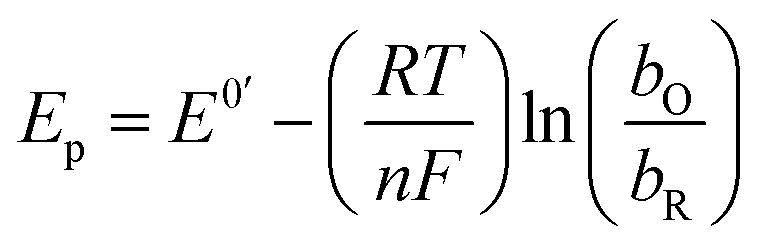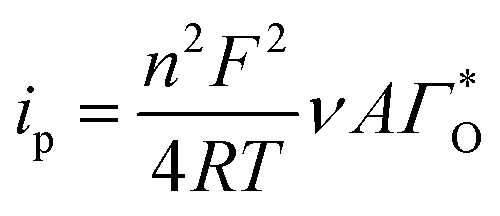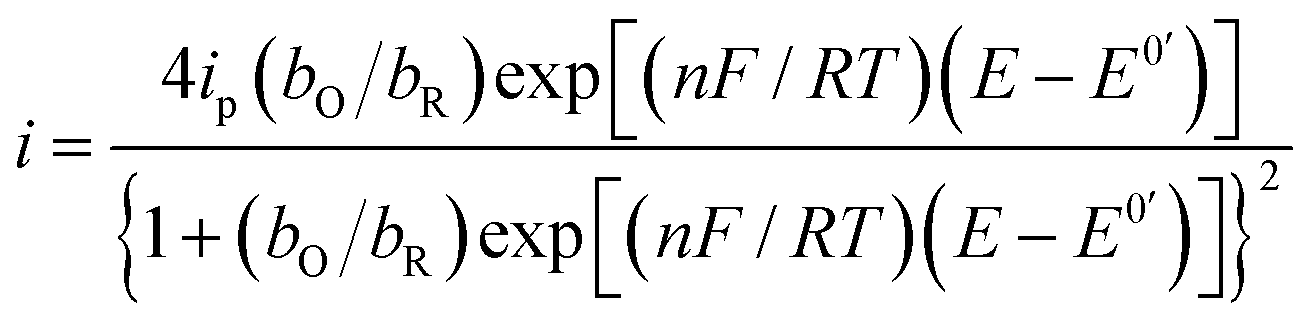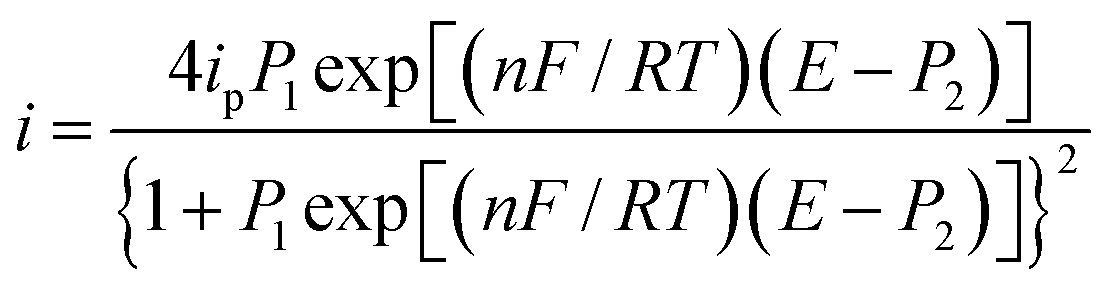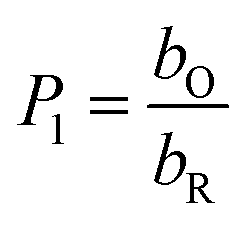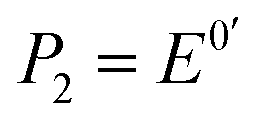DOI:
10.1039/C7RA07269D
(Paper)
RSC Adv., 2017,
7, 40401-40410
Taking advantage of CTAB micelles for the simultaneous electrochemical quantification of diclofenac and acetaminophen in aqueous media†
Received
1st July 2017
, Accepted 10th August 2017
First published on 18th August 2017
Abstract
From spectrophotometric and electrochemical techniques, it was shown that cetyltrimethylammonium bromide, CTAB, hemimicelles, formed on the surfaces of a carbon paste electrode (CPE), selectively adsorbed diclofenac, DCF, molecules from a neutral aqueous solution of DCF and acetaminophen, ACT. This CTAB–DCF interaction is so important that it modified the DCF electrochemical oxidation mechanism from a mass-transferred controlled one (in the absence of CTAB) to an adsorption controlled mechanism which allows the simultaneous quantification of both drugs. This novel methodology was used for the individual and simultaneous quantification of DCF and ACT in both aqueous media and synthetic urine. Moreover, it is shown that the analytical method proposed in this work is similar or even better than other more sophisticated and expensive techniques available.
1 Introduction
Anionic, cationic or neutral surfactant molecules have the ability to spontaneously form molecular aggregates termed micelles when they are dissolved in water, depending on the ions present and their concentration in solution.1 This way, the surfactants have been used to increase the solubility, improve the bioavailability or to prolong the release of various drugs, the stability of analytes associated with exposure under ambient light and normal laboratory atmospheric conditions, or of different types of organic molecules.2–5 Surfactants have also been used as masking agents for the simultaneous determination of neurotransmitters6 and to promote the percutaneous absorption of some analgesics.7 Two of the most important non-steroidal anti-inflammatory drugs (NSAIDS) are diclofenac (DCF), (2-[(2,6-dichlorophenyl)amino]phenyl acetate), and acetaminophen (ACT) commonly known as paracetamol (N-acetyl-p-amino-phenol), due to their analgesic, antipyretic and anti-inflammatory properties. Sophisticated analytical methods have been developed for their individual quantification, such as those involving HPLC techniques with UV detector,8 diffuse reflectance photometry,9 spectrofluorimetry,10 HPLC with electrochemical detector,11 aptasensors,12 membrane sensors with rhodamine B,13,14 molecularly-imprinted solid-phase extraction and adsorptive differential pulse voltammetry15 and modifying an edge-plane pyrolytic graphite electrode with single-wall carbon nanotubes.16
The mixture of DCF and ACT has been shown to decrease morphine consumption during heart surgeries and to reduce postoperative nausea and vomiting17 and to display greater efficiency than separate drugs, to relieve pain in cesarean patients.18 On the other hand, these drugs have been classified as emerging contaminants in both wastewater and drinking water,19,20 making imminent the development of affordable, simpler, analytical methods for their simultaneous determination.
In this work, a simple methodology based on the electrochemical oxidation of the DCF and ACT drugs will be shown for the first time, for individual and/or simultaneous quantification (as in mixtures) in aqueous solution, using a modified carbon paste electrode, CPE, with hemimicelles of the cationic surfactant cetyltrimethylammonium bromide (CTAB).
2 Experimental
All reactants were analytical grade; the drugs diclofenac and acetaminophen (Sigma) were purchased under the form of sodium salt and acid respectively, phosphate salts and sodium hydroxide (J. T. Baker) and CTAB (Aldrich). All solutions were prepared with deionized water from a Milli-Q (Millipore) instrument with 18.0 MΩ cm resistivity. When DCF and ACT were mixed in real samples, they were made from Tempra® I.V., solution 500 mg of acetaminophen 10 mg mL−1 (ACT) and AMSA® injectable 75 mg/3 mL (DCF). The synthetic urine was prepared according to Deroco et al.21 Thus, 0.73 g of NaCl, 0.40 g of KCl, 0.28 g of CaCl2·2H2O, 0.56 g of Na2SO4, 0.35 g of KH2PO4, 0.25 g of NH4Cl, and 6.25 g of urea were placed in a 250 mL volumetric flask and the volume was completed with water. The samples thus prepared were used immediately after. Also, these samples were added with 33 μM ACT (from Tempra®), 22.7 μM DCF (from AMSA®). The potentiometer was a pH/Ion Analyzer (HACH) coupled to a glass electrode (HACH series 5010T) to carry out pH measurements in the 0–14 range. Throughout the experiments, the drug solution in the cell was maintained under a nitrogen atmosphere, precluded from light at 25 °C.
A Perkin Elmer lambda 20 spectrophotometer was used to obtain the UV-Vis spectra with 1 cm optical path length quartz cells.
A potentiostat Epsilon BASi was used to perform electrochemical studies, aided by a conventional three-electrode cell comprising a bare CPE (displaying a geometric area of 0.071 cm2) as the working electrode, a Pt wire auxiliary electrode (BASi MW-1032) and Ag/AgCl (BASi MF-2021) as the reference electrode, to which all potentials in this work are quoted. The CPE was prepared by mixing graphite powder (Johnson Matthey 1 mm, 99.9%) and mineral oil (Nujol) from Sigma-Aldrich in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w ratio; the resulting mix was placed in a polyethylene tube (10 cm long and 3 mm in diameter) containing a piston; the tip of the tube was then placed on a flat surface and the piston was pressed to achieve the elimination of trapped air. Electric contact was attained soldering a Pt wire to an external Cu connector; further details can be found in Ramírez et al..22,23
:1 w/w ratio; the resulting mix was placed in a polyethylene tube (10 cm long and 3 mm in diameter) containing a piston; the tip of the tube was then placed on a flat surface and the piston was pressed to achieve the elimination of trapped air. Electric contact was attained soldering a Pt wire to an external Cu connector; further details can be found in Ramírez et al..22,23
3 Results and discussion
Fig. 1 shows a linear predominance zones diagram constructed following the methodology proposed by Rojas-Hernández et al.,24–27 for DCF, Fig. 1a, and for ACT, Fig. 1b, using the respective pKa values.27–29 For DCF it is observed that at pH values less than 4.15 the neutral species predominates and at higher pHs the anionic species predominates, whereas for ACT at pH less than 9.50, the neutral species predominates and for higher pH the anionic species. From these diagrams, it is clear that at pH 7 the predominant DCF species is anionic while for ACT it will be neutral. Since CTAB is a cationic surfactant, see Fig. 1c, in aqueous solution will be as ionic CTA+ molecules when [CTA+] is less than the respective critical micelle concentration, CMC, though micelles (CTA+)n will form when [CTA+] > CMC.
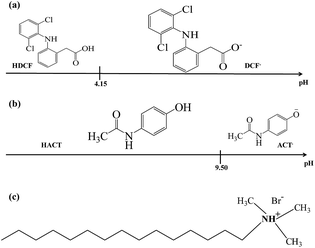 |
| | Fig. 1 Linear predominance zones diagram: (a) DCF and (b) ACT constructed following the methodology proposed Rojas-Hernández et al.24–27 (c) Cetyltrimethylammonium bromide's molecule. | |
3.1 Spectrophotometric study of interaction of DCF− and HACT with CTA+
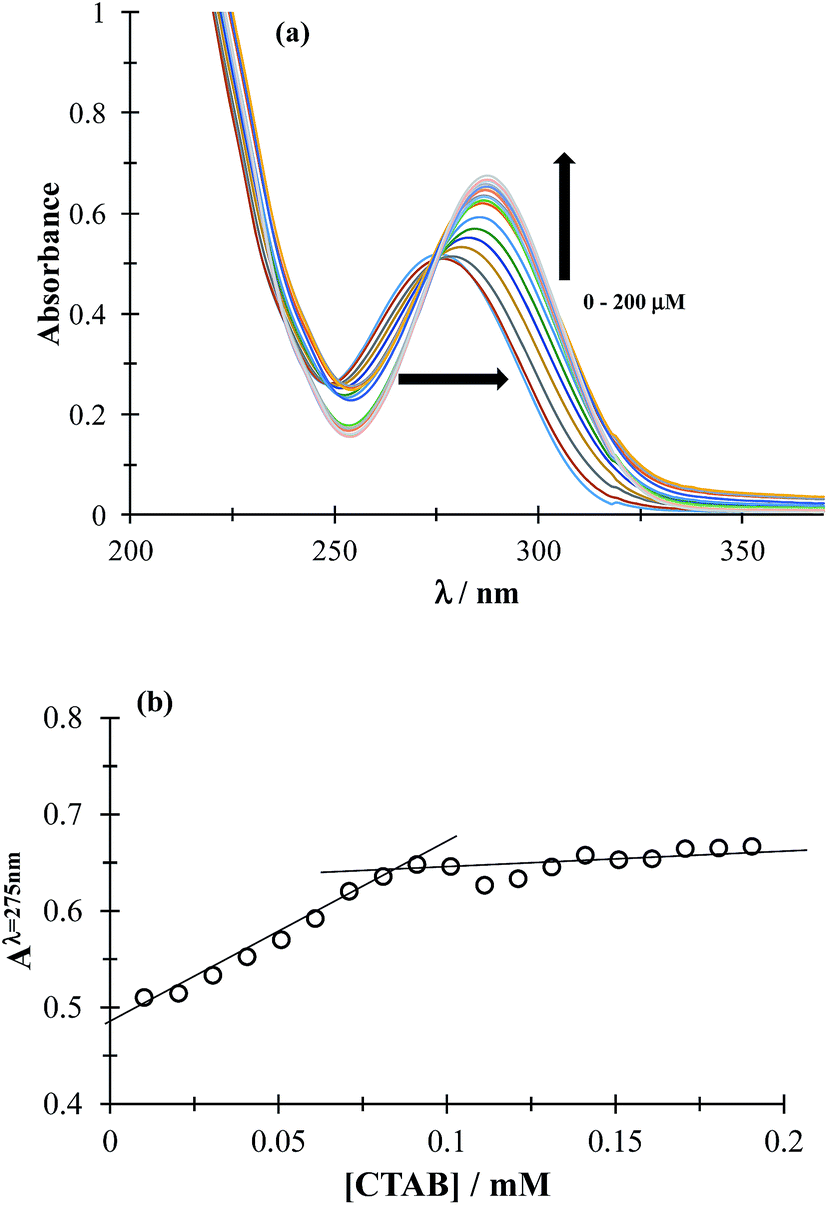
3.1.1 CMC determination. The CMC determines the concentration (c) region in which the surfactant molecules present in the solution tend to form micelles (c > CMC) or where they are mainly present as the monomer (c < CMC). To determine CMC, UV-Vis absorption spectra of DCF were obtained by varying the concentration of CTAB, see Fig. 2a. It can be observed that increasing CTAB concentration, the spectra displayed both bathochromic and hyperchromic effects. Fig. 2b shows the variation of absorbance recorded at 275 nm, as a function of CTAB concentration, a slope change can be observed at 0.09 mM CTAB concentration, corresponding to the CMC. It is important to stress that the CTAB CMC value may change with different factors, including the electrolyte nature and concentration, for instance Fuguet et al.30 found (0.91 ± 0.05) mM and (0.33 ± 0.09) mM in pure water and 5 mM phosphate buffer (pH 7.0) at 25 °C, respectively while Sánchez-Rivera et al.31 found a CMC value of 0.6 mM in 0.1 M NaCl aqueous solution at 20 °C. Furthermore, we have also estimated the CMC value in the presence of both drugs and it was practically the same as that found when there was only DCF, which means that when CTAB concentration is higher than 0.09 it is in the form of micelles even in the presence of ACT.
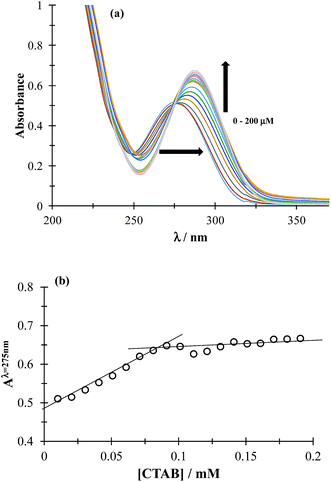 |
| | Fig. 2 (a) Family of UV-Vis spectra obtained in the 0.05 mM DCF system, in phosphate buffer at pH 7 0.1 M for different CTAB concentrations shown in the figure. (b) Absorbance variation recorded at 275 nm, as a function of CTAB concentration. | |
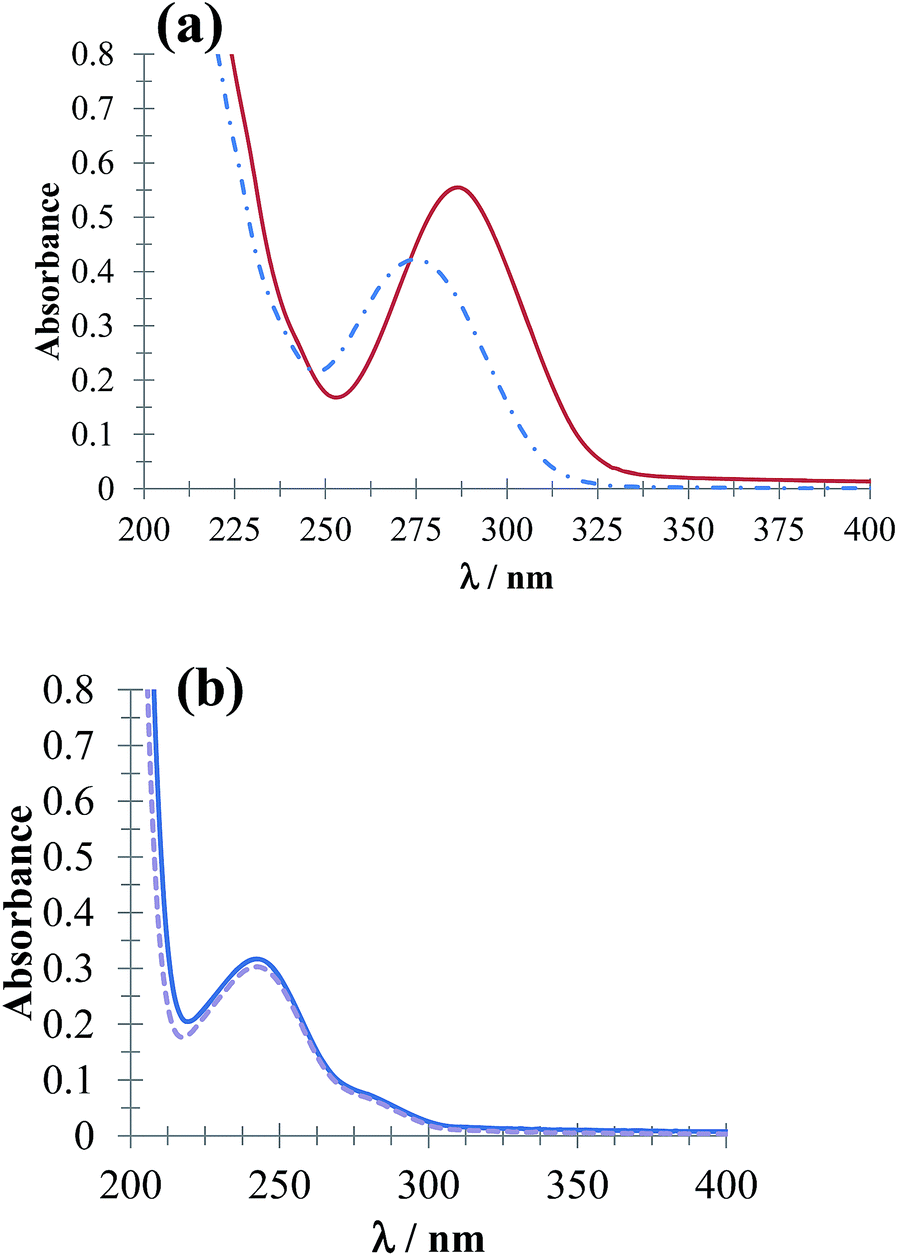
Fig. 3 shows UV-Vis absorption spectra of DCF− molecules (Fig. 3a) and HACT (Fig. 3b) obtained in the presence and absence of CTAB for a c > CMC. The spectrophotometric behavior of DCF− evidenced micelle formation (CTA+)n, whereas for the HACT, these micelles did not modify the behavior of their absorption spectrum.
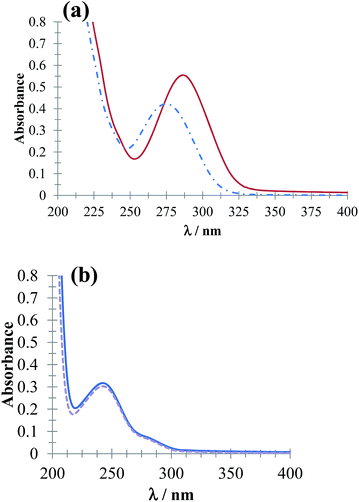 |
| | Fig. 3 UV-Vis absorption spectra obtained in the systems (a) 64 μM DCF and (b) 64 μM ACT in phosphate buffer solution at pH 7.01 in the presence of 0.1 mM CTAB (solid line) and absence (broken line). | |
3.2 Electrochemical study of the DCF−–(CTA+)n interaction
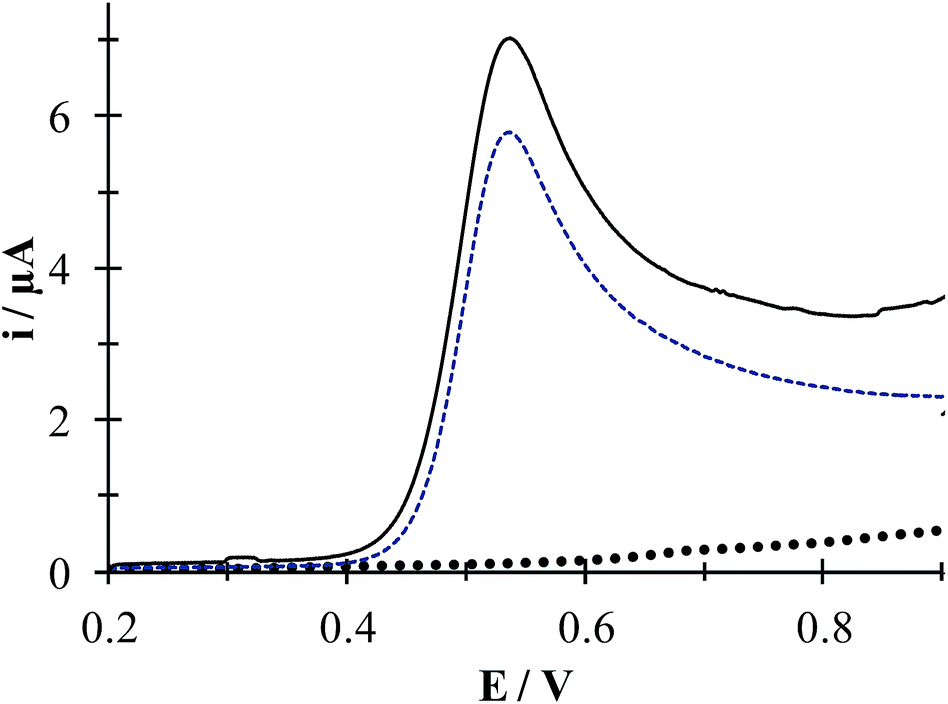
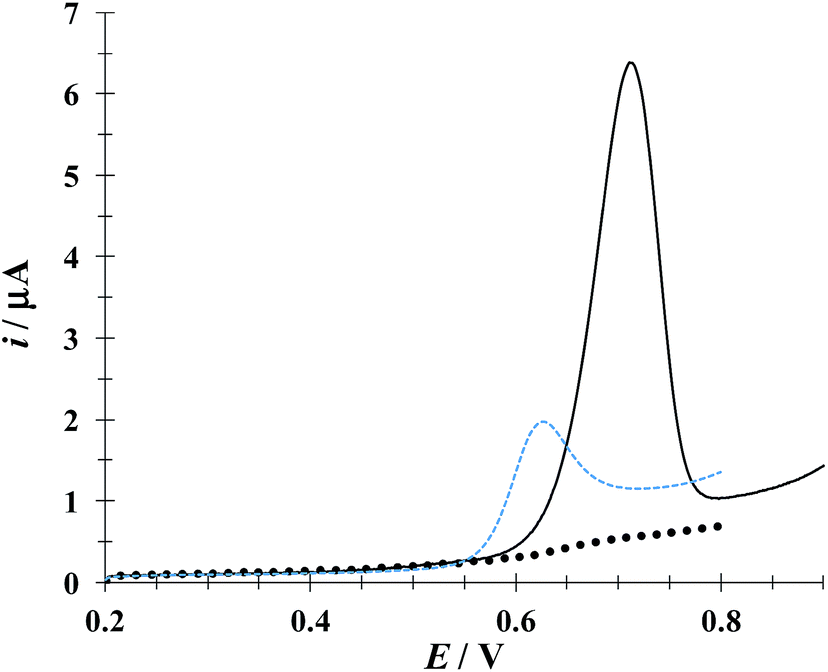
Fig. 4 depicts linear sweep voltammograms (LSVs) obtained during the oxidation of DCF− in the presence and absence of CTAB. In the CTAB presence, the plot exhibits an anodic peak at 0.713 V, while in its absence the anodic peak occurs at 0.630 V. The presence of (CTA+)n not only shifts the anodic peak 83 mV towards more positive potentials but also increases the anodic peak current from 2 to 6.41 μA. This confirms the existence of a “supramolecular” interaction between the CTAB micelles and the DCF anionic form (see Fig. 1a). To deepen on this interaction, a study was performed considering different potential sweep speeds, see below.
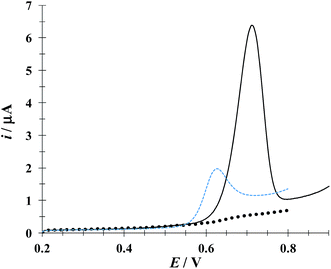 |
| | Fig. 4 Comparison of experimental LSVs recorded in the system CPE/16.34 μM DCF, (phosphate buffer 0.1 M, pH 7) in the presence of 0.1 mM CTAB (solid line) and in the absence (broken line) The LSV of the system CPE/0.1 mM CTAB (solid circles) is also shown. In all cases the potential sweep started at 0.2 V at 100 mV s−1 rate. | |
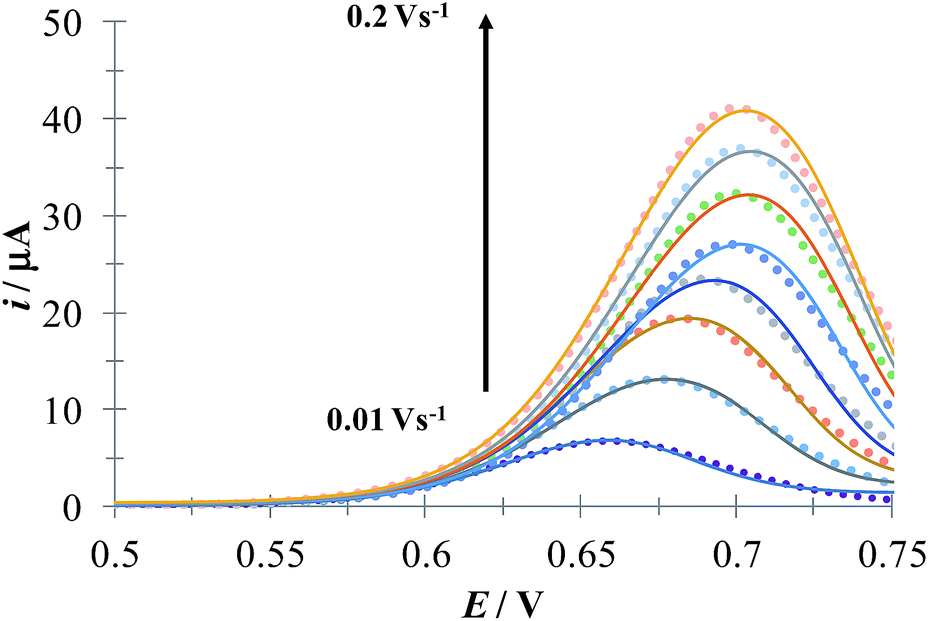
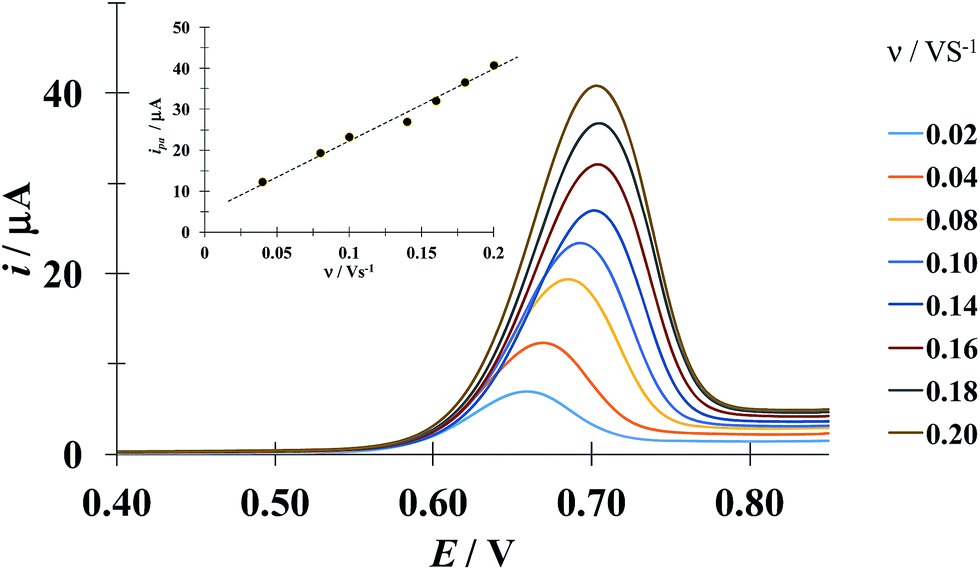
Fig. 5 shows a LSVs family obtained during the electrochemical oxidation of DCF− applying different potential sweep speeds. It was found that the anodic peak current varies linearly with the sweep potential rate, see inset in Fig. 5. This indicates that this process is limited by the adsorption of DCF− on the surface of the electrode.30 Cid-Cerón et al.,32 showed that in the absence of CTAB the electrochemical oxidation process of DCF on a CPE is limited by diffusion, therefore the CTA+ hemimicelles formed on the surface of the CPE31 provoke the strong adsorption of DCF−, most probably through a supramolecular-type interaction, see Fig. 1a. This same effect has already been documented in the case of the electrochemical oxidation of dopamine in the presence of SDS by Corona-Avendaño et al.33
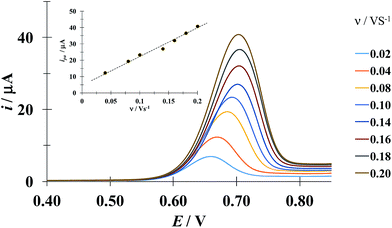 |
| | Fig. 5 Experimental LSVs recorded in the system CPE/100 μM DCF, 0.1 mM CTAB (phosphate buffer 0.1 M, pH 7). The potential sweep started at 0.4 V in the anodic direction for different rates indicated in the figure. The inset shows the anodic peak current (ipa) as a function of the potential sweep rate (ν). The broken line is the linear regression of the experimental data (solid circles). | |
Considering the above features observed on the LSVs recorded at [CTAB] = 0.1 mM, see Fig. 5, and the case where the adsorbed species is electroactive,34–36 eqn (1) has been proposed to describe the experimental i–E curves.
| |
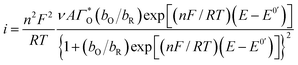 | (1) |
where
A is the electrode surface area,
n is the number of electrons transferred during the heterogeneous reaction,
ν is the potential scan rate,
R,
T and
F are the universal gas constant, absolute temperature and Faraday constant, respectively.
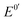
is the formal potential,
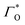
is the surface coverage,
bO and
bR are related to the adsorption free energy through
eqn (2) and
(3), respectively.
| | |
bO = exp(−ΔG0ads,O/RT)
| (2) |
| | |
bR = exp(−ΔG0ads,R/RT)
| (3) |
The peak's potential and current are:
| |
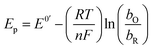 | (4) |
| |
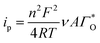 | (5) |
Substituting eqn (4) and (5) in (1) yields eqn (6)
| |
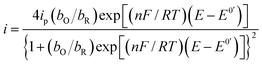 | (6) |
A parameterized form of eqn (6) is eqn (7)
| |
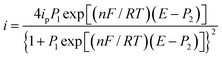 | (7) |
where:
| |
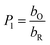 | (8) |
| |
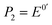 | (9) |
Fig. 6 depicts a comparison of the experimental LSVs shown in Fig. 5 with their corresponding theoretical LSVs plots generated by non-linear fit of eqn (7) to the experimental data, the number of electrons for DCF n = 1 was taken from Cid-Cerón et al.32 From this figure it can be concluded that the model on which eqn (1) is based, adequately described the experimental evidence and that when CTAB is present in the system, in concentrations greater that the CMC, the reduced form of DCF−, is strongly adsorbed. For systems where strong adsorption of reactant and product are involved, the full peak's width at half-height (FWHM) should be equal to 90.6/n where n is the number of electrons transferred during the electrochemical reaction. n was calculated for each LSV using this characteristic and n ≈ 1 was also obtained. The relatively small values for P2 parameter suggest that the interaction between CTAB micelles and DCF− make the DCF− electrochemical oxidation a sluggish kinetic process.
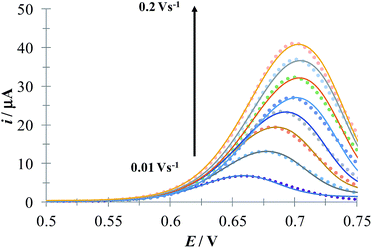 |
| | Fig. 6 Comparison of the experimental LSVs (○○○) reported in Fig. 5 and the theoretical LSVs (lines) generated by non-linear fit of eqn (7) to the experimental data. The best-fit parameters are reported in Table 1. | |
3.3 Electrochemical study of the HACT−(CTA+)n interaction
Fig. 7 shows experimental LSVs recorded during the HACT oxidation in the presence and absence of CTAB. In both cases one anodic peak was obtained at 0.538 V, however, the anodic peak current increases only by 1.23 μA when the CTAB is present. Fig. S1, in the ESI,† shows the effect of the potential sweep rates on the HACT oxidation in the absence, Fig. S1a,† and in the presence, Fig. S1b,† of CTAB. In both cases the peak current varies linearly with ν1/2, see insets in Fig. S1a and S1b,† indicating that the process is limited by the diffusion of HACT.35 As shown in Section 3.1.1 in the case of HACT, CTAB also shows a smaller effect on their electrochemical properties compared to DCF−, this is because unlike DCF, the ACT predominates in neutral form (see Fig. 1b) thus, its interaction with CTA+ is negligible.
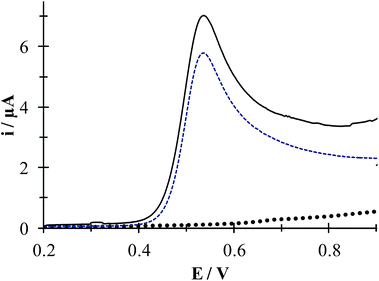 |
| | Fig. 7 Experimental LSVs recorded in system CPE/64.9 μM ACT, (0.1 M phosphate buffer, pH 7) in the presence (solid line) and the absence (broken line) of 0.1 mM CTAB. The LSV of the system CPE/0.1 mM CTAB is shown by solid black circles. In all cases, the potential sweep started at 0.2 V at 100 mVs−1 rate. | |
3.4 Individual quantification of the drugs
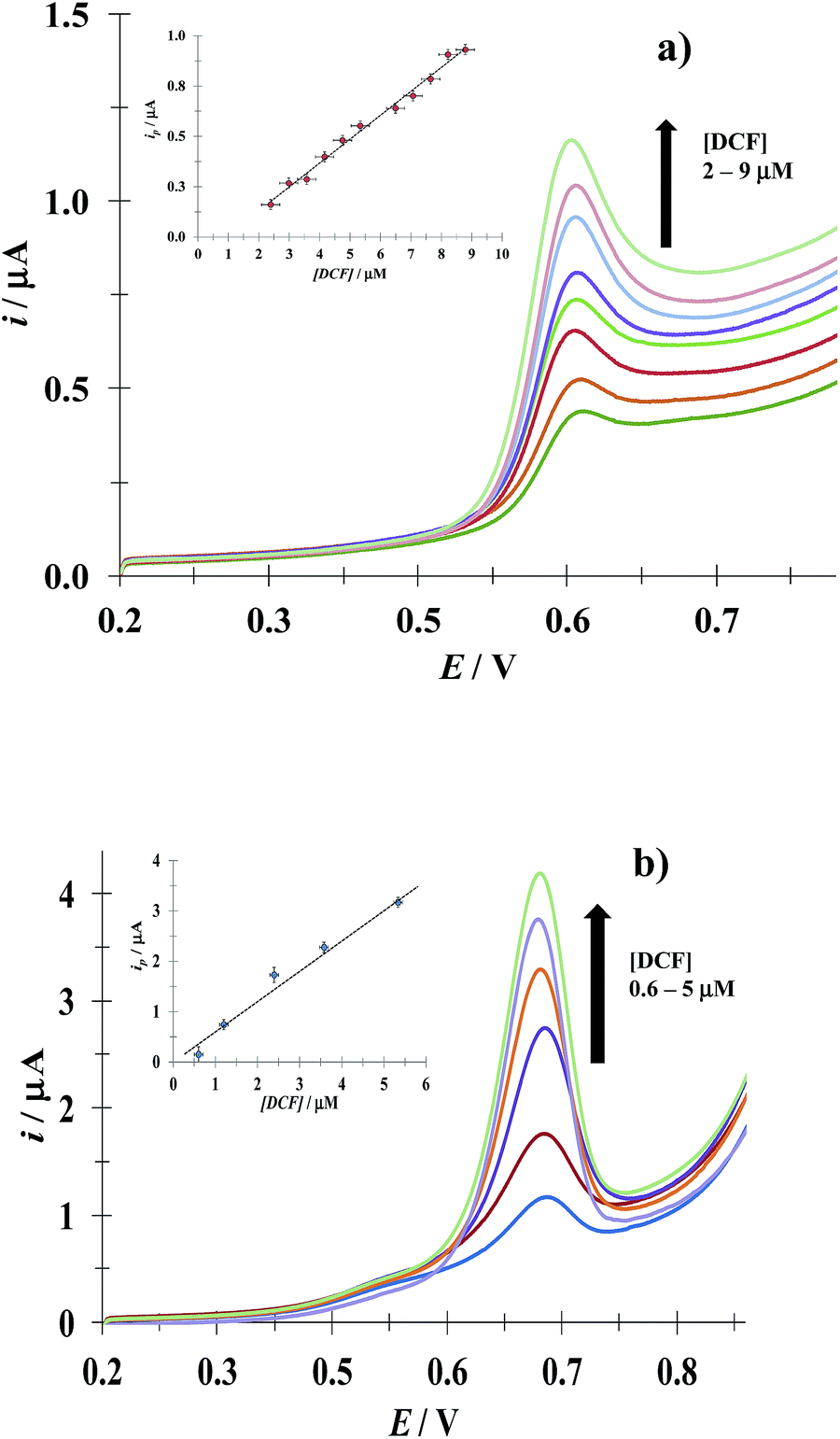
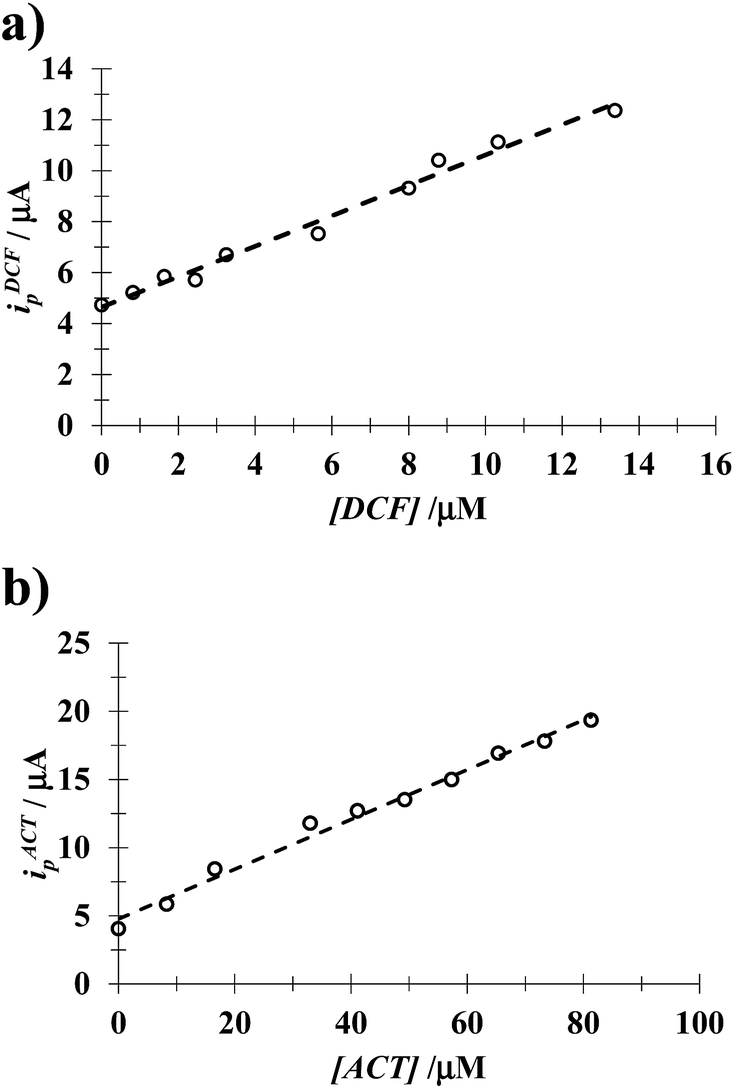
3.4.1 DCF. Fig. 8 shows LSVs recorded during the DCF electrochemical oxidation in the absence, Fig. 8a, and presence, Fig. 8b, of CTAB. In both cases, the respective anodic peak current (ip) varies linearly with the DCF concentration; see the insets in Fig. 8. From these calibration plots it was possible to estimate the influence of CTAB on the analytical features, namely: the limits of detection (LOD) and quantification (LOQ) and the respective sensitivity as well, using the methodology reported by Swartz and Krull,37 see Table 2; it becomes plain that the CTAB presence improves the analytical features of the electrode. Furthermore, as can be noted from Table 3, this rather simple electrode depicts similar or better analytical features towards DCF quantification as compared with other more sophisticated electrodes and/or techniques.
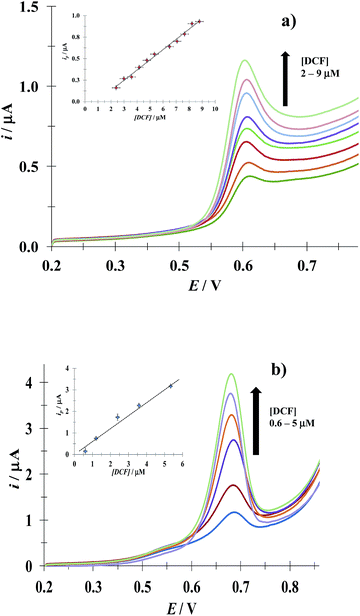 |
| | Fig. 8 Experimental LSVs recorded in the system CPE/(0.1 M phosphate buffer, pH 7) with different DCF concentrations (indicated in the figures) and two different CTAB concentrations: (a) 0 and (b) 0.1 mM. The potential scan rate started at 0.2 V, in the positive direction at 100 mV s−1. The figures inset depict the variation of the respective anodic peak current (ip) as a function of the DCF concentration. The lines were obtained from the linear fit into the experimental data (solid circles). | |
Table 1 Best-fit parameters obtained by non-linear fit of eqn (7) to the LSVs recorded in the system CPE/DCF 100 μM, CTAB 0.1 mM (phosphate buffer 0.1 M, pH 7), see Fig. 6
| ν/V s−1 |
109 P1 |
P2/V |
| 0.02 |
6.8 |
0.19 |
| 0.04 |
16.0 |
0.22 |
| 0.06 |
4.7 |
0.20 |
| 0.08 |
12.0 |
0.24 |
| 0.14 |
7.6 |
0.24 |
| 0.16 |
11.0 |
0.24 |
| 0.18 |
7.30 |
0.23 |
| 0.2 |
22.0 |
0.25 |
Table 2 Analytical features depicted for the CPE toward DCF and ACT quantification as a function of [CTAB] recorded from the calibration plots shown as insets in Fig. 8 and S2 respectively
| |
[CTAB]/mM |
Sensitivity/μA μM−1 |
LOD/μM |
LOQ/μM |
R2 |
| DCF |
0 |
0.120 ± 0.004 |
0.70 ± 0.30 |
2.4 ± 0.27 |
0.998 |
| 0.1 |
0.71 ± 0.007 |
0.73 ± 0.30 |
2.4 ± 0.28 |
0.999 |
| ACT |
0 |
0.085 ± 0.001 |
3.8 ± 1.5 |
12.8 ± 1.4 |
0.998 |
| 0.1 |
0.11 ± 0.01 |
7.6 ± 3.0 |
25.4 ± 2.7 |
0.997 |
Table 3 Comparison of the analytical features towards DCF quantification reported for different experimental techniques n. r. = not reported
| Technique |
LOD/M |
Sensitivity |
Ref. |
| LSV using a CPE immersed in 0.1 mM CTAB aqueous solution (pH 7) |
(7.3 ± 3.0) × 10−7 |
(0.71 ± 0.007) μA μM−1 |
This work |
| Diffuse reflectance in the visible region |
(2 ± n. r.) × 10−3 |
n. r. |
9 |
| Spectrofluorimetry using β-CD |
(2.20 ± n. r.) × 10−7 |
(16 ± n. r.) mL μg−1 |
10 |
| Electrochemical impedance spectroscopy using artificial nucleic acids ligands (aptasensor) |
(2.7 ± n. r.) × 10−7 |
(15.66 ± n. r.) kΩ M−1 |
12 |
| Potentiometric using and ISE with crystal violet |
(2.5 ± n. r.) × 10−5 |
n. r. |
13 |
| DPV using an edge-plane pyrolytic graphite electrode with single-wall carbon nanotubes. |
(0.82 ± n. r.) × 10−9 |
(224 ± n. r.) nA μM−1 |
16 |
| DPV using a tyrosine-modified CPE |
(3.28 ± n. r.) ×10−6 |
n. r. |
38 |
| DPV using a GCE modified with Cu(OH)2 nanoparticles, hydrophobic ionic liquid and multiwalled carbon nanotubes. |
(4 ± n. r.) × 10−8 |
(0.0147 ± n. r.) μA μM−1 |
39 |
3.4.2 ACT. From the calibration plots obtained using the respective experimental LSVs, see Fig. S2 in the ESI† of this work, it was possible to evaluate the influence of CTAB on the analytical features of the CPE towards ACT quantification, see Table 2. Once again, the presence of CTAB improves the analytical features of the electrode. Moreover, as can be noted from Table 4, this simple electrode depicts similar or better analytical features towards ACT quantification as compared with other more sophisticated electrodes and/or techniques.
Table 4 Comparison of the analytical features towards ACT quantification reported for different experimental techniquesa
| Technique |
LOD/M |
Sensitivity |
Ref. |
| n. r. = not reported. |
| LSV using a CPE immersed in 0.1 mM CTAB aqueous solution (pH 7) |
(0.76 ± 0.03) × 10−7 |
(0.11 ± 0.01) μA μM−1 |
This work |
| Spectrofluorimetry using ethylacetoacetate |
(3.77 ± n.r.) × 10−7 |
n. r. |
40 |
| Spectrophotometry without chemical reagents |
(6.62 ± n. r.) × 10−7 |
n. r. |
41 |
| Chronoamperometric determination using a vaseline/graphite modified avocado tissue |
(8.8 ± n. r.) × 10−5 |
n. r. |
42 |
| DPV using GCE at pH 3.29 |
(4.31 ± n. r.) × 10−6 |
n. r. |
43 |
| Amperometry using screen-printed electrodes modified with carbon nanotubes |
(1 ± n. r.) × 10−7 |
n. r. |
44 |
3.5 Simultaneous quantification of DCF and ACT
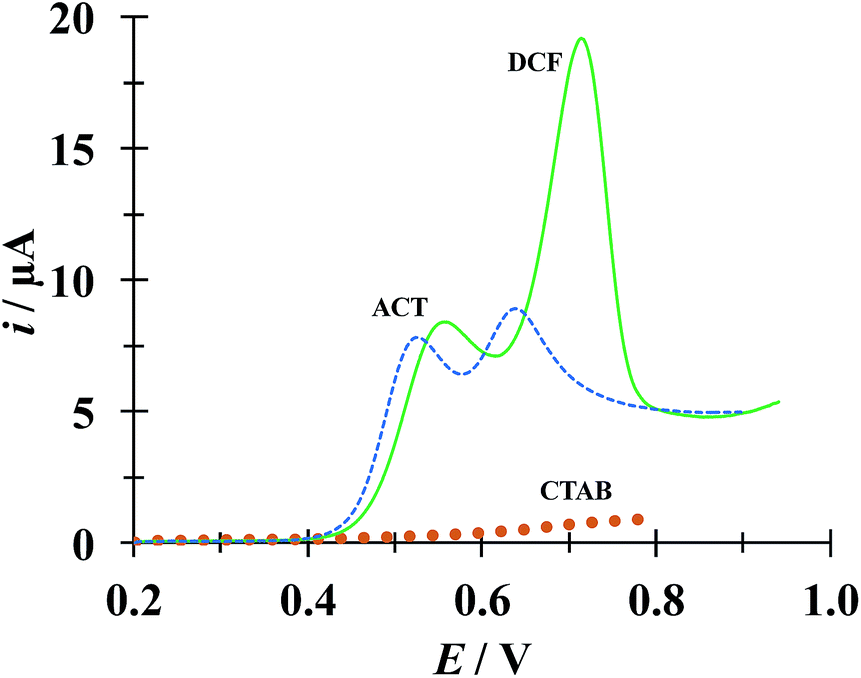
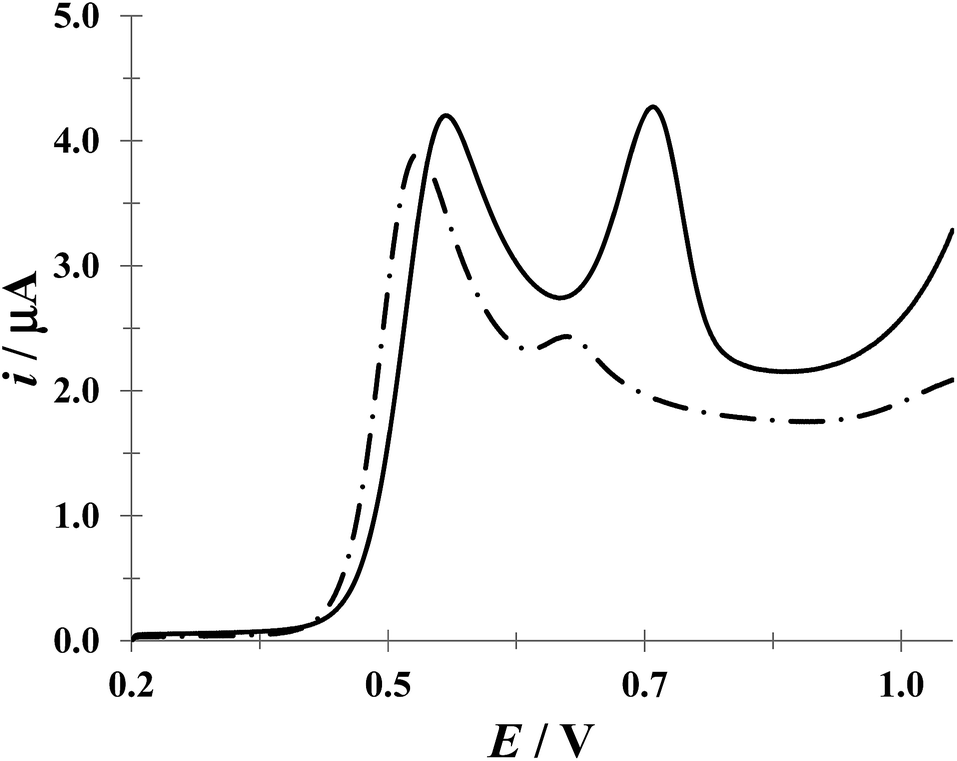
Fig. 9 shows a comparison of LSVs recorded during the electrochemical oxidation of HACT and DCF−, using a CPE, both present in the same aqueous solution at the same concentration, in the absence and presence of CTAB. Even when in the absence of CTAB two voltammetric peaks corresponding to HACT and DCF− could be distinguished with a ΔEp = EpDCF − EpACT = 630 − 525 = 105 mV, the presence of CTAB changed this potential difference to 158 mV, which is not a drastic change, however the current peak associated to DCF− went from 8.81 to 19.1 μA (117% increase) while that corresponding to HACT solely depicted a slight increase from 7.52 to 8.35 μA. It is important to note that even when the CTAB alone showed some electrochemical activity in this potential region, it can be considered negligible (for instance, it is 0.76 μA at the EpDCF value in the presence of CTAB).
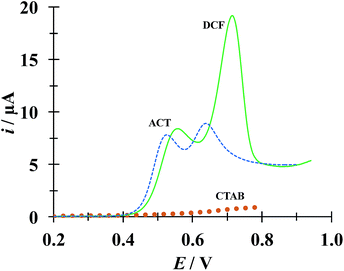 |
| | Fig. 9 Comparison of LSVs recorded in the systems: CPE/51.98 μM DCF, 51.98 μM ACT (0.1 M phosphate buffer, pH 7) with different CTAB concentrations 0 (broken line) and 0.1 mM (solid line). The LSV corresponding to the system CPE/0.1 mM CTAB (solid circles) was also included. In all cases, the potential scan rate started at 0.2 V, in the positive direction at 100 mV s−1. | |
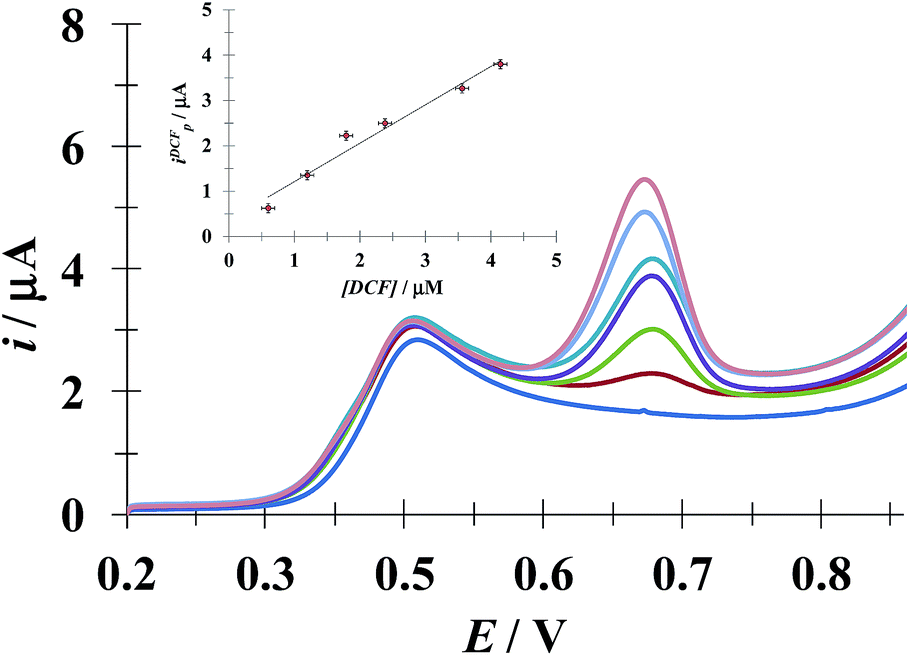
3.5.1 DCF quantification in the presence of a fixed [ACT]. Fig. 10 shows the LSVs recorded during DCF− electrochemical oxidation in the presence of fixed concentration of HACT and CTAB but varying the DCF− concentration. The calibration plot for DCF− quantification was obtained from this plot, see the inset in Fig. 10. The analytical parameter depicted for this electrode towards DCF− quantification in the presence of HACT are reported in Table 5 along with those obtained in this same system but in the absence of CTAB, see Fig. S3.†
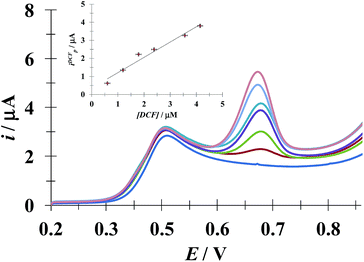 |
| | Fig. 10 Family of LSVs recorded in the systems: CPE/48.8 μM ACT, 0.1 mM CTAB (0.1 M phosphate buffer, pH 7) with different DCF concentrations indicated in the figure, in all cases the potential scan rate started at 0.2 V, in the positive direction at 100 mV s−1. The insert depicts the variation of the DCF anodic current peak (ipDCF) as a function of DCF concentration. The line was obtained from the linear fit into the experimental data (solid circles). | |
Table 5 Analytical features depicted for the CPE toward DCF and ACT quantification, as a function of [CTAB] recorded from the calibration plots shown as insets in Fig. 10, S3 and S4 respectively
| |
[CTAB]/mM |
Sensitivity/μA μM−1 |
LOD/μM |
LOQ/μM |
R2 |
| DCF |
0 |
0.10 ± 0.013 |
1.6 ± 0.62 |
5.6 ± 0.67 |
0.998 |
| 0.1 |
0.86 ± 0.04 |
0.50 ± 0.21 |
1.7 ± 0.18 |
0.997 |
| ACT |
0 |
0.062 ± 0.002 |
2.8 ± 1.8 |
9.3 ± 1.7 |
0.998 |
| 0.1 |
0.054 ± 0.001 |
2.5 ± 0.9 |
8.3 ± 0.98 |
0.997 |
3.5.2 ACT quantification in the presence of a fixed [DCF]. From the calibration plots obtained from the respective experimental LSVs, see Fig. S4 in the ESI† of this work, it was possible to evaluate the influence of CTAB on the analytical features of the CPE towards ACT quantification in the presence of a fixed concentration of DCF, see Table 5. Once more it is possible to note that the presence of CTAB improves the analytical features of the electrode.From Table 6 it is possible to note that the simultaneous electrochemical, LSV, quantification of DCF and ACT using a simple CPE immersed in an aqueous solution containing (CTA+)n micelles displays similar or better analytical features towards ACT quantification as compared with other more sophisticated electrodes and/or techniques.
Table 6 Comparison of the analytical features towards DCF quantification in the presence of ACT and its absence reported for different experimental techniquesa
| Technique |
DCF |
ACT |
Ref. |
| LOD/M |
Sensitivity |
LOD/M |
Sensitivity |
| n. r. = not reported. |
| LSV with a CPE immersed in 0.1 mM CTAB aqueous solution (pH 7) |
(5.0 ± 2.1) × 10−7 |
(0.86 ± 0.04) μA μM−1 |
(2.5 ± 0.9) × 10−6 |
(0.054 ± 0.001) μA μM−1 |
This work |
| Chemometric method (processing of absorbance data) |
n. r. |
(5.31 ± n. r.) L mg−1 |
n. r. |
(3.03 ± n. r.) L mg−1 |
45 |
| Reverse phase high performance liquid chromatography |
(1.57 ± n. r.) × 10−8 |
n. r. |
(6.62 ± n. r.) × 10−8 |
n. r. |
46 |
| Reverse phase high performance liquid chromatography |
(8.80 ± n. r.) × 10−9 |
n. r. |
(6.3 ± n. r.) × 10−6 |
n. r. |
47 |
| UV-Vis spectrophotometry |
n. r. |
n. r. |
n. r. |
n. r. |
48 |
| Reverse phase high performance liquid chromatography |
(3.14 ± n. r.) × 10−9 |
n. r. |
(2.65 ± n. r.) × 10−8 |
n. r. |
49 |
| Differential pulse voltammograms using a 4-phosphatephenyl-modified glassy carbon electrode |
n. r. |
n. r. |
n. r. |
n. r. |
50 |
| Differential pulse voltammograms using a poly(diallyldimethylammonium chloride) functionalized graphene |
(0.609 ± n. r.) × 10−6 |
n. r. |
(0.221 ± n. r.) × 10−6 |
n. r. |
51 |
4 Conclusions
It was shown that CTAB hemimicelles, formed on the surfaces of a CPE, could be successfully used for the simultaneous electrochemical quantification of DCF and ACT dissolved in aqueous solution. The analytical performance, namely: LOD, LOQ and sensitivity, depicted by this inexpensive and rather simple manufacture, CTAB-modified CPE towards DCF and ACT quantification was evaluated, showing high selectivity and sensitivity even in real samples formed by commercial drugs and synthetic urine.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
MMCC and MTRS thank CONACYT for support through project 237327 and DSGH and MTRS thanks CONACYT for cathedra 2159. MTRS, SCA, MEPH, MRR and MEPP like to thank SEP PRODEP for financial support through the RedNIQAE. DSGH, MTRS, SCA, MEPH, MRR and MPP wish to thank the SNI for the distinction of their membership and the stipend received.
Notes and references
- M. J. Rosen and J. T. Kunjappu, Surfactants and Interfacial Phenomena, Wiley, 2012 Search PubMed.
- W. Liu and R. Guo, The interaction between morin and CTAB aggregates, J. Colloid Interface Sci., 2005, 290(2), 564–573 CrossRef CAS PubMed.
- S. Schreier, S. V. P. Malheiros and E. De Paula, Surface active drugs: Self-association and interaction with membranes and surfactants. Physicochemical and biological aspects, Biochim. Biophys. Acta, Biomembr., 2000, 1508(1–2), 210–234 CrossRef CAS.
- T. F. Tadros, Surfactants in Pharmaceutical Formulations. Applied Surfactants, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 433–501 Search PubMed.
- N. Vučinić-Milanković, S. Savić, G. Vuleta and S. Vučinić, Natural surfactant-based emulsion vehicles: A correlation between colloidal structure and in vitro release of diclofenac diethylamine, J. Dispersion Sci. Technol., 2010, 31(8), 1077–1084 CrossRef.
- G. Alarcón-Angeles, S. Corona-Avendaño, M. Palomar-Pardavé, A. Rojas-Hernández, M. Romero-Romo and M. T. Ramírez-Silva, Selective electrochemical determination of dopamine in the presence of ascorbic acid using sodium dodecyl sulfate micelles as masking agent, Electrochim. Acta, 2008, 53(6), 3013–3020 CrossRef.
- A. Arellano, S. Santoyo, C. Martn and P. Ygartua, Surfactant effects on the in vitro percutaneous absorption of diclofenac sodium, Eur. J. Drug Metab. Pharmacokinet., 1998, 23(2), 307–312 CrossRef CAS PubMed.
- P. L. Kole, J. Millership and J. C. McElnay, Determination of diclofenac from paediatric urine samples by stir bar sorptive extraction (SBSE)-HPLC-UV technique, Talanta, 2011, 85(4), 1948–1958 CrossRef CAS PubMed.
- M. Tubino and R. L. De Souza, Determination of diclofenac in pharmaceutical preparations by diffuse reflectance photometry, Talanta, 2006, 68(3), 776–780 CrossRef CAS PubMed.
- J. A. Arancibia and G. M. Escandar, Complexation study of diclofenac with β-cyclodextrin and spectrofluorimetric determination, Analyst, 1999, 124(12), 1833–1838 RSC.
- A. Chmielewska, L. Konieczna, A. Plenis, M. Bieniecki and H. Lamparczyk, Determination of diclofenac in plasma by high-performance liquid chromatography with electrochemical detection, Biomed. Chromatogr., 2006, 20(1), 119–124 CrossRef CAS PubMed.
- L. Kashefi-Kheyrabadi and M. A. Mehrgardi, Design and construction of a label free aptasensor for electrochemical detection of sodium diclofenac, Biosens. Bioelectron., 2012, 33(1), 184–189 CrossRef CAS PubMed.
- Z. Kormosh, I. Hunka, Y. Bazel, N. Kormosh, A. Laganovsky and I. Mazurenko, A new diclofenac membrane sensor based on its ion associate with crystal violet. Application to diclofenac determination in urine and pharmaceuticals, J. Iran. Chem. Soc., 2007, 4(4), 408–413 CrossRef CAS.
- Z. Kormosh, I. Hunka, Y. Bazel, A. Laganovsky, I. Mazurenko and N. Kormosh, Determination of diclofenac in pharmaceuticals and urine samples using a membrane sensor based on the ion associate of diclofenac with Rhodamine B., Cent. Eur. J. Chem., 2007, 5(3), 813–823 CAS.
- L. Fernández-Llano, M. C. Blanco-López, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tuñón-Blanco, Determination of diclofenac in urine samples by molecularly-imprinted solid-phase extraction and adsorptive differential pulse voltammetry, Electroanalysis, 2007, 19(15), 1555–1561 CrossRef.
- R. N. Goyal, S. Chatterjee and A. R. S. Rana, The effect of modifying an edge-plane pyrolytic graphite electrode with single-wall carbon nanotubes on its use for sensing diclofenac, Carbon, 2010, 48(14), 4136–4144 CrossRef CAS.
- M. K. Fayaz, R. J. Abel, S. C. Pugh, J. E. Hall, G. Djaiani and J. S. Mecklenburgh, Opioid-sparing effects of diclofenac and paracetamol lead to improved outcomes after cardiac surgery, J. Cardiothorac. Vasc. Anesth., 2004, 18(6), 742–747 CrossRef CAS PubMed.
- B. Munishankar, P. Fettes, C. Moore and G. A. McLeod, A double-blind randomised controlled trial of paracetamol, diclofenac
or the combination for pain relief after caesarean section, Int. J. Obstet. Anesth., 2008, 17(1), 9–14 CrossRef CAS PubMed.
- K. Fent, A. A. Weston and D. Caminada, Ecotoxicology of human pharmaceuticals, Aquat. Toxicol., 2006, 76(2), 122–159 CrossRef CAS PubMed.
- B. Petrie, R. Barden and B. Kasprzyk-Hordern, A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring, Water Res., 2014, 72, 3–27 CrossRef PubMed.
- P. B. Deroco, F. C. Vicentini, G. G. Oliveira, R. C. Rocha-Filho and O. Fatibello-Filho, Square-wave voltammetric determination of hydroxychloroquine in pharmaceutical and synthetic urine samples using a cathodically pretreated boron-doped diamond electrode, J. Electroanal. Chem., 2014, 719, 19–23 CrossRef CAS.
- R. Martínez, M. T. Ramírez and I. González, Voltammetric Characterization of Carbon Paste Electrodes with a Nonconducting Binder. Part I: Evidence of the Influence of Electroactive Species Dissolution into the Paste on the Voltammetric Response, Electroanalysis, 1998, 10(5), 336–342 CrossRef.
- M. T. Ramírez, M. E. Palomar, I. González and A. Rojas-Hernández, Carbon paste electrodes with electrolytic binder: Influence of the preparation method, Electroanalysis, 1995, 7(2), 184–188 CrossRef.
- A. Rojas-Hernández, M. T. Ramirez, J. G. Ibánez and I. Gonzalez, Construction of multicomponent pourbaix diagrams using generalized species, J. Electrochem. Soc., 1991, 138(2), 365–371 CrossRef.
- A. Rojas-Hernández, M. T. Ramírez, J. G. Ibáñez and I. González, Relationship of multidimensional predominance-zone diagrams with multiconditional constants for complexation equilibria, Anal. Chim. Acta, 1991, 246(2), 435–442 CrossRef.
- A. Rojas-Hernández, M. T. Ramírez, I. González and J. G. Ibáñez, Multi-dimensional predominance-zone diagrams for polynuclear chemical species, Anal. Chim. Acta, 1992, 259(1), 95–104 CrossRef.
- A. Rojas-Hernández, M. T. Ramírez and I. González, Equilibria among condensed phases and a multi-component solution using the concept of generalized species. Part II. Systems with polynuclear species, Anal. Chim. Acta, 1993, 278(2), 335–347 CrossRef.
- S. Babić, A. J. M. Horvat, D. Mutavdžić Pavlović and M. Kaštelan-Macan, Determination of pKa values of active pharmaceutical ingredients, TrAC, Trends Anal. Chem., 2007, 26(11), 1043–1061 CrossRef.
- P. Norouzi, F. Dousty, M. R. Ganjali and P. Daneshgar, Dysprosium nanowire modified carbon paste electrode for the simultaneous determination of naproxen and paracetamol: Application in pharmaceutical formulation and biological fluid, Int. J. Electrochem. Sci., 2009, 4(10), 1373–1386 CAS.
- E. Fuguet, C. Ràfols, M. Rosés and E. Bosch, Critical micelle concentration of surfactants in aqueous buffered and unbuffered systems, Anal. Chim. Acta, 2005, 548, 95–100 CrossRef CAS.
- A. E. Sánchez-Rivera, V. Vital-Vaquier, M. Romero-Romo, M. T. Ramírez-Silva and M. Palomar-Pardavé, Electrochemical deposition of cetyltrimethylammonium surface hemimicelles at the Hg/0.1 M NaCl(aq) interface, J. Electrochem. Soc., 2004, 151(10), C666–C673 CrossRef.
- M. M. Cid-Cerón, D. S. Guzmán-Hernández, M. T. Ramírez-Silva, A. Galano, M. Romero-Romo and M. Palomar-Pardavé, New insights on the kinetics and mechanism of the electrochemical oxidation of diclofenac in neutral aqueous medium, Electrochim. Acta, 2016, 199, 92–98 CrossRef.
- S. Corona-Avendaño, G. Alarcón-Angeles, M. T. Ramírez-Silva, G. Rosquete-Pina, M. Romero-Romo and M. Palomar-Pardavé, On the electrochemistry of dopamine in aqueous solution. Part I: The role of [SDS] on the voltammetric behavior of dopamine on a carbon paste electrode, J. Electroanal. Chem., 2007, 609(1), 17–26 CrossRef.
- E. Laviron, General Expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- M. Palomar-Pardavé, S. Corona-Avendaño, M. Romero-Romo, G. Alarcón-Angeles, A. Merkoçi and M. T. Ramírez-Silva, Supramolecular interaction of dopamine with β-cyclodextrin: An experimental and theoretical electrochemical study, J. Electroanal. Chem., 2014, 717–718, 103–109 CrossRef.
- M. E. Swartz and I. S. Krull, Analytical Method Development and Validation, Marcel Dekker, New York, 1997, p. 92 Search PubMed.
- B. K. Chethana, S. Basavanna and Y. Arthoba Naik, Voltammetric determination of diclofenac sodium using tyrosine-modified carbon paste electrode, Ind. Eng. Chem. Res., 2012, 51(31), 10287–10295 CrossRef CAS.
- M. Arvand, T. M. Gholizadeh and M. A. Zanjanchi, MWCNTs/Cu(OH) 2 nanoparticles/IL nanocomposite modified glassy carbon electrode as a voltammetric sensor for determination of the non-steroidal anti-inflammatory drug diclofenac, Mater. Sci. Eng., C, 2012, 32(6), 1682–1689 CrossRef CAS PubMed.
- A. De Los, M. Oliva, R. A. Olsina and A. N. Masi, Selective spectrofluorimetric method for paracetamol determination through coumarinic compound formation, Talanta, 2005, 66(1), 229–235 CrossRef PubMed.
- K. A. R. Sirajuddin, A. Shah, M. I. Bhanger, A. Niaz and S. Mahesar, Simpler spectrophotometric assay of paracetamol in tablets and urine samples, Spectrochim. Acta, Part A, 2007, 68(3), 747–751 CrossRef PubMed.
- O. Fatibello-Filho, K. O. Lupetti and I. C. Vieira, Chronoamperometric determination of paracetamol using an avocado tissue (Persea americana) biosensor, Talanta, 2001, 55(4), 685–692 CrossRef CAS PubMed.
- I. Baranowska and M. Koper, The preliminary studies of electrochemical behavior of paracetamol and its metabolites on glassy carbon electrode by voltammetric methods, Electroanalysis, 2009, 21(10), 1194–1199 CrossRef CAS.
- P. Fanjul-Bolado, P. J. Lamas-Ardisana, D. Hernández-Santos and A. Costa-García, Electrochemical study and flow injection analysis of paracetamol in pharmaceutical formulations based on screen-printed electrodes and carbon nanotubes, Anal. Chim. Acta, 2009, 638(2), 133–138 CrossRef CAS PubMed.
- P. M. Castellano, S. E. Vignaduzzo, R. M. Maggio and T. S. Kaufman, Application of a chemometric method for simultaneous determination of acetaminophen and diclofenac in content-uniformity and drug-dissolution studies, Anal. Bioanal. Chem., 2005, 382(7), 1711–1714 CrossRef CAS PubMed.
- B. Gowramma, S. Rajan, S. Muralidharan, S. N. Meyyanathan and B. Suresh, A validated RP-HPLC method for simultaneous estimation of paracetamol and diclofenac potassium in pharmaceutical formulation, Int. J. PharmTech Res., 2010, 2(1), 676–680 CAS.
- K. Jana, L. Adhikari, S. K. Moitra and A. Behera, Analysis of multicomponent drug formulations (Diclofenac and paracetamol), Asian J. Pharm. Clin. Res., 2011, 4(2), 41–43 CAS.
- S. Beeravolu, R. K. Vejendla and S. K. Nagula, Estimation and validation of UV-visible spectrophotometric method for combined tablet dosage form of paracetamol and diclofenac sodium using extraction technique, Int. J. Pharm. Technol., 2012, 4(3), 4733–4740 CAS.
- S. N. Nag, B. Gouthami, L. Madhuri, N. Krishnaveni, S. N. Meyyanathan and B. Suresh, Development and validation of a RP-HPLC method for the simultaneous determination of paracetamol and diclofenac potassium on stainless steel surface of pharmaceutical manufacturing equipments, J. Chem. Pharm. Res., 2012, 4(3), 1670–1675 Search PubMed.
- T. Zhou, L. Li, J. Wang, X. Chen, G. Yang and Y. Shan, 4-Phosphatephenyl-modified glassy carbon electrode for real-time and simultaneous electrochemical monitoring of paracetamol and diclofenac release from electrospun nanofibers, Anal. Methods, 2015, 7(21), 9289–9294 RSC.
- O. K. Okoth, K. Yan, L. Liu and J. Zhang, Simultaneous Electrochemical Determination of Paracetamol and Diclofenac Based on Poly(diallyldimethylammonium chloride) Functionalized Graphene, Electroanalysis, 2016, 28(1), 76–82 CrossRef CAS.
- L. Fernández-Llano, M. C. Blanco-López, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tuñón-Blanco, Determination of Diclofenac in Urine Samples by Molecularly-Imprinted Solid-Phase Extraction and Adsorptive Differential Pulse Voltammetry, Electroanalysis, 2007, 19(15), 1555–1561 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07269d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
M. T. Ramírez-Silva
c,
M. T. Ramírez-Silva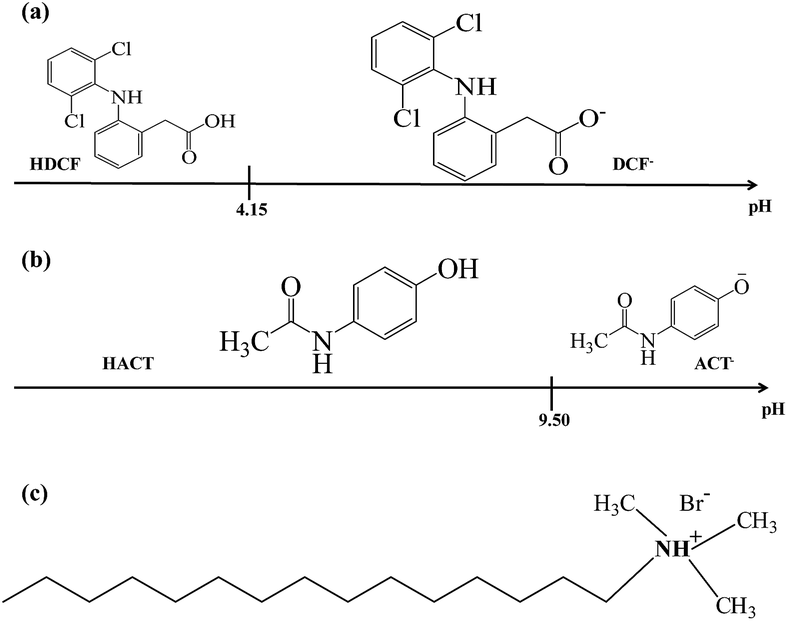
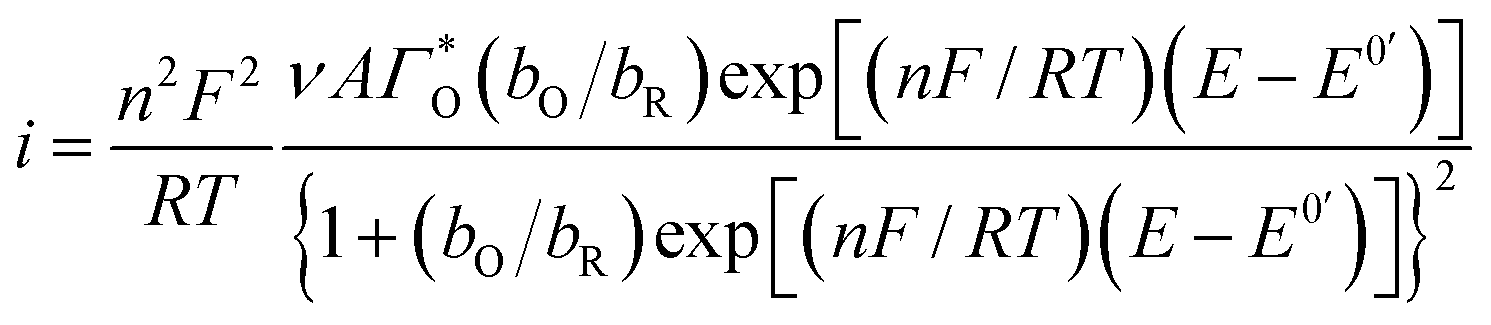
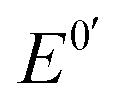 is the formal potential,
is the formal potential, 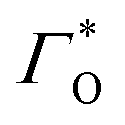 is the surface coverage, bO and bR are related to the adsorption free energy through eqn (2) and (3), respectively.
is the surface coverage, bO and bR are related to the adsorption free energy through eqn (2) and (3), respectively.