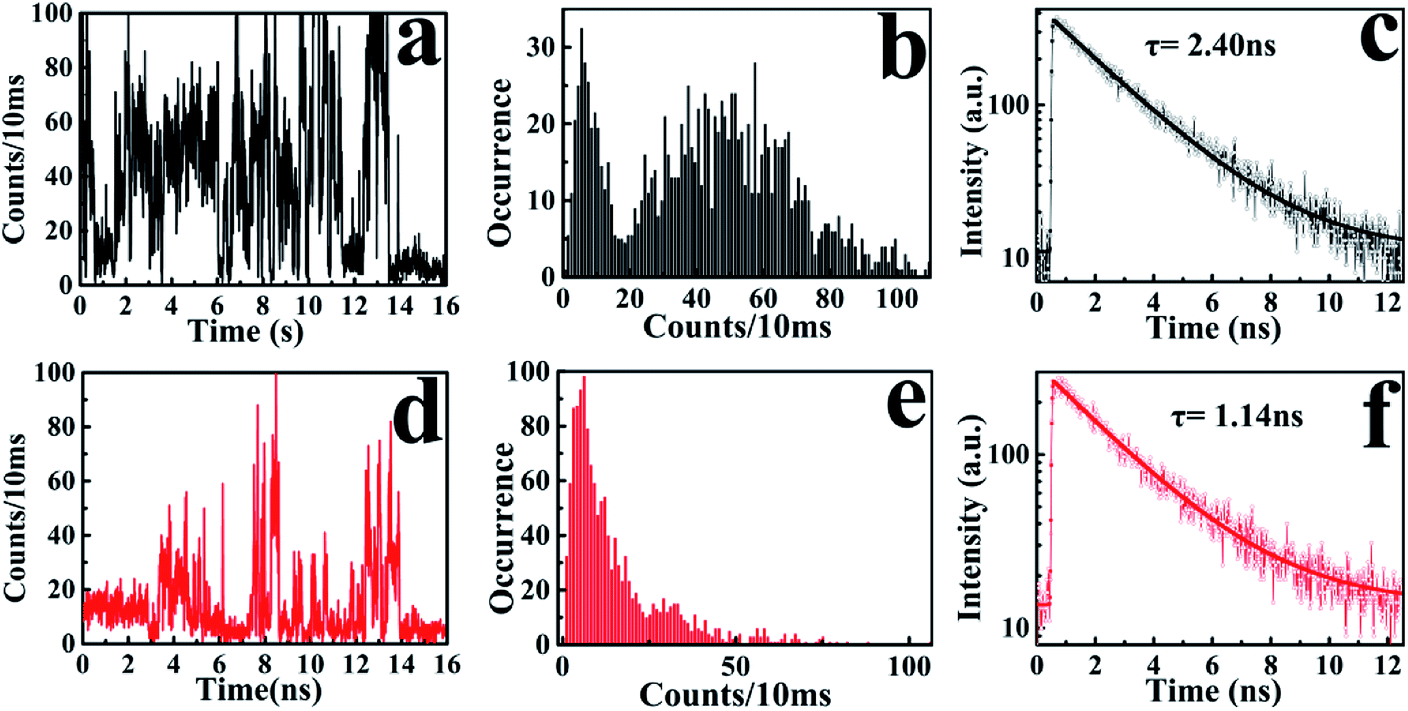 | ||
| Fig. 5 (a) Bright and (d) dark states of single-molecule fluorescence intensity trajectories; (b) bright and (e) dark states of emission intensity histograms to determine the threshold for the bright and dark states in the RhB/Ag3PO4 system, (c) bright and (f) dark states of the fluorescence lifetime decays. | ||
Single-molecule observation of the photodegradation intermediates
According to the above-mentioned results and discussion, the photogenerated deethylation intermediate by Ag3PO4 nanoparticles under visible irradiation shows a longer fluorescence lifetime compared with that of the RhB molecule in aqueous condition. Without visible irradiation, the RhB/Ag3PO4 on the cover glass (without water) demonstrates a shorter fluorescence lifetime due to the injection electrons from RhB into the CB of the Ag3PO4 semiconductor. Considering the co-existence of these features, we were inspired to observe the whole photodegrading dynamics of a single RhB molecule on a Ag3PO4 nanoparticle in aqueous conditions. Specifically, we expected to derive the precedence relation between the photosensitizing and photocatalytic degradation from the single-molecule emission fluctuation and lifetime sequencing with time, and to be able to correlate the degradation of RhB with the adsorbing sites on the Ag3PO4 surface. As the generation of deethylation intermediate involves the formation of O2˙− radicals,11 an aqueous atmosphere microenvironment for RhB/Ag3PO4 was created before performing the single-molecule measurements. It should be mentioned that the excitation laser at 488 nm worked as the visible irradiation source to generate electron–hole pairs in the Ag3PO4 nanoparticles, and also was used to excite the adsorbed RhB molecule and the intermediate deethylation of photocatalysis. Fig. 6a shows a representative single-molecule fluorescence time trajectory, which can be decomposed into two subintervals: the first half, section a1 and the second half, section a2. At the beginning, the emission intensity level was around 60 counts/10 ms (section a1), while a higher level of about 120 counts/10 ms was observed before the end of photodegradation (section a2). From the time correlation analysis of photons in section a1, a 1.06 ns fluorescence lifetime was obtained by mono-exponential fitting the decay curve (Fig. 6b), demonstrating an efficient photosensitization process. However, a longer fluorescence lifetime of 2.88 ns was acquired by analyzing the photons in section a2, clearly indicating the formation of the deethylation intermediate. Similarly, Fig. 6c shows another representative single-molecule fluorescence trajectory and the derived lifetime traces from sections c1 and c2. The intensity level was around 100 counts/10 ms and the lifetime was 2.08 ns in section c1, while the corresponding values were 150 counts/10 ms and 3.09 ns in section c2, respectively (Fig. 6d). Apparently, section a1 shows a shorter lifetime than section c1, reflecting the efficient IET dynamics at the active sites on the Ag3PO4 surface. For some single-molecule events, it was difficult to identify the discrete intensity-lifetime level in the emission trajectory, especially for the intensity fluctuation occurring on a time scale faster than the binning time, or where the intensity change is not large enough to be separated into discrete levels. Therefore, the most efficient photosensitization or IET process with a much shorter lifetime is hard to derive from the single trajectory of RhB/Ag3PO4 in the aqueous microenvironment.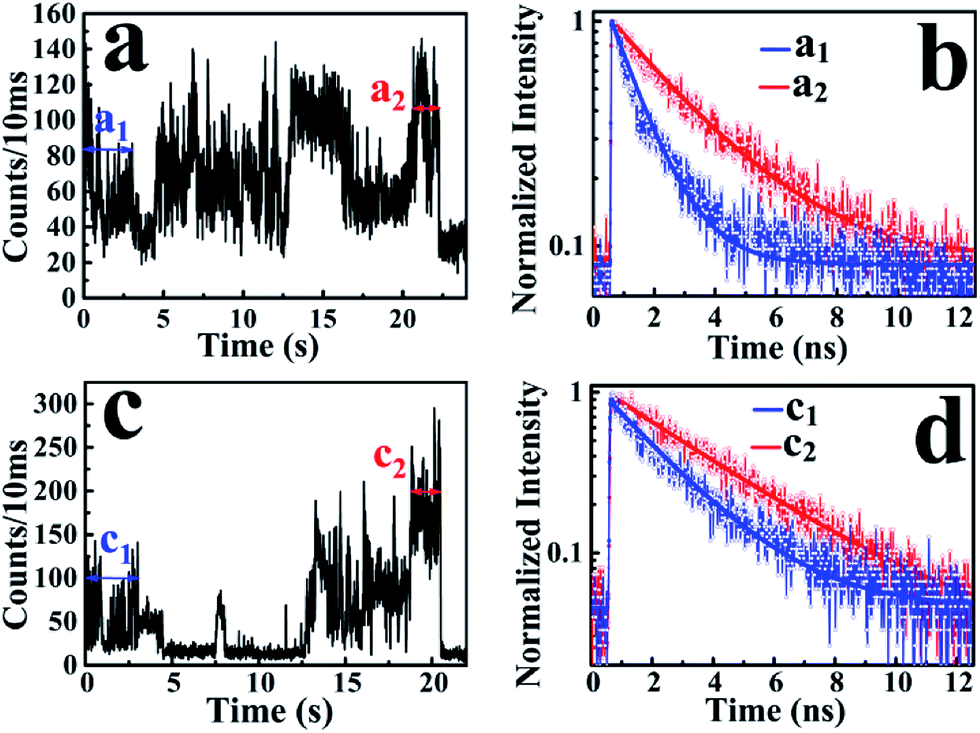 | ||
| Fig. 6 Subsections of two representative single-molecule fluorescence trajectories and lifetime curves of RhB on Ag3PO4. (a) and (b) RhB adsorbed on the active site, where the time trace starts with a lifetime of 1.06 ns and ends with 2.80 ns. (c) and (d) Physically adsorbed RhB, where the time trace starts with a lifetime of 2.08 ns and ends with 3.09 ns. | ||
To confirm the extensive existence of photosensitized degradation and intermediate generation, we performed lifetime distribution analysis by monitoring hundreds of single-molecule emission trajectories. As shown in Fig. 7, there are two obviously separated lifetime distributions. The collected photons in the first half section demonstrate a broad lifetime distribution, consistent with the observations in single-molecule photosensitization, indicating different electron transfer efficiencies and inhomogeneous interactions of RhB molecules with different adsorption sites on the Ag3PO4 surface. In the tail section of the emission trajectory, the photons demonstrate a longer lifetime, reflecting the generation of the photocatalytic intermediate deethylation. These results clearly indicate that when the Ag3PO4 photocatalyst and the adsorbed RhB molecule are excited by visible light at the same time, the photosensitization occurs prior to the photocatalytic degradation of RhB. Combining the results from the ensemble to a single molecule, we successfully observed the heterogeneous photosensitization and intermediate generation in the photocatalysis of the RhB/Ag3PO4 system.
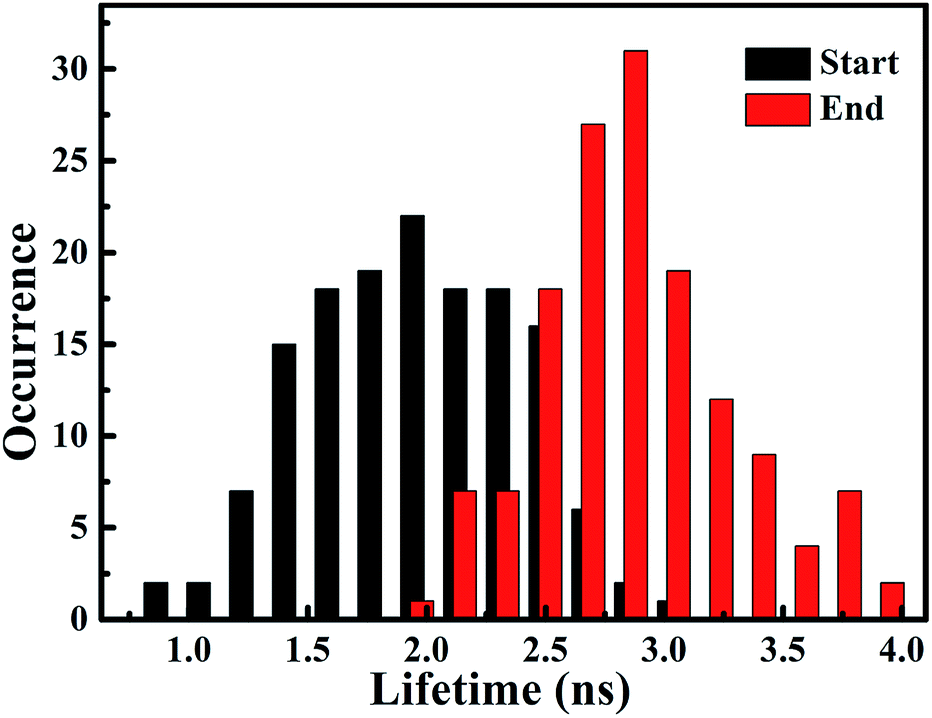 | ||
| Fig. 7 Distribution histogram of single molecule fluorescence lifetime derived from start section (black column) and end section (red column) of intensity trajectories. | ||
Mechanism interpretation
Both photosensitization and photocatalysis in the RhB/Ag3PO4 system can generate fluorescence quenching of adsorbed dye molecules, causing a simultaneous change in emission intensity and fluorescence lifetime but reflecting different interaction mechanisms. Under visible light irradiation, both Ag3PO4 and RhB molecules can be excited in an aqueous environment. The electron in the singlet excited state of RhB is injected into the conduction band of Ag3PO4, where the IET efficiency depends on the interaction strength or the adsorption sites on the Ag3PO4 surface. Therefore, the fluorescence lifetime of RhB is shortened compared to that without Ag3PO4, corresponding to the photosensitization process. This injected electron on Ag3PO4 from the excited RhB can be captured by O2 to form O2˙−, which can attack the excited RhB molecule to generate the intermediate product with a longer lifetime. Meanwhile, Ag3PO4 particle can also be excited to produce electron–hole pairs by visible light irradiation. In this case, the holes in the valence band of Ag3PO4 can directly attack the adsorbed RhB molecule via the destruction of its conjugated structure, resulting in the photobleaching or complete degradation of RhB. In other words, electron injection is the principal pathway as an initiation step to observe the generation of the photochemical deethylation intermediate,10 which has a blue-shifted emission spectrum and a longer fluorescence lifetime.For clarity, the two competitive photodegradation processes, the so-called N-deethylation and chromophore cleavage, occur simultaneously in this RhB/Ag3PO4 system under visible light irradiation and in aqueous environment, as illustrated in Fig. 8. For the chromophore cleavage case, the electron–hole pair in the Ag3PO4 nanoparticle can be generated under visible light irradiation, that is, the electron is excited into the conduction band and a hole is left in the valence band. As one of most efficient reactive species in catalysis, this hole (h+) can directly attack and oxidize the adsorbed RhB on the surface of the Ag3PO4 nanoparticle, leading to the photodegradation of RhB through the destruction of its conjugated structure.27,40 In this case, the product of the photocatalytic degradation was not observed in our single molecule fluorescence measurements, even though the ensemble fluorescence quenching partially came from this degradation contribution. In particular, this hole-oxidizing photodegradation plays a major role in non-aqueous environments, and it is not surprising to observe a shorter emission duration time in the single-molecule photosensitizing measurement. In principle, the photogenerated active species, such as OH˙ radicals, could also directly attack the central carbon of the RhB molecule to some extent. However, a previous study indicated that the OH˙ radical scavenger has no observable effect on the photodegradation rate of RhB in the Ag3PO4 system.41 Thus, the contribution from OH˙ radicals could be negligible in this work. Since the absorption cross-section of Ag3PO4 nanoparticles is much smaller than that of RhB molecules at the excitation wavelength, the photosensitization process prior to degradation plays an important role in this RhB/Ag3PO4 model system. When the adsorbed RhB molecule is excited from the ground state (S0) to the excited state (S1) by visible light, the electron will be injected into the CB of Ag3PO4 catalyst, with the inhomogeneous efficiency associated with the adsorption sites and interaction strength. This photosensitized interfacial electron transfer will generate a temporal RhB˙+ before recombining with an electron. In the meantime, the O2 molecule around the Ag3PO4 catalyst surface in the aqueous environment can be reduced by the transferred electron or photogenerated electron–hole pair in Ag3PO4 to form O2˙− radicals, which is a critical step in the photosensitized degradation of RhB molecules. Once the active O2˙− radical is created, the temporal RhB˙+ generated after electron injection into Ag3PO4 will be attacked to produce N-deethylation intermediates, rather than to attack the conjugated structure in the oxygen-rich aqueous atmosphere.11 These deethylation intermediates will be degraded completely by the surrounding radicals. Therefore, an important photodegradation pathway of RhB by Ag3PO4 nanoparticles in aqueous environment consists of a pre-photosensitization process and the intermediate generation of N-deethylation, which demonstrates a clear inhomogeneity and dynamical characteristics.
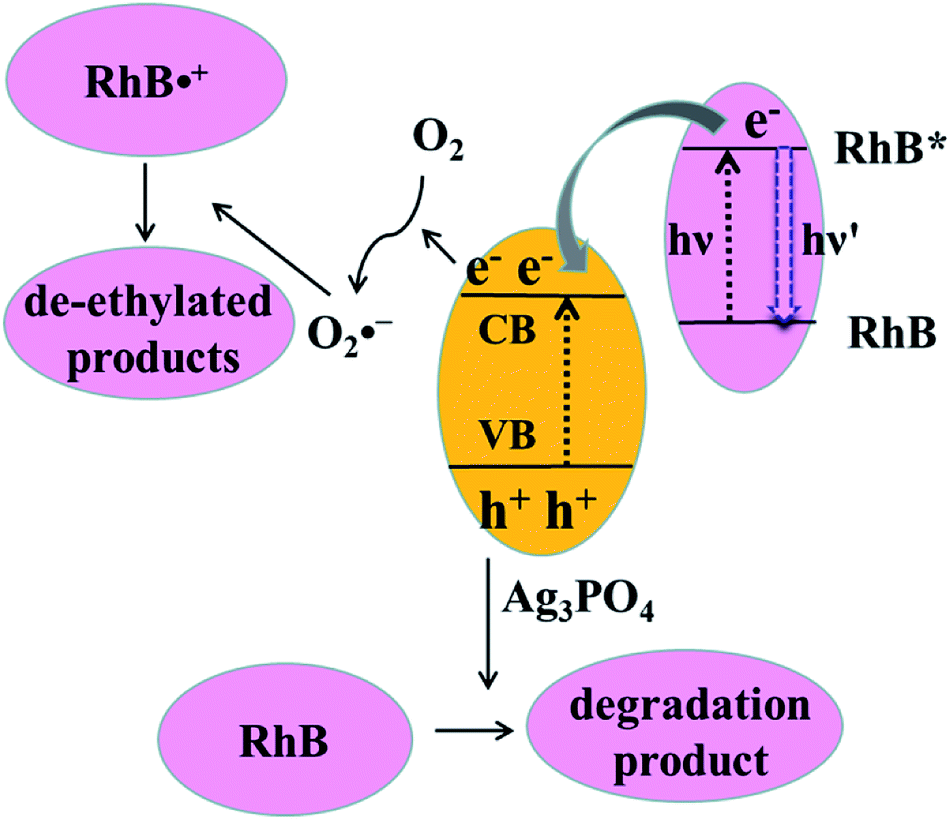 | ||
| Fig. 8 Photodegradation diagram of RhB molecules on the surface of the Ag3PO4 nanocatalyst. | ||
Conclusions
We systematically investigated the inhomogeneous interactions between the dye molecule RhB and semiconductor Ag3PO4 nanoparticle, as well as the intermediates generation in the photodegradation process, by using ensemble-averaged spectroscopy and single-molecule, single-particle FLIM spectroscopy. The as-prepared Ag3PO4 nanoparticles demonstrated good catalytic activity under visible light irradiation in aqueous environment. Besides the direct oxidization of RhB by photogenerated holes in Ag3PO4 nanoparticles, the degradation process via intermediate generation plays an important role in the photodegradation. The intermediate product deethylation has a blue-shifted absorption and fluorescence bands, and a longer fluorescence lifetime compared with that of RhB molecules in aqueous solution. The fluorescence quenching and lifetime shortening of a RhB molecule on the Ag3PO4 surface suggest photosensitization in the RhB/Ag3PO4 system prior to the photodegradation of the RhB molecule.The photosensitized photodegradation process of RhB on the Ag3PO4 nanoparticle surface was successfully characterized by analyzing the single-molecule fluorescence intensity fluctuation and lifetime distribution. The single-molecule fluorescence intensity fluctuations of RhB were closely associated with the photosensitization activities and IET efficiencies. The dominant dark states in the fluorescence intensity trajectory with a shorter lifetime demonstrate a higher charge injection process taking place at the active sites. The observed broad distribution of the single-molecules lifetimes reflects the heterogeneous interactions between RhB molecules and Ag3PO4 nanoparticles, correlated to the distribution of active sites on the Ag3PO4 nanoparticles. The shorter fluorescence lifetime derived from the starting subsection than that from the end subsection of a single-molecule intensity trajectory clearly reveals the photosensitized photodegradation of RhB and the intermediate generation in the aqueous environment. Moreover, this time dependency of the subsection fluorescence lifetime indicates that the electron injection from the adsorbates to catalyst occurs prior to radical attack in photosensitized degradation. These findings at a single-molecule level provide new insights into understanding the inhomogeneous photosensitization and photodegradation processes involved in solar energy conversion and photocatalysis.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was financially supported by the National Science Foundation Committee of China (Project No. U1604129, U1404619, U1504510 and 21173068) and Natural Science Foundation of Henan Province 162300410028.Notes and references
- J. Romao and G. Mul, ACS Catal., 2016, 6, 1254–1262 CrossRef CAS.
- Y. Lin, D. Li, J. Hu, G. Xiao, J. Wang, W. Li and X. Fu, J. Phys. Chem. C, 2012, 116, 5764–5772 CAS.
- M. T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, L. Servant, M. M. Mueller, H.-J. Kleebe, J. Ziegler and W. Jaegermann, Inorg. Chem., 2012, 51, 7764–7773 CrossRef CAS PubMed.
- A. Mukherji, B. Seger, G. Q. Lu and L. Wang, ACS Nano, 2011, 5, 3483–3492 CrossRef CAS PubMed.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed.
- J. Liu, Y. Zhao, L. Shi, S. Yuan, J. Fang, Z. Wang and M. Zhang, ACS Appl. Mater. Interfaces, 2011, 3, 1261–1268 CAS.
- S. Ardo, D. Achey, A. J. Morris, M. Abrahamsson and G. J. Meyer, J. Am. Chem. Soc., 2011, 133, 16572–16580 CrossRef CAS PubMed.
- S. Balasubramanian, P. Wang, R. D. Schaller, T. Rajh and E. A. Rozhkova, Nano Lett., 2013, 13, 3365–3371 CrossRef CAS PubMed.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- J. Zhuang, W. Dai, Q. Tian, Z. Li, L. Xie, J. Wang, P. Liu, X. Shi and D. Wang, Langmuir, 2010, 26, 9686–9694 CrossRef CAS PubMed.
- H. Fu, S. Zhang, T. Xu, Y. Zhu and J. Chen, Environ. Sci. Technol., 2008, 42, 2085–2091 CrossRef CAS PubMed.
- J. B. Asbury, E. Hao, Y. Q. Wang, H. N. Ghosh and T. Q. Lian, J. Phys. Chem. B, 2001, 105, 4545–4557 CrossRef CAS.
- D. A. Gaal and J. T. Hupp, J. Am. Chem. Soc., 2000, 122, 10956–10963 CrossRef CAS.
- V. Biju, M. Micic, D. H. Hu and H. P. Lu, J. Am. Chem. Soc., 2004, 126, 9374–9381 CrossRef CAS PubMed.
- M. B. J. Roeffaers, B. F. Sels, H. Uji-i, F. C. De Schryver, P. A. Jacobs, D. E. De Vos and J. Hofkens, Nature, 2006, 439, 572–575 CrossRef CAS PubMed.
- P. C. Sevinc, X. Wang, Y. Wang, D. Zhang, A. J. Meixner and H. P. Lu, Nano Lett., 2011, 11, 1490–1494 CrossRef CAS PubMed.
- Y. Park, W. Kim, D. Monllor-Satoca, T. Tachikawa, T. Majima and W. Choi, J. Phys. Chem. Lett., 2013, 4, 189–194 CrossRef CAS PubMed.
- J. Li, I. Kondov, H. Wang and M. Thoss, J. Phys. Chem. C, 2010, 114, 18481–18493 CAS.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197–7204 CrossRef CAS PubMed.
- Y. Wang, X. Wang, S. K. Ghosh and H. P. Lu, J. Am. Chem. Soc., 2009, 131, 1479–1487 CrossRef CAS PubMed.
- V. G. Rao, B. Dhital, Y. He and H. P. Lu, J. Phys. Chem. C, 2014, 118, 20209–20221 CAS.
- L. Damalakiene, V. Karabanovas, S. Bagdonas and R. Rotomskis, Int. J. Mol. Sci., 2016, 17, 473 CrossRef.
- L. Zhang, J. Lei, J. Liu, F. Ma and H. Ju, Biomaterials, 2015, 67, 323–334 CrossRef CAS PubMed.
- K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705 CrossRef PubMed.
- A. Orte, J. M. Alvarez-Pez and M. J. Ruedas-Rama, ACS Nano, 2013, 7, 6387–6395 CrossRef CAS PubMed.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo and Z. Li, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- M. Ge, Chin. J. Catal., 2014, 35, 1410–1417 CrossRef CAS.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- L. Wang, J. Shang, W. Hao, S. Jiang, S. Huang, T. Wang, Z. Sun, Y. Du, S. Dou, T. Xie, D. Wang and J. Wang, Sci. Rep., 2014, 4, 7384 CrossRef PubMed.
- K. Yu, S. G. Yang, H. He, C. Sun, C. G. Gu and Y. M. Ju, J. Phys. Chem. A, 2009, 113, 10024–10032 CrossRef CAS PubMed.
- C. C. Chen, W. Zhao, J. Y. Li and J. C. Zhao, Environ. Sci. Technol., 2002, 36, 3604–3611 CrossRef CAS PubMed.
- Z.-R. Tang, Y. Zhang, N. Zhang and Y.-J. Xu, Nanoscale, 2015, 7, 7030–7034 RSC.
- Z. Xiong, L. L. Zhang, J. Ma and X. S. Zhao, Chem. Commun., 2010, 46, 6099–6101 RSC.
- T. Wu, G. Liu, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845–5851 CrossRef CAS.
- A.-H. Liang, S.-M. Zhou and Z.-L. Jiang, Talanta, 2006, 70, 444–448 CrossRef CAS PubMed.
- S. Saraswat, A. Desireddy, D. Zheng, L. Guo, H. P. Lu, T. P. Bigioni and D. Isailovic, J. Phys. Chem. C, 2011, 115, 17587–17593 CAS.
- Y. He, V. G. Rao, J. Cao and H. P. Lu, J. Phys. Chem. Lett., 2016, 7, 2221–2227 CrossRef CAS PubMed.
- L. Guo, Y. Wang and H. P. Lu, J. Am. Chem. Soc., 2010, 132, 1999–2004 CrossRef CAS PubMed.
- S.-C. Cui, T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2008, 112, 19625–19634 CAS.
- W. Teng, X. Li, Q. Zhao, J. Zhao and D. Zhang, Appl. Catal., B, 2012, 125, 538–545 CrossRef CAS.
- M. Ge, N. Zhu, Y. Zhao, J. Li and L. Liu, Ind. Eng. Chem. Res., 2012, 51, 5167–5173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07163a |
| This journal is © The Royal Society of Chemistry 2017 |