
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThixotropic stiff hydrogels from a new class of oleoyl-D-glucamine-based low-molecular-weight gelators†
Yutaka Ohsedo *abc,
Masashi Oonod,
Kowichiro Saruhashid,
Hisayuki Watanabecd and
Nobuyoshi Miyamotoa
*abc,
Masashi Oonod,
Kowichiro Saruhashid,
Hisayuki Watanabecd and
Nobuyoshi Miyamotoa
aDepartment of Life, Environment and Materials Science, Fukuoka Institute of Technology, 3-30-1, Wajiro-Higashi, Higashi-ku, Fukuoka 811-0295, Japan. E-mail: josedo@bene.fit.ac.jp; Fax: +81 92-606-3977; Tel: +81 92-606-3977
bComprehensive Research Organization, Fukuoka Institute of Technology, 3-30-1, Wajiro-Higashi, Higashi-ku, Fukuoka 811-0295, Japan
cGlobal Innovation Center, Kyushu University, 6-1 Kasuga-koen Kasuga-city, Fukuoka 816-8580, Japan
dNissan Chemical Industries, Ltd., 2-10-1 Tsuboinishi, Funabashi, Chiba 274-8507, Japan
First published on 25th August 2017
Abstract
A series of new compounds comprising oleoyl, amino acid and D-glucamine moieties were synthesised using mild reaction conditions, and their hydrogelation ability was evaluated. The gels exhibited improved stiffness and thixotropic properties dependent on the chemical structure of the amino acid linker and the number of potential hydrogen bonding sites.
Introduction
Low-molecular-weight gelators (LMWGs) have received much attention owing to their ability to be tuned by chemical synthesis to form soft matter (i.e. molecular gels) with desired properties.1,2 In the last two decades, there have been numerous attempts to synthesise molecular gels comprising LMWGs and organic (organogel) or aqueous (hydrogel) media for soft matter applications in fields such as medical science and optoelectronics.3,4 Therefore, LMWGs that form stimuli-responsive molecular gels have been extensively studied.5 To improve the mechanical properties of gels, the relationship between thixotropic behaviour, i.e. mechanically reversible sol-to-gel transitions, and their network structure has been extensively studied.6 In particular, thixotropic behaviour is required for injectable and ointment-type gels for biomaterials in medical applications.3For the creation of molecular hydrogels, fibre micelle and hydrogel formation of N-alkylaldonamides such as n-octyl- and n-dodecyl-D-glucamides was extensively studied.7 These molecules, which can be obtained by simple amidation, are efficient hydrogelators. Several widely studied derivatives of N-alkylaldonamides exhibit unique nano- and micro-metre scale architectures such as tubular micelles.7–11 The original N-alkylaldonamides tend to crystallise in aqueous solution, unless a surfactant is added to the hydrogel system.9
Recently, we reported that the onset of thixotropic behaviour in molecular organogels can be induced simply by mixing two alkyl-chain substituted homologues of commercially available hydrazides,12a,b amides,12c,e,h ureas12d and hydrogelators12f,g such as N-alkyl-D-glucamides. Although mixing induced thixotropy is observed in other systems,13,14 newer gelators that form functional gels are needed to promote the study of molecular gels.
In this study, we evaluate the potential of a new class of D-glucamine derivatives for their hydrogelation abilities. Our design of the hydrogelator constitutes three structural motifs: a D-glucamine moiety (head), an amino acid (linker), and an oleoyl moiety (tail). The three moieties are linked by two amide bonds (OG-G, OG2-G, OG3-G, and OA-G, Scheme 1). We consider that the oleoyl group, which is more hydrophobic than a stearoyl group and the long alkyl chain present in surfactants, can facilitate gelation of water owing to its increased hydrophobicity relative to linear alkyl chains.15 Furthermore, the covalent amide bond linking the components can provide better chemical stability than the corresponding esters moieties present in other gelators. Finally, the hydrogen bonding ability of the molecules can be chemically tuned by introducing glycine(s) or its homologue as the linker between the oleoyl and D-glucamine groups.
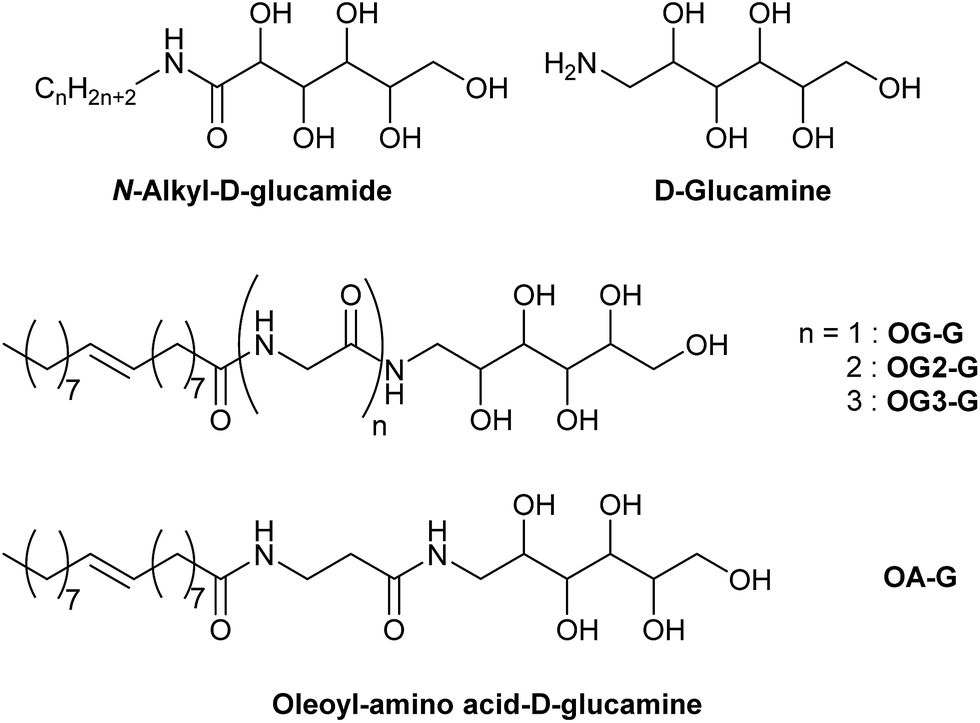 | ||
| Scheme 1 Chemical structures of oleoyl-D-gluconamide derivatives. | ||
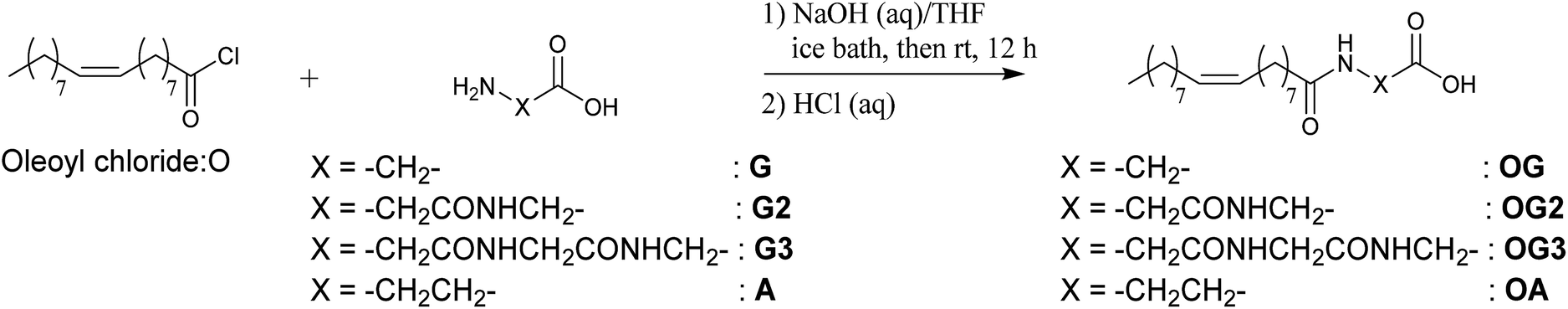 | ||
| Scheme 2 Synthesis of oleoyl amino acid derivatives. | ||
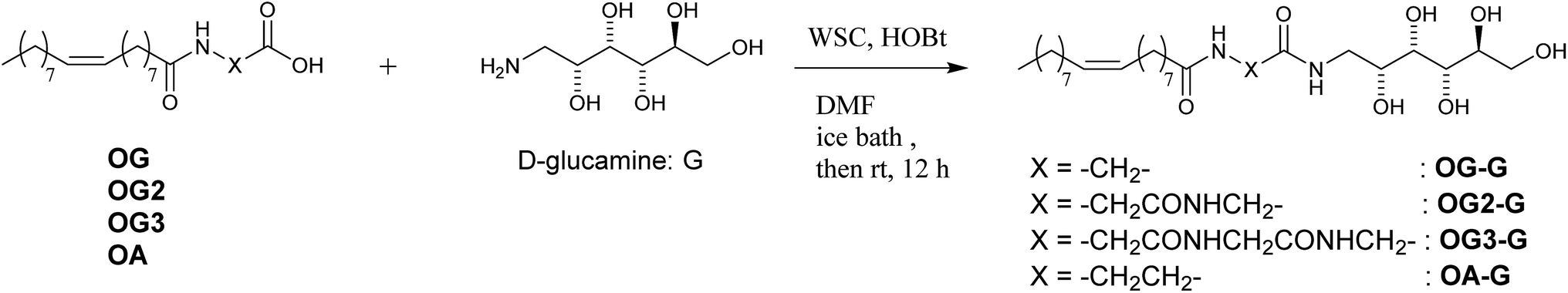 | ||
| Scheme 3 Coupling of oleoyl amino acids with D-glucamine. | ||
Experimental
Materials and methods
β-Alanine (99%), D-glucamine (95%), glycine (99%), glycylglycine (99%), glycylglycylglycine (98%) and 1-hydroxybenzotriazole monohydrate (HOBt, 97%) were purchased from Tokyo Chemical Industry Co., Ltd. and used without further purification. All organic solvents, buffer aqueous solutions, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (water soluble carbodiimide (WSC)), hydrochloric acid, oleoyl chloride (97%) and sodium hydroxide were purchased from Wako Pure Chemical Industries, Ltd. and used without further purification. Water was deionised with an Elix UV 3 Milli-Q integral water purification system (Nihon Millipore K.K.). Phosphate buffered saline (1× PBS (−)) was obtained by diluting 10× PBS (−) buffer (Wako Pure Chemical Industries, Ltd.) with water.1H-NMR and 13C-NMR spectra were acquired using an AVANCE 500 (500 MHz, Bruker BioSpin K.K.) spectrometer. Mass spectra were recorded on an LTQ Orbitrap spectrometer (ESI FTMS, Thermo Fisher Scientific K.K.) or a 3100 Mass detector equipped with e2695 Separations module and an s2489 UV/Visible detector (Nihon Waters K.K.). Elemental analysis was performed with a JM10 elemental analyzer (J-SCIENCE LAB CO., Ltd.).
The gelation tests were performed using the vial inversion method. A crystal of gelator was placed in a vial with a solvent at a set concentration (wt%) and capped. The vial was heated in a dry bath of 100 °C until the crystal of gelator was dissolved. Gelator solution was left for 1 h at room temperature, and gelation was checked with naked eyes by inverting the vial.
The thixotropic behaviour was evaluated using the vial inversion method. The hydrogel in vial was shaken and collapsed by a vortex genie (Scientific Industries, Inc). Then the obtained sol was left for the set time at room temperature, the recovery of the gel state from the sol state was checked with naked eyes by inverting the vial.
Thermal analysis was performed with an EXSTAR6000 differential scanning calorimeter DSC (Seiko Instruments Inc.) using an Ag-made closable sample pan. Tgel → sol and Tsol → gel of gels were determined as extrapolated onset temperatures from the DSC curves.
SEM images were recorded with a SU-8000 scanning electron microscope (Hitachi High-Technologies Corporation) at 1.0 kV; the SEM sample was vacuum dried and placed on a conductive tape on the SEM sample stage. Pt, as a conductive material, was used as a coating on the sample (Pt coating is 10 nm thick). The xerogel for SEM sample was obtained from freeze-drying of the hydrogel at the concentration of critical gelation concentration by using dry ice/acetone as a refrigerant.
Rheological measurements of frequency sweep were performed with an MCR-301 rheometer (Anton Paar Japan K.K.) with a parallel plate (8 mm diameter) at a gap of 0.50 mm and γ of 0.01% (measurement temperature: 25 °C). Rheological measurements of strain sweep were performed with an MCR-301 rheometer with a parallel plate (8 mm diameter) at a gap of 0.50 mm and constant angular frequency 1 rad s−1 (measurement temperature: 25 °C). For rheological measurements, the hydrogel sample was applied onto the parallel plate and sample stage (the overflow gel was swept). The hydrogel sample for rheological measurements was placed on a parallel plate and a sample stage (the overflow gel was swept). Step-shear measurement was carried out by applying normal strain (strain amplitude 0.01% and frequency 1 Hz) and large strain (shear rate 3000 s−1 for 0.1 s), repeatedly.
X-ray diffraction data were recorded on a D8 Discover X-ray diffractometer (Bruker AXS K.K.) with CuKα at 26 °C (the sample was filled in a quartz glass capillary tube of 1 mm diameter).
Infrared spectroscopy was performed with an FT/IR-620 (JASCO Corporation) using the ATR method (ZnSe prism).
Synthesis of OG-G, OG2-G, OG3-G and OA-G
Results and discussion
Molecules containing glycine (OG-G, OG2-G, and OG3-G) and β-alanine (OA-G) as the linkers are synthesised by condensation of N-glucamine with alkyloylamino acid in DMF under mild conditions. After DMF is removed in vacuo, recrystallisation affords the pure compound. Note that column chromatography is unnecessary for the purification. Elemental analysis and comparison with the predicted values further confirmed the purity. The alkyloylamino acids used in the above condensation are synthesised from the appropriate amino acid and the alkyloyl chloride in aqueous NaOH.16The hydrogelation ability of these new compounds are examined at various pH values and in the presence of salts to obtain the critical gelation concentration (CGC) (Fig. 1, S1, S2, ESI† and Table 1). These newly synthesised compounds, OG2-G and OG3-G, can gelate water in a pH range from 2 to 10, as well as in the presence of a high concentration of NaCl (1 M) and phosphate buffered saline (PBS), which is generally used in biological studies. In Table 1, OG-G showed hydrogelation, except with aq. NaCl and PBS, and OA-G formed a hydrogel in deionised water and carbonate buffer. The CGCs in buffer solutions become higher than those in deionised water. Furthermore, in higher salt concentrations (1 M NaCl), the CGCs become higher than those in buffer solutions; the presence of salt and acid or base may change the polarity of aqueous solutions, making hydrogelation difficult by disturbing intermolecular hydrogen bonding between gelators. Although hydrogelation in deionised water and buffered solutions occurs at moderate CGCs for all gelators, the gel is maintained for over 6 months without onset of crystallisation. N-alkyl-D-glucamines, compounds where the polar head is directly connected to the alkyl tail (i.e. in the absence of an amino acid linker), demonstrate poor gel stability in the absence of a surfactant. Therefore, the better gel stability observed in this study is attributed to the presence of the peptide moiety in these molecules. In addition, it is thought that the balance of hydrogen bonding (involving the peptide moieties) and hydrophobicity of gelators play an important role in hydrogelation. The better CGC observed in the case of OG2-G and OG3-G may be the result of a higher number of hydrogen bonding sites when compared OG-G and OA-G Moreover, the changes in the CGC with the introduction of a single methylene group (OA-G has one more methylene unit than OG-G) show that an appropriate chemical structure can enhance the gelation properties in this family of LMWGs (OA-G hydrogels showed inferior CGCs due to increased hydrophobicity in this system). Additionally, the CGC of OG-G hydrogels are superior to those of SG-G (strearoylamido-D-glucamine),15 likely due to the increased hydrophobic interactions of the oleoyl group.
 | ||
| Fig. 1 Evaluation of hydrogelation in deionised water: (a) OG-G, (b) OG2-G, (c) OG3-G, and (d) OA-G. | ||
| Solvent | OG-G | OG2-G | OG3-G | OA-G | SG-G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a pH = 1.68.b pH = 6.86.c pH = 10.11.d Turbid gel.e Clear gel.f Crystal gel.g Partial gel.h Opaque gel.i PBS: phosphate buffered saline. The CGC values are expressed as wt%. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Deionised water | 3.0d | 2.0d | 2.0d | 3.0e | 3.0e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxalate buffera | 4.0d | 3.0d | 3.0d | 5.0d | 5.0g | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phosphate bufferb | 4.0d | 3.0d | 3.0d | 5.0f | 5.0g | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Carbonate bufferc | 4.0d | 3.0d | 3.0d | 5.0d | 5.0g | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 M NaCl aq. | 5.0f | 4.0d | 4.0d | 5.0f | 5.0f | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PBS(−)i | 5.0f | 3.0d | 3.0h | 5.0f | 5.0f | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The rheometric results (Fig. 2) show the existence of a gel state, similar to that observed in polymer gels.17 Because of an increase in the applied stress, the relationship between storage modulus, G′, and the loss modulus, G′′, changed relative to G′ > G′′ systems.17 In the frequency sweep, a pseudo plateau with G′ > G′′ (gel state) is observed. On comparing the response of the hydrogels to applied shear stress, stable values with a modulus of a higher magnitude (104 to 105 Pa) was observed compared to the SG-G analogue with the same number of alkyl carbons (stearoyl group). The only exception was for the weak hydrogels formed with OA-G, likely caused by unbalanced hydrophobicity resulting in a weaker gel fibre or network. In the stiffness of hydrogels, OG-G and OG3-G showed similar stiffness; OG2-G formed the stiffest hydrogel, likely due to balanced hydrophobicity and hydrogen bonding yielding a stronger gel fibre or network. In recent studies of molecular gel systems, higher G′ values near 105 Pa were obtained by Yu (over 106 Pa for 1.5% (w/v) organogels),18 Steed (over 105 Pa for 1% (w/v) organogels)19 and Shinkai (over 104 Pa for 1 mM organogels).20 Importantly, our molecular hydrogel systems demonstrated a similar degree of high stiffness to those shown in the studies of Yu, Steed and Shinkai. This indicates that a stiff and stable hydrogel can be obtained by introducing an oleoyl group, instead of stearoyl chain, and glycine linker between the polar head and alkyl tail of N-alkyl-D-glucamines (note that the structure of glycine is more rigid than that of β-alanine).
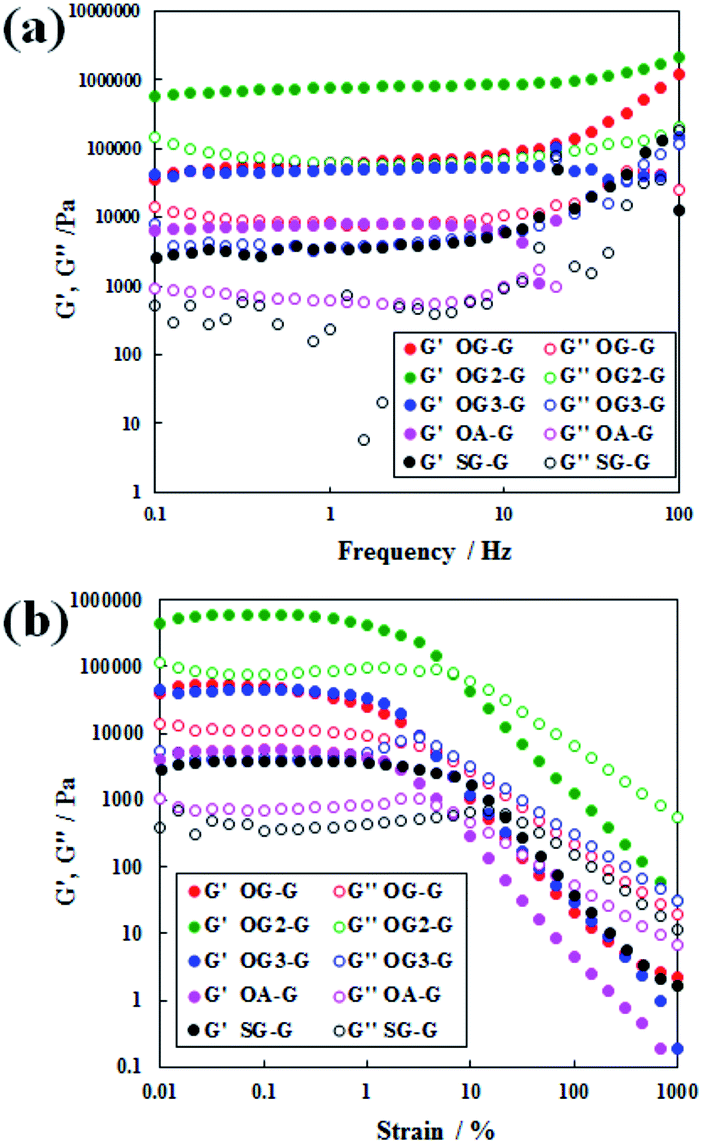 | ||
| Fig. 2 Dynamic rheological properties of hydrogels formed with OG-G (3.0 wt%), OG2-G (2.0 wt%), OG3-G (2.0 wt%), and OA-G (3.0 wt%): (a) frequency sweep and (b) strain sweep. | ||
Temperature-dependent data of sol-to-gel and gel-to-sol changes of the hydrogels measured by differential scanning calorimetry (DSC, Fig. S3 and Table S1, ESI†) show that similar ΔH values are involved in these reversible transitions. Sol-to-gel and gel-to-sol transition temperatures of hydrogels increase with the increase in the number of glycine repeat units, i.e. from OG-G to OG3-G, probably due to an increase in the interaction between glycine moieties, in addition to the increased molecular weight. Furthermore, the effect on thermal transitions by introduction of a methylene group in OA-G is similar to the addition of a second glycine unit, as in OG2-G. Considering the results of the gelation test, the transition temperatures may reflect the fibre properties, rather than the network properties of gel in this system (Table S1†).
Investigations into the mechanical properties of hydrogels show reveal thixotropic behaviour, even at the CGC. For example, re-gelation of a shaken OG3-G (2.0 wt%) hydrogel is clearly observed (Fig. 3). Evaluation of the qualitative thixotropic behaviour of hydrogels is performed with rheometry (Fig. 4, full version shown in Fig. S4, ESI†). The hydrogels show better recovery for G′ than G′′ even after being repeatedly subjected to a large deformation shear. Higher recoveries are observed for the stiffer and more stable hydrogels obtained from OG-G, OG2-G, and OG3-G compared to OA-G and SG-G15 hydrogels; an observation that may result from the introduction of the hydrogen bonding amino acid moieties and more hydrophobic oleoyl group that can facilitate the rebuilding of a damaged network. In addition, thixotropic behaviour was observed for OG3-G 3.0 wt% PBS hydrogel (Fig. S5, ESI†), whereas OG2-G 3.0 wt% PBS hydrogel did not show such behaviour. Further investigations on thixotropic properties of hydrogels, including other types of aqueous solutions, are underway. The results indicate that these hydrogels will be applicable in the healthcare and medical fields.
 | ||
| Fig. 3 Thixotropic behaviour of hydrogel formed with OG3-G (2.0 wt%). | ||
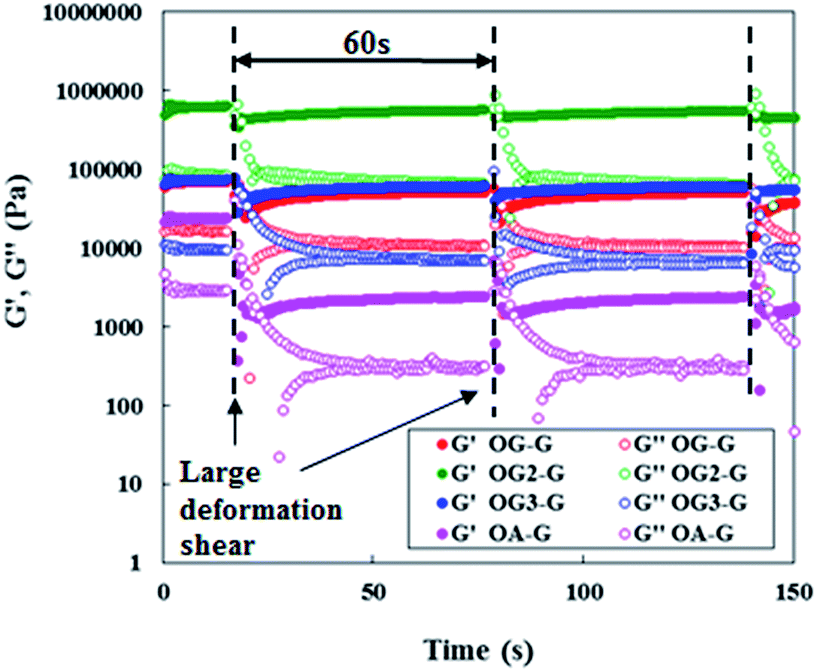 | ||
| Fig. 4 Periodical step-shear test results for OG-G (3.0 wt%), OG2-G (2.0 wt%), OG3-G (2.0 wt%), and OA-G (3.0 wt%) hydrogels.21 For full version see Fig. S4, ESI.† | ||
Scanning electron microscope (SEM) images of xerogels prepared by freeze-drying the corresponding hydrogels at the CGC show a network structure composed primarily of sub-micrometre thick fibres (Fig. 5). Considering that freeze-drying promotes the bundling of fibres, it is possible that the hydrogels consist of a finer network than what is observed in xerogels. Subsequently, these networks in the hydrogel can exhibit polymer-like functions, as demonstrated by the observed thixotropic behaviour.
 | ||
| Fig. 5 SEM images of xerogels prepared from the corresponding hydrogels: (a and b) OG-G (3.0 wt%), (c and d) OG2-G (2.0 wt%), (e and f) OG3-G (2.0 wt%), and (g and h) OA-G (3.0 wt%). The values in parenthesis denote the concentration of hydrogels before drying. | ||
To study the molecular packing structure in the fibre, X-ray diffraction (XRD) measurements were performed. The XRD results (Fig. 6a and b), when analysed in the context of the contour length of the gelators (Fig. 6c), suggests that the peak positions of hydrogels are different from those of corresponding xerogel samples and larger than the corresponding contour length of gelators (for example, 31.1 Å for OG-G). As previously shown, N-alkyl-D-aldonamide molecules with similar structures to the gelators in this study form fibre micelles and lamellar structures, based on the belayed structures of the molecules. By extension, in the gel state, it is possible that the molecules of the gelators can form bilayered or lamellar structures in the fibre (Fig. 6d). Also, in the xerogels, gelator molecules can form bilayered or lamellar structures. As shown in the xerogels of OG-G, OG2-G, and OG3-G, gelator molecules can also form tight bilayered or lamellar structures likely due to the absence of water molecules, and the XRD peaks might occur from a larger periodic structure corresponding to the length of two gelators.
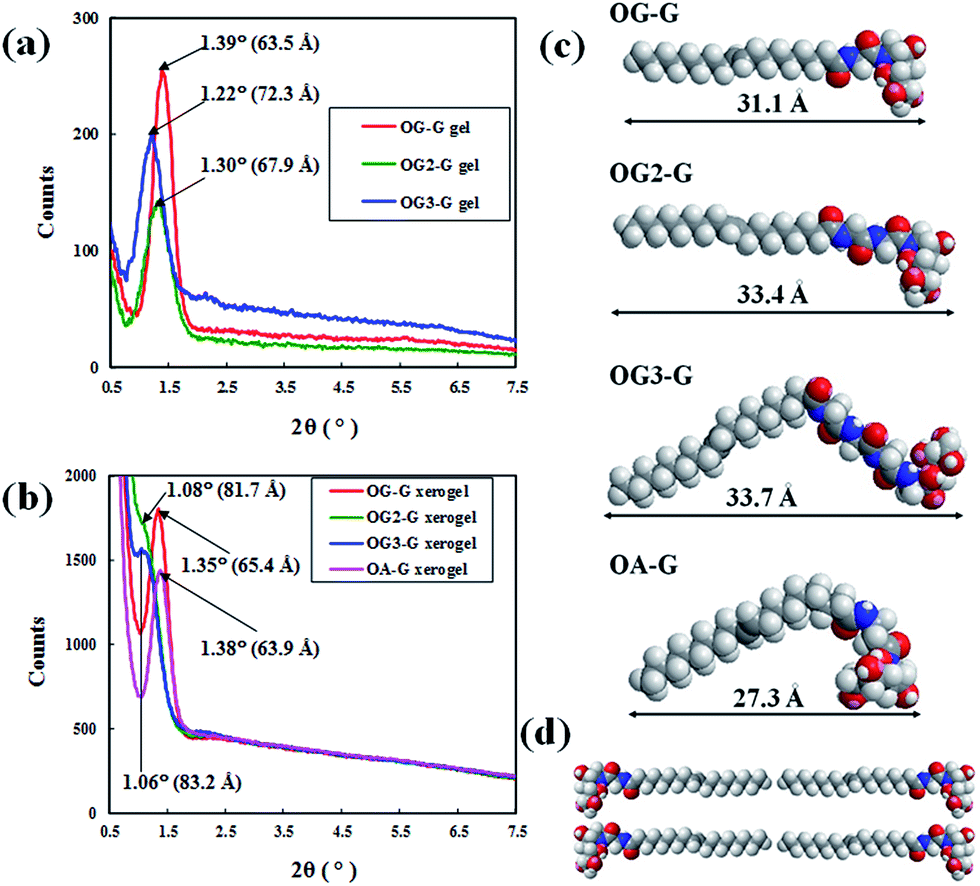 | ||
| Fig. 6 XRD data of (a) hydrogels at CGC and (b) xerogels formed with OG-G, OG2-G, OG3-G, and OA-G, (c) contour length and the length of OG-G, OG2-G, OG3-G and OA-G obtained from MM2 calculation in ChemDraw and (d) schematic illustration of possible lamella packing structure of OG-G. | ||
To investigate the interaction of gelators, IR measurements were carried out. As seen in the IR spectra of different states of gelator systems, hydrogen bonding of gelators may play an important role in the driving force of fibre formation (Fig. S6, ESI†).
Conclusions
In conclusion, we have created a new family of oleoyl- and D-glucamine-based hydrogelators by introducing glycine-based linkers connecting the two moieties. The molecules, prepared under mild reaction conditions, form hydrogels that demonstrate thixotropic properties and improved stiffness compared to the stearoyl analogue. These results suggest that N-alkylaldonamides can be chemically tuned to generate functional hydrogels by introducing an amino acid linker and an oleoyl group, rather than the stearoyl group.15 Their thixotropic nature suggests that these hydrogels will have extensive applications in the healthcare and medical fields for use as biomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Nissan Chemical Industries, Ltd. for financial and technical supports. A part of this work was supported by Research Center for Materials and Energy Devices of Fukuoka Institute of Technology (FIT-ME) (Strategic Research Foundation Grant-Aided Project for Private University from MEXT, Japan) as well as by Canon Foundation, KEKENHI (#24104005, #15K05610 and #15K05657) and Network Joint Research Center for Materials and Devices (#201507 and #20166009). A part of this work was also supported by the Nanotechnology Network Project (Kyushu-area Nanotechnology Network) of the MEXT, Japan. The SEM measurement was performed at the Center of Advanced Instrumental Analysis, Kyushu University. We are indebted to the Service Center of the Elementary Analysis of Organic Compounds, Kyushu University for elemental analysis. We would like to thank Enago (http://www.enago.jp) for the English language review.Notes and references
- For books: (a) Molecular Gels: Materials With Self-assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer Netherlands, Dordrecht, 2006 Search PubMed; (b) Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
- For recent reviews: (a) A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem. Asian J., 2011, 6, 266 CrossRef CAS PubMed; (b) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519 CrossRef CAS PubMed; (c) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed; (d) M. A. Rogers and R. G. Weiss, New J. Chem., 2015, 39, 785 RSC; (e) Y. Ohsedo, Polym. Adv. Technol., 2016, 27, 704 CrossRef CAS; (f) M. Yamanaka, Chem. Rec., 2016, 16, 768 CrossRef CAS PubMed; (g) H. Ihara, M. Takafuji and Y. Kuwahara, Polym. J., 2016, 48, 843 CrossRef CAS; (h) D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404 RSC; (i) R. Miao, J. Peng and Y. Fang, Langmuir, 2017 DOI:10.1021/acs.langmuir.6b04655.
- (a) J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642 CrossRef CAS PubMed; (b) Y. Ohsedo, Gels, 2016, 2, 13 CrossRef; (c) H. Wang, Z. Feng and B. Xu, Chem. Soc. Rev., 2017, 46, 2421 RSC.
- (a) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS PubMed; (b) S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766 CrossRef CAS PubMed.
- For recent examples of stimuli-responsive LMWG gels: (a) A. Dawn, T. Shiraki, H. Ichikawa, A. Takada, Y. Tahakashi, Y. Tsuchiya, L. T. N. Lien and S. Shinkai, J. Am. Chem. Soc., 2012, 134, 2161 CrossRef CAS PubMed; (b) Y. Ohsedo, M. Miyamoto, A. Tanaka and H. Watanabe, New J. Chem., 2013, 37, 2874 RSC; (c) H. Yu, Y. Liu, X. Chen, K. Liu and Y. Fang, Soft Matter, 2014, 10, 9159 RSC; (d) P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137(770), 1887 CrossRef CAS PubMed; (e) A. Maity, F. Ali, H. Agarwalla, B. Anothumakkool and A. Das, Chem. Commun., 2015, 51, 2130 RSC; (f) J. Schiller, J. V. Alegre-Requena, E. Marqups-Léóez, R. P. Herrera, J. Casanovas, C. Alemán and D. D. Díaz, Soft Matter, 2016, 12, 4361 RSC; (g) V. K. Pandey, M. K. Dixit, S. Manneville, C. Bucher and M. Dubey, J. Mater. Chem. A, 2017, 5, 6211 RSC.
- For recent examples of thixotropic LMWG gels: (a) N. Yan, Z.-Y. Xu, K. K. Diehn, S. R. Raghavan, Y. Fang and R. G. Weiss, Langmuir, 2013, 29, 793 CrossRef CAS PubMed; (b) A. Baral, S. Roy, A. Dehsorkhi, I. W. Hamley, S. Mohapatra, S. Ghosh and A. Banerjee, Langmuir, 2014, 30, 929 CrossRef CAS PubMed; (c) M. Suzuki, Y. Hayakawa and K. Hanabusa, Gels, 2015, 1, 81 CrossRef; (d) V. Ajay Mallia and R. G. Weiss, Soft Matter, 2016, 12, 3665 RSC; (e) B. K. Srivastava and M. K. Manheri, Chem. Commun., 2017, 53, 4485 RSC; (f) N. Zanna, S. Focaroli, A. Merlettini, L. Gentilucci, G. Teti, M. Falconi and C. Tomasini, ACS Omega, 2017, 2, 2374 CrossRef CAS.
- (a) J. Fuhrhop, P. Schnieder, J. Rosenberg and E. Boekema, J. Am. Chem. Soc., 1987, 109, 3387 CrossRef CAS; (b) J. Fuhrhop, P. Schnieder, E. Boekema and W. Helfrich, J. Am. Chem. Soc., 1988, 110, 2861 CrossRef CAS; (c) J. Fuhrhop and C. Boettcher, J. Am. Chem. Soc., 1990, 112, 1768 CrossRef CAS; (d) J. Fuhrhop, S. Svenson, C. Boettcher, E. Rössler and H. Vieth, J. Am. Chem. Soc., 1990, 112, 4307 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS PubMed.
- L. E. Buerkle, R. Galleguillos and S. J. Rowan, Soft Matter, 2011, 7, 6984 RSC.
- S. Sun and P. Wu, Soft Matter, 2011, 7, 6451 RSC.
- C. J. Capicciotti, M. Leclère, F. A. Perras, D. L. Bryce, H. Paulin, J. Harden, Y. Liu and R. N. Ben, Chem. Sci., 2012, 3, 1408 RSC.
- (a) Y. Ohsedo, M. Miyamoto, H. Watanabe, M. Oono and A. Tanaka, Bull. Chem. Soc. Jpn., 2013, 86, 671 CrossRef CAS; (b) Y. Ohsedo, H. Watanabe, M. Oono and A. Tanaka, Chem. Lett., 2013, 42, 363 CrossRef CAS; (c) Y. Ohsedo, H. Watanabe, M. Oono and A. Tanaka, RSC Adv., 2013, 3, 5803 RSC; (d) Y. Ohsedo, M. Oono, A. Tanaka and H. Watanabe, New J. Chem., 2013, 37, 2250 RSC; (e) Y. Ohsedo, M. Taniguchi, M. Oono, K. Saruhashi and H. Watanabe, RSC Adv., 2014, 4, 35484 RSC; (f) Y. Ohsedo, M. Oono, K. Saruhashi and H. Watanabe, RSC Adv., 2014, 4, 43560 RSC; (g) Y. Ohsedo, M. Oono, K. Saruhashi, H. Watanabe and N. Miyamoto, RSC Adv., 2014, 4, 44837 RSC; (h) Y. Ohsedo, M. Taniguchi, M. Oono, K. Saruhashi and H. Watanabe, New J. Chem., 2015, 39, 6482 RSC.
- For reviews of multi-component LMWG gels: (a) A. R. Hirst and D. K. Smith, Chem.–Eur. J., 2005, 11, 5496 CrossRef CAS PubMed; (b) C. A. Dreiss, Soft Matter, 2007, 3, 956 RSC; (c) L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089 RSC; (d) D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279 RSC; (e) J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170 RSC.
- For recent examples of multi-component LMWG gels; (a) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116 CrossRef CAS PubMed; (b) E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Commun., 2015, 7, 848 CAS; (c) E. Ressouche, S. Pensec, B. Isare, G. Ducouret and L. Bouteiller, ACS Macro Lett., 2016, 5, 244 CrossRef CAS; (d) S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743 CrossRef CAS PubMed; (e) K. Hanabusa, T. Ueda, S. Takata and M. Suzuki, Chem.–Eur. J., 2016, 22, 16939 CrossRef CAS PubMed; (f) E. R. Draper, M. Wallace, R. Schweins, R. J. Poole and D. J. Adams, Langmuir, 2017, 33, 2387 CrossRef CAS PubMed.
- For N-alkylamido-D-glucamine-based hydrogelators: Y. Ohsedo, M. Oono, K. Saruhashi and H. Watanabe, RSC Adv., 2014, 4, 48554 RSC.
- Synthesis of alkyloylamino acid: Y. M. Osornio, P. Uebelhart, S. Bosshard, F. Konrad, J. S. Siegel and E. M. Landau, J. Org. Chem., 2012, 77, 10583 CrossRef CAS PubMed.
- G. M. Kavanagh and S. B. Ross-Murphy, Prog. Polym. Sci., 1998, 23, 533 CrossRef CAS.
- Z. Xu, J. Peng, N. Yan, H. Yu, S. Zhang, K. Liu and Y. Fang, Soft Matter, 2013, 9, 1091 RSC.
- J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem.–Eur. J., 2014, 20, 279 CrossRef CAS PubMed.
- M. Shirakawa, N. Fujita, A. Takada and S. Shinkai, Chem. Lett., 2014, 43, 1330 CrossRef CAS.
- In the measurements, there had been observed the oscillations of G′ value in recovery process, so they seemed like to have two curves of G′ in Fig. 4.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07244a |
| This journal is © The Royal Society of Chemistry 2017 |