
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fine dispersion morphology of polystyrene/poly(ethylene terephthalate glycol) blending generation for controlled foaming behavior
Wenzhao Wang *ab,
Liancai Wangb,
Yang Jiaob,
Xinmiao Zengb,
Xiangdong Wang*c,
Yongjun Lub,
Anren Chengb,
Pei Daib and
Xuena Zhaob
*ab,
Liancai Wangb,
Yang Jiaob,
Xinmiao Zengb,
Xiangdong Wang*c,
Yongjun Lub,
Anren Chengb,
Pei Daib and
Xuena Zhaob
aBeijing Radiation Center, Beijing 100875, China. E-mail: wwzhero1@126.com
bBeijing Key Laboratory of Radiation Advanced Materials, Beijing Research Center for Radiation Application, Beijing 100015, China
cSchool of Materials and Mechanical Engineering, Beijing Technology and Business University, Beijing 100048, China. E-mail: wangxid@th.btbu.edu.cn
First published on 10th August 2017
Abstract
Polystyrene/poly(ethylene terephthalate glycol) (PS/PETG) blends with different PETG contents were prepared using a Haake internal mixer at 190 °C. Morphologies of the PS/PETG blends exhibited a sea-island structure with homogeneity and a fine degree of dispersion. Additionally, it was found that the size of spherical PETG particles in a PS matrix maintained an unchanged scale but the density of dispersed particles increased with a PETG content from 10 wt% to 25 wt%. The dispersion density or interface density could be tuned by the added PETG content. Furthermore, the rheological properties and melt strength of PS and PS/PETG blends were also tested and discussed. The addition of PETG in a PS matrix makes little difference in the PS/PETG blends. Finally, foaming behaviors of the PS and PS/PETG blends were investigated and the mechanism of these blends foaming was analyzed. We concluded that addition of the PETG phase not only facilitated foaming nucleation but also increased the CO2 in solution for all the blends during the foaming process. This resulted in foam with a higher expansion ratio and better uniform cell structure in comparison with PS. However, PETG addition contents should be controlled to a certain degree, or a serious open-cell phenomenon will occur.
1. Introduction
Polymeric foams have been widely used in many applications ranging from packaging and insulation to scaffolds for tissue engineering, due to their advantages such as light weight, cushioning performance, thermal and acoustic insulation, and impact resistance.1,2 Polystyrene (PS) foam is a well-established material for industrial applications and occupies the second largest segment of the foam market.3,4 With more and more concerns about the environment, traditional blowing agents like hydrogen-containing chlorofluorocarbons (HCFCs) have been eliminated in the past few years because of their detrimental effects to the environment.5,6 Supercritical carbon dioxide (sc-CO2) has emerged as a “green” physical foaming agent for replacing traditional foaming agents in the production of PS foams.7,8 Nevertheless, CO2 is a small molecule and faces many challenges to replace traditional foaming agents due to its low solubility and high diffusivity in PS melts.9,10 Consequently, foam density and cell morphology are more difficult to control, and this has an unfavorable influence on final properties of the foaming products.11–13Presently, several methods for CO2 as a physical blowing agent have been developed to improve foam performance by controlling the foaming processes: (1) employing a co-blowing agent,14 (2) adding inorganic particles,15 (3) grafting groups,16 (4) inducing two or more foaming stage processes,17 (5) generating a particular cellular structure,18 and (6) using a polymer blending system.19 Richard et al. studied the PS foaming process with a mixture of CO2 and ethanol (EtOH). The EtOH was found to contribute significantly to the overall plasticization of the system, in which cells would start to nucleate at a decreased pressure, and thus make the mixture less ‘explosive’.14 Zhu et al. prepared PS–silicate nanocomposites having a good dispersion of clay particles. The dispersion acted as nucleation sites to facilitate formation of nucleation centers for the gaseous phase and a PS foam having the morphology of reduced cell size and increased cell density was obtained.20
Thus, research on PS blending foaming processes is very active and all kinds of PS blending systems21–29 (e.g., polystyrene (PS)/polypropylene (PP), PS/polyethylene (PE), PP/PE, PS/poly(methyl methacrylate) (PMMA), PS/polyethylene oxide (PEO), PS/polylactic acid (PLA), PS/poly(vinylidene fluoride) (PVDF), and so on) were proposed to investigate foaming behavior. The idea of fabricating polymer blends is applied to combine the properties of their phases in a unique product, and therefore to improve foamability of the polymer. Properties of polymer blends are directly related to the quality of their morphology, such as cell density, cell size, and size distribution. Until now, the effect of a blending system on foaming behavior was mainly focused on two aspects: (1) generating numerous homogeneous interfaces to improve nucleation efficiency during the initial foaming process. Since interfacial tension is high in the polymer blend interfaces, it could reduce the Gibbs free energy, nucleation energy barrier according to classical nucleation theory, and that should facilitate the foaming process. For example, Zhai et al. prepared a series of polypropylene (PP)/polystyrene (PS) blends with PS-grafted PP copolymers (PP-g-PS) as compatibilizers and it was found that the phase size decreased and more homogeneous interfaces were obtained with PP-g-PS added. This finally resulted in a PP/PS blend foam with a higher expansion ratio and superior cell morphology.27 (2) Increasing CO2 solubility in PS by adding a CO2-philic polymer to increase foamability of the PS foam. For example, Ruiz et al. blended PS and PMMA with triblock polymer (MAM). They found that the CO2-philic PMMA had a much higher CO2 solubility than PS; PMMA had about 15% CO2 uptake and the PS only 7.5% CO2 uptake when both reached equilibrium at 300 bar and 40 °C. The added PMMA phase acted as a CO2 reservoir and the poor affinity of PS was ameliorated.30 However, no matter what the polymer blend systems are, extra compatibilizers and/or chemical synthesis methods are used to improve the phase morphologies for superior cellular morphologies. This is because a majority of polymers are immiscible and if only two polymers are blended it will result in inferior phase morphologies.31,32
In this study, we prepared PS/poly(ethylene-co-1,4-cyclohexanedimethanol terephthalate) (PETG) blends using an internal mixer, without adding any additives or disposing of chemicals. The aim of adding PETG to a PS matrix was also to improve its foamability, which favors the polymer foaming process. It was found that the PETG phase was homogeneous with a fine degree of dispersion in the PS matrix, although the PS/PETG blends were immiscible. Prior to our study, research on PETG was rarely reported in the literature, especially the PETG foaming process.33,34 Compared with large-scale PET, PETG has the favorable mechanical properties of PET, but the viscous flow temperature of PETG is no more than 130 °C and this feature makes PETG easier to process.35–37 Furthermore, PETG is an amorphous polymer and has a high affinity for CO2. It was reported that almost 15 wt% CO2 could be dissolved in PETG at 35 °C and 6.0 MPa.38,39 The induced PETG phase not only formed stable interfaces in the PS/PETG blends, which facilitated the foaming nucleation, but also improved CO2 solubility in the polymer blends. Compared with PS foam, the foams of PS/PETG blends had a higher expansion ratio and more irregular cellular structure. However, the open-cellular phenomenon was easily observed when the fraction of PETG in PS/PETG blends was higher than 20 wt%.
2. Experimental
2.1 Materials
PS (158K) was purchased from BASF (Nanjing) Industry Co., Ltd., with a melt flow rate (200 °C/5.0 kg) about 3.00 g/10 min according to ASTM D1238. PETG (S2008) with number-averaged molecular weight (Mn) of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, intrinsic viscosity of 0.78 dl g−1, and glass-transition temperature (Tg) of 80 °C was supplied by SK chemical corporation (Seoul, South Korea). The molecular structure of PETG is presented in Fig. 1.
000 g mol−1, intrinsic viscosity of 0.78 dl g−1, and glass-transition temperature (Tg) of 80 °C was supplied by SK chemical corporation (Seoul, South Korea). The molecular structure of PETG is presented in Fig. 1.
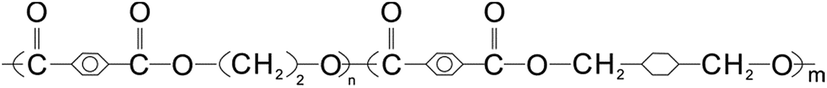 | ||
| Fig. 1 The molecular structure of PETG. | ||
2.2 Preparation of PS/PETG blend
The PS and PS/PETG blends were prepared in a Haake internal mixer at 190 °C, with mixing time of 8 min and mixing speed of 40 rpm. Prior to melt mixing, PETG was dried at 60 °C for 4 hours to remove excess moisture. Afterwards, the obtained samples were processed into sheets (100 × 100 × 2 mm3) by preforming for subsequent characterizations and batch foaming. The formulations are listed in Table 1.| Samples | PS | PETG |
|---|---|---|
| G0 | 100 | 0 |
| G10 | 90 | 10 |
| G15 | 85 | 15 |
| G20 | 80 | 20 |
| G25 | 75 | 25 |
2.3 Preparation of PS and PS/PETG blending foams
The PS and PS/PETG blending foams were prepared through a batch foaming process using CO2 as the physical blowing agent. For the foaming process, samples were first put into an autoclave at 170 °C and a pressure of 13 MPa for 4 h. After saturating at the saturation pressure and foaming temperature, the pressure was dropped to ambient pressure by releasing the CO2 which provided the driving force to induce cell nucleation and growth. The depressurization process was finished within 8 s.2.4 Characterization
δ), and complex melt viscosity (η*) were measured at various frequencies.
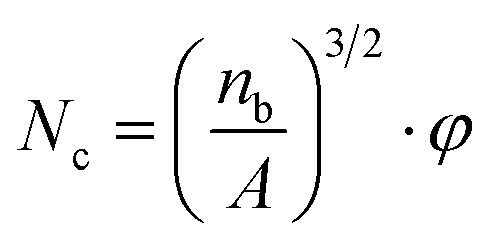 | (1) |
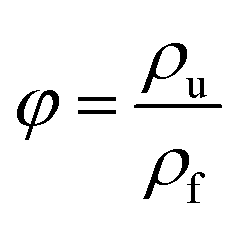 | (2) |
The ρu and ρf are the bulk densities of the unfoamed samples and the foamed ones, respectively. They were measured using a density balance (Sartorius RDK-01).
Furthermore, particle density of PETG in PS/PEGT blends was also calculated according to formula (1). The value of φ was regarded as 1.
3. Results and discussion
3.1 The PS/PETG blend morphologies
Fig. 2 shows the SEM micrographs of G10, G15, G20, and G25 blends, respectively. To give an integral description of the blend morphology, magnifications of 3000× and 12000× were used. In Fig. 2(a1)–(d1), countless sea-island structures are observed, in which the disperse phase is PETG and the matrix is PS. With a magnification of 12,000×, the PETG particles are distinctly presented in Fig. 2(a2)–(d2). It is obvious that the PETG mostly presents as a spherical shape and the domain of PETG increases with increasing PETG content. Meanwhile, with the samples from G10 to G25, the size of spherical PETG particles doesn't change but the density of the dispersed particles increases.
 | ||
| Fig. 2 SEM photographs of PS/PETG blends with different PETG contents. (a) 90/10; (b) 85/15; (c) 80/20; (d) 75/25, in which the 1 represents 3000× and 2 represents 12000×, respectively. | ||
The average dispersion size of a PETG particle and the dispersion number were obtained through the analysis of the above SEM photographs by Image-Pro Plus. And the dispersion density of PETG particles was calculated according to formula (1) and presented in Table 1. From Table 1, the average dispersion size of PETG particles all maintained around 0.8 μm and the dispersion density increased from 2.26 × 1011 cells per cm3 to 6.57 × 1011 cells per cm3, with PETG content from 10 wt% to 25 wt%. The PS/PETG blend is immiscible because of poor interfacial adhesion according to our previously published paper.18 It also can be observed that, from Fig. 2(a2)–(d2), the boundary between the PETG domain and PS matrix is clear. This infers that the interfacial tension between PS and PETG is high, which would be beneficial to the foaming nucleation stage because of a low nucleation barrier.41 Cell nucleation in the initial foaming stage was significantly influenced by phase morphology (the size, the size distribution, and the shape of the dispersed phase in the matrix) and the interfacial area.42 The larger interface domain of a PS/PETG blend would lead to more nucleation sites for bubble formation with micro-level PETG dispersion. Furthermore, the nucleation process of PS/PETG blend foaming could be tuned by changing the ratio, based on the stable dispersed particles in a PS matrix. Before our study, there were also many researchers studying the morphology of polymer blends and morphological photographs are presented in their literature.43,44 Most of their results showed that the size of dispersed droplets became larger with a higher disperse content since the phenomenon of coalescence happened more easily (Table 2).
| Samples | G10 | G15 | G20 | G25 |
|---|---|---|---|---|
| Dispersion average size (μm) | 0.80 | 0.79 | 0.77 | 0.80 |
| Dispersion density (cm−3) | 2.26 × 1011 | 3.82 × 1011 | 5.17 × 1011 | 6.57 × 1011 |
N. Tokita proposed some theoretical considerations about polymer blending processes, based on either an internal mixer or a mill, in his literature.45 Basically, in a given stress field, the processes of breaking down and coalescence happen simultaneously.
K1 = Ė/EDb = Ė/(EDk + EDf) = η![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) 2/(EDk + 3σ/R) 2/(EDk + 3σ/R)
| (3) |
is a shear rate, σ is the interfacial tension, and R is the radius of the spherical particles.|
K2 = (4/π)pΦD
| (4) |
At equilibrium, when the rates of breaking-down and coalescence are balanced,
| K1 − K2 = 0 | (5) |
Combining with eqn (3)–(5), the equilibrium particles size (R*) can be obtained and expressed approximately as following:
|
R* ≈ [12pσΦD/(πη)][1 + 4pΦDEDk/(πη)]
| (6) |
From the above equation, the equilibrium particle size could be finally determined by many factors which are closely related to the intrinsic properties of a polymer. Eqn (8) indicates that the R* becomes smaller when (1) the apparent stress field increases (mainly due to the matrix viscosity); (2) EDk becomes smaller (the rate of breaking-down increases); (3) σ decreases (more compatibility between matrix and dispersed phase) and (d) the ΦD is smaller. Therefore, the final value of R* is an aggregate result of the above factors. Generally, the size of the dispersed phase increases as a function of increasing concentration of disperses content. However, in the PS/PETG blend system, the size of the dispersed PETG phase maintained an unchanged dispersion size, although the added PETG content was from 10 wt% to 25 wt%. This mainly depends on the intrinsic properties of polymer blends, PS and PETG, and will be theoretically studied in a later research project. In this paper, the specific morphology of PS/PETG blends offers stable interfaces for the subsequent foaming process. Moreover, the dispersion density or interface density could be tuned by the added PETG content and thus effect the foaming process.
3.2 The rheological properties of PS and PS/PETG blends
Dynamic shear rheological testing was carried out to study the rheological properties of PS and PS/PETG blends. Shear rheological behavior was relative and sensitive to the topological structure and blend ratio. Fig. 3 shows the curves of the storage modulus (G′), loss modulus (G′′), loss factor (tanδ), and complex melt viscosity (η*) at different shear rates when the temperature was set at 190 °C. All G′–ω curves exhibited a similar trend, that storage modulus increased with increasing frequency. The improvement of melt elasticity could be reflected by an increase in storage modulus. From Fig. 3(a), it is seen that the PS/PETG blends have a higher storage modulus at low frequency than PS, implying that the melt elasticity of PS/PETG blends could be improved by adding PETG and that the PS/PETG blends had a longer relaxing time. This was due to the immiscible PS/PETG blends, which could be attributed to a relaxation process of the dispersed droplets of minor phase when slightly deformed, and this phenomenon was also reported in other PS/SAN and PS/PMMA blends.46,47 In the low frequency region, the storage modulus of PS/PETG blends slightly increased with PETG content, but the improved storage modulus made little difference to foaming behavior.
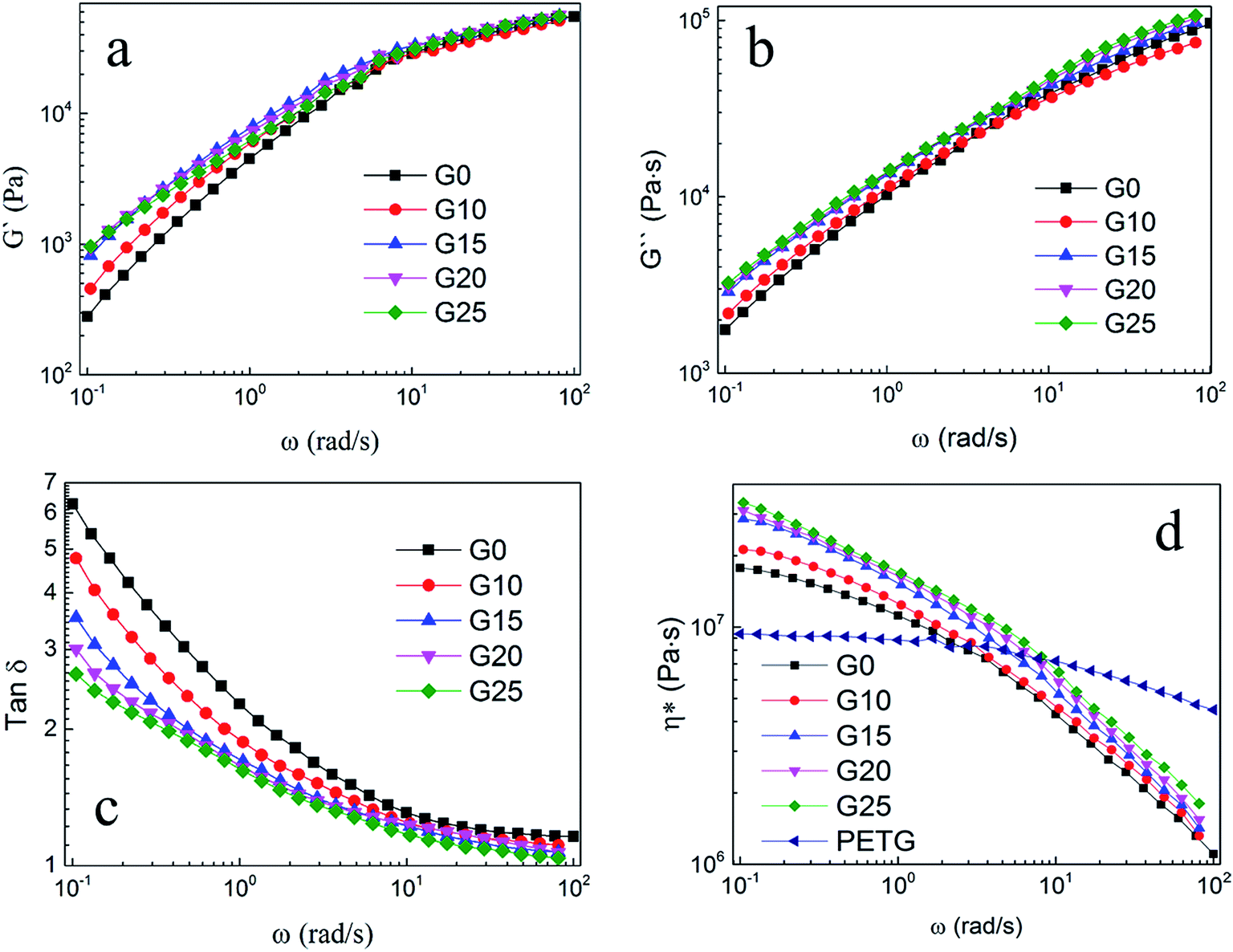 | ||
| Fig. 3 Dynamic shear rheological properties of PS and PS/PETG blends with various PETG contents. (a) G′–ω, (b) G′′–ω, (c) tanδ–ω, (d) η*–ω. | ||
Furthermore, Fig. 3(b)–(d) show the relationships of G′′–ω, tanδ–ω and η*–ω, respectively. The values of G′′ and η* also gradually increased with increasing PETG content, but the tanδ decreased. These results also could be associated with the dispersed droplets of the minor phase. In Fig. 3(d), the complex viscosity of PETG was much lower than that of PS and PS/PETG blends because the viscous flow temperature of PETG is no more than 130 °C.
3.3 The PS, PETG, and PS/PETG blend molten strength
Melt elongational flow properties of PS, PETG, and PS/PETG blends can be characterized by a Rheotens elongational rheometer and the melt strength can be measured by a rheological test. The polymer melt was extruded through a round die with 190 °C, and the resulting strand was carried off at 20 mm s−1. Fig. 4 exhibits the melt strength of PS, PETG, and PS/PETG blends. The curve of neat PS (G0) is higher than the other curves and the PS melt ruptured when the force approximately reached 0.139 N. Conversely, melt strength of the PETG melt was the lowest and the melt ruptured at almost 0.06 N. Fig. 3(d) shows that the complex viscosity of PETG was much lower than that of PS, owing to its low viscous flow temperature. When the testing temperature was 190 °C, the PETG melt was much weaker.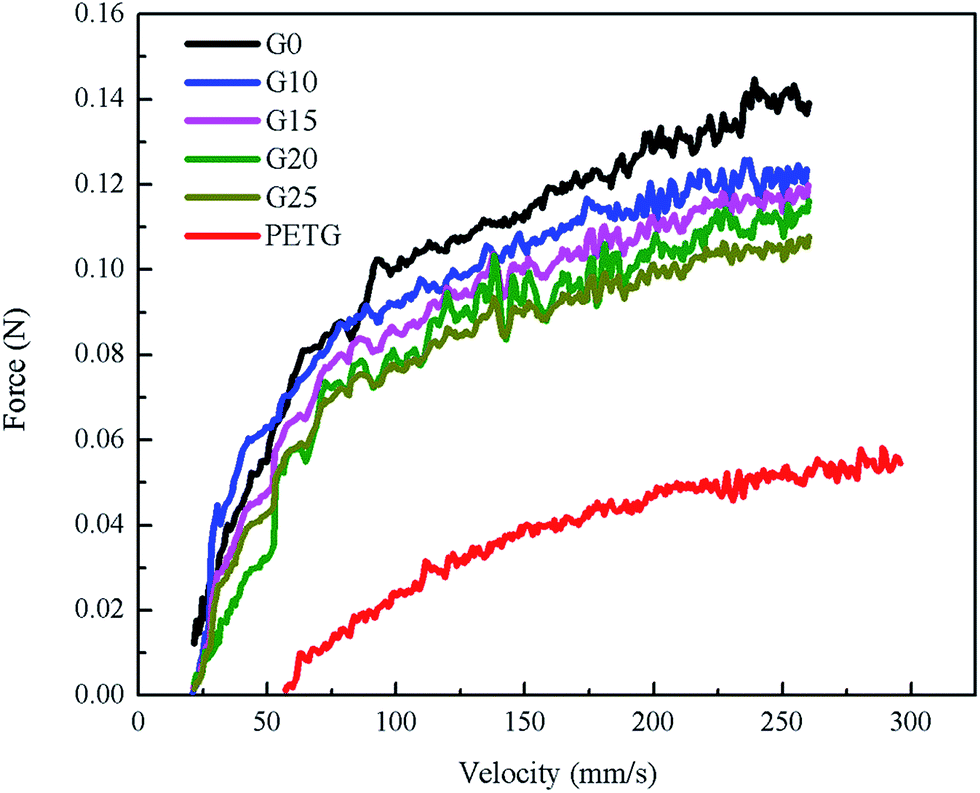 | ||
| Fig. 4 The melt strength of PS, PETG, and PS/PETG blends with different PETG content. | ||
From Fig. 4, the melt strength of PS/PETG blends consistently decreased with increasing PETG content, mainly because of the negative effect caused by the PETG phase. The added PETG in a PS matrix would decrease the melt strength of all the blends. In the PS/PETG blending system, the more PETG that was added, the smaller was the melt strength of the PS/PETG blend that was obtained. When the PETG content added increased to 25 wt%, the melt strength reached the lowest level among the PS/PETG blends, which was about 0.109 N.
3.4 The foaming morphologies of PS and PS/PETG blends
In classical nucleation theory, cell nucleation contains both homogeneous nucleation and heterogeneous nucleation. Heterogeneous nucleation represents critical pores formed on the surfaces of some additives (inorganic fillers or a second polymer phase) in a polymeric matrix, and the activation energy of heterogeneous nucleation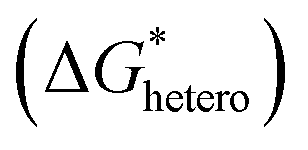 is much lower than that of homogeneous nucleation
is much lower than that of homogeneous nucleation 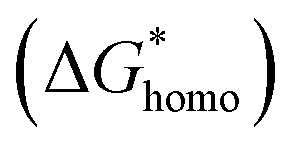 . According to research of Colton and Suh,48 the
. According to research of Colton and Suh,48 the 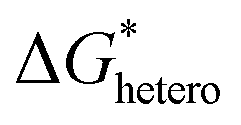 was derived from
was derived from 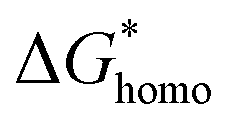 by the heterogeneity factor, as shown in the following formulas:
by the heterogeneity factor, as shown in the following formulas:
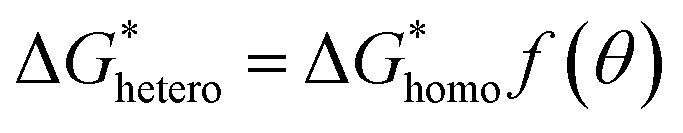 | (7) |
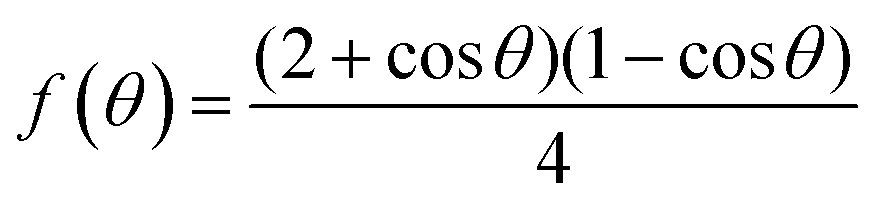 | (8) |
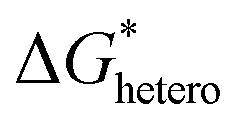 has a smaller value than that of
has a smaller value than that of 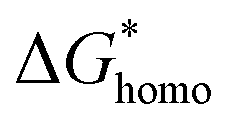 , because the value of f(θ) is no more than 1. Cell nucleation would prefer to happen in the interfaces because of the smaller nucleation barrier. With a higher PETG content, more stable interfaces formed, which favored cell nucleation. However, a bubble collapse phenomenon was observed when the PETG ratio increased to 20 wt% and became even more severe when the PETG was increased to 25 wt%. Note a collapsed bubble in Fig. 5(e) was magnified and presented in Fig. 5(f). It was found that many particles stuck to the cell walls, which inferred that would be the PETG phase because the particles had similar shapes and sizes of unfoamed PS/PETG blends, as seen in Fig. 2(d2) and 5(f). The PETG phase did not foam and maintained its initial status during the foaming process; this phenomenon has also been reported in other literature.49 Foam growth mostly happened in the PS phase and the PETG phase encased with the PS matrix. When too many cells formed with added high PETG content, the cell walls became much thinner and the PETG phase would not be encased in the cell walls. Cell collapse would easily take place in the PETG position because the PETG melt was hard to resist the drawing force during the bubble growing stage. Therefore, the PETG phase would contribute to the foaming process because it had a higher CO2 solution than the PS melt and tended to permeate into the PS melt phase which then could promote bubble growth of the PS phase.
, because the value of f(θ) is no more than 1. Cell nucleation would prefer to happen in the interfaces because of the smaller nucleation barrier. With a higher PETG content, more stable interfaces formed, which favored cell nucleation. However, a bubble collapse phenomenon was observed when the PETG ratio increased to 20 wt% and became even more severe when the PETG was increased to 25 wt%. Note a collapsed bubble in Fig. 5(e) was magnified and presented in Fig. 5(f). It was found that many particles stuck to the cell walls, which inferred that would be the PETG phase because the particles had similar shapes and sizes of unfoamed PS/PETG blends, as seen in Fig. 2(d2) and 5(f). The PETG phase did not foam and maintained its initial status during the foaming process; this phenomenon has also been reported in other literature.49 Foam growth mostly happened in the PS phase and the PETG phase encased with the PS matrix. When too many cells formed with added high PETG content, the cell walls became much thinner and the PETG phase would not be encased in the cell walls. Cell collapse would easily take place in the PETG position because the PETG melt was hard to resist the drawing force during the bubble growing stage. Therefore, the PETG phase would contribute to the foaming process because it had a higher CO2 solution than the PS melt and tended to permeate into the PS melt phase which then could promote bubble growth of the PS phase.
 | ||
| Fig. 5 The cellular morphology of (a) PS, (b) PS/PETG (90/10), (c) PS/PETG (85/15), (d) PS/PETG (80/20), (e) PS/PETG (75/25), and (f) PS/PETG (75/25). Note that (a) to (e) represent 3000× and (f) represents 12000×, respectively. | ||
Foam parameters like foam density, foam expansion ratio, and cell size distribution were obtained by using a software image tool and calculation with eqn (1) and (2). The results are presented in Fig. 6 and Table 3. According to Fig. 6, the size range of bubbles in PS (nearly from 100 μm to 500 μm) is much wider than that of PS/PETG blends. This is attributed to the homogeneous nucleation type in the PS matrix. In PS/PETG blends, the existing interfaces lead to much more nucleus formation and finally resulted in smaller bubbles. From Fig. 6(b)–(d), the overall cell sizes became smaller with increasing PETG ratio. The larger the PETG ratio was, the more interfaces between PS and PETG there were; the more interfaces that existed, the more foaming nucleation happened. Furthermore, stable interfaces would lead to a uniform cell size with a narrow cell size distribution. However, when the PETG ratio increased from 20 wt% to 25 wt%, the tendency of the whole bubble size moved to a higher scale, as seen in Fig. 6(d) and (e). This was due to a more severe bubble collapse and it made a difference to the cell morphology, as shown in Fig. 5(e).
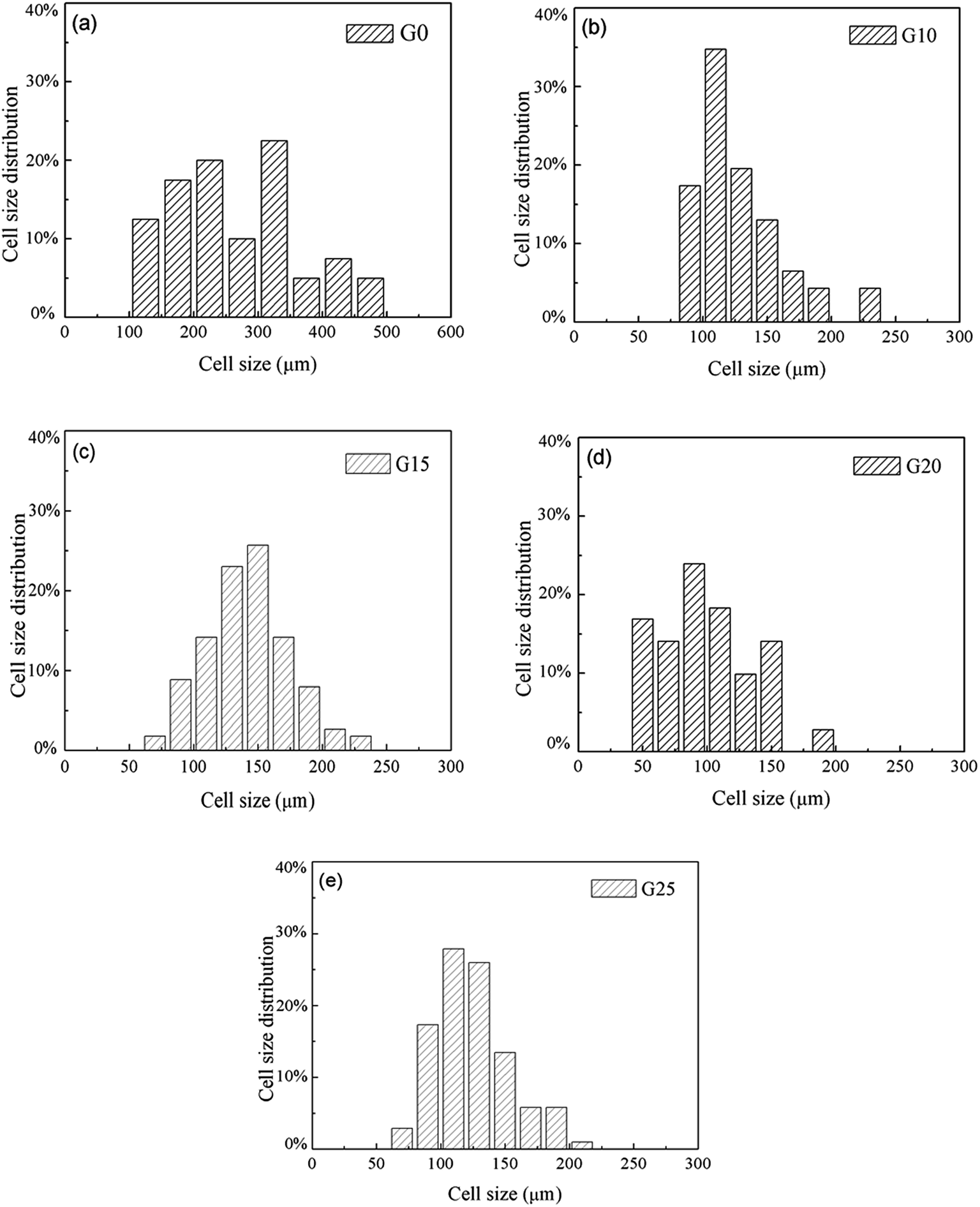 | ||
| Fig. 6 The cell size distribution of (a) PS, (b) PS/PETG (90/10), (c) PS/PETG (85/15), (d) PS/PETG (80/20), and (e) PS/PETG (75/25). | ||
| Samples | Foam density (g cm−3) | Expansion ratio | Mean cell size (μm) | Cell density (cells per cm3) |
|---|---|---|---|---|
| G0 | 0.082 | 12.7 | 275 | 1.23 × 106 |
| G10 | 0.062 | 16.9 | 158 | 6.95 × 106 |
| G15 | 0.055 | 19.2 | 142 | 1.24 × 107 |
| G20 | 0.046 | 22.7 | 100 | 1.89 × 107 |
| G25 | 0.051 | 20.6 | 123 | 1.47 × 107 |
From Table 3, detailed information about the foams was represented. G0 had the highest foam density and the smallest expansion ratio corresponding to the values of 0.082 g cm−3 and 12.7, respectively. For the foams in PS/PETG blends, the foam density decreased and expansion ratio increased, compared with PS foam. This could be explained by the following reasons. On one hand, the interfaces between PS and PETG facilitated nucleation and promoted cell formation. On the other hand, the PETG had a high CO2 concentration and this would improve the CO2 concentration of all the melts in the PS/PETG blends. The dissolved CO2 in the PETG phase would permeate into the PS phase and thus accelerate the PS phase foam. When PETG was added, it not only improved the foam expansion ratio but also increased cell density from smaller cell sizes. Furthermore, with a PETG content from 10 wt% to 20 wt%, the foam expansion ratio and cell density gradually increased. The foam expansion ratio and cell morphology thus could be tuned by the addition of PETG. The foam had the lowest foam density and largest expansion ratio, where the average cell size and cell density were 100 μm and 1.89 × 107 cells per cm3, when the PETG content was 20 wt%. When the PETG content further increased to 25 wt%, the foam expansion ratio decreased and the cell size increased. This could be explained by a severe bubble collapse occurring when the added PETG content was too high. We concluded that too much PETG addition would not further improve the foamability of PS/PETG blends.
3.5 Mechanism of the PS/PETG blends foaming
In this paper, our aim was to investigate the foaming process of PS/PETG blends. Usually, this process involves four stages:50 (1) the immersing stage, (2) cell nucleation, (3) cell growth stage, and (4) cell stabilization. In this work, the foaming process was illustrated and presented by the following schematic diagram.As shown in Fig. 7(a), the polymer had been immersed in the autoclave for 4 h and CO2 was diffused into the PS/PETG blends melt in this stage. The CO2 has a high diffusion coefficient and low solubility in the PS melt in comparison with that of traditional blowing agents. Consequently, a PS foam with low density is difficult to obtain. However, PETG has a high affinity for CO2 and thus addition of PETG could increase whole CO2 solution during the gas dissolution stage. The ability of CO2 dissolution improves with increasing PETG content.
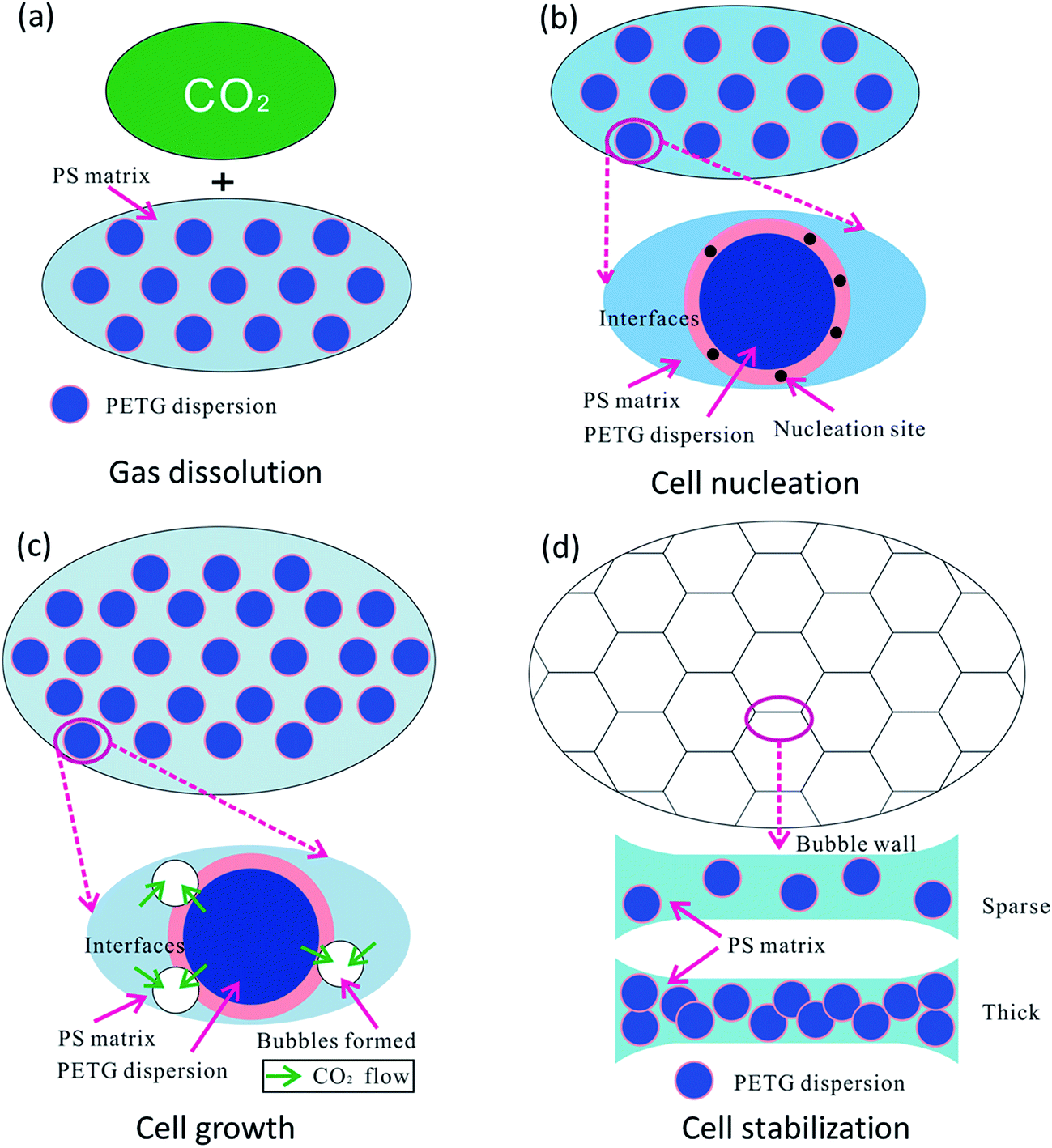 | ||
| Fig. 7 Schematic diagram of cell formation in polymer blends, with (a) gas dissolution, (b) cell nucleation, (c) cell growth, and (d) cell stabilization. | ||
Sudden thermal instability, mainly by releasing the pressure and/or increasing temperature, could lead to bubble nucleation in PS/PETG blends, as shown in Fig. 7(b). From Fig. 2, the obvious interfaces observed indicated that interface tension is high in PS/PETG blends and this makes the barrier energy decrease according to formulas (7) and (8). Bubble nucleation would easily happen and mostly concentrate on the interfaces between PS and PETG. In the PS/PETG blends, the PETG phase was homogeneous and had a fine degree of dispersion in the PS matrix. This offered a foaming process with abundant nucleation sites. Moreover, with increased PETG content, the size of PETG dispersion in the PS matrix maintained an unchanged scale with proportional increasing of PETG dispersion density. This was a different condition in comparison with most polymer blending systems. In the PS/PETG blends, the cell nucleation sites could be tuned to a certain extent by the addition of PETG.
Once cell nucleation was completed, the gas would diffuse into the spherical tiny cells and this made them expand, as shown in Fig. 7(c). The main driving force contributed to super-saturation which was a consequence of releasing pressure and/or increasing temperature. During the cell growth process in a polymer blending system, the cell was prone to grow on the polymer melt with lower viscosity. It seemed that the cell would grow on the PETG phase, compared with the higher-melt-viscosity PS phase. However, the melt strength of PETG was too small to support cell formation, seen in Fig. 4, and the PETG melt could not expand during the cell growth stage, as seen in Fig. 5(f). The PS phase would support cell growth and the CO2 dissolved in PS and PETG melt prompted cells grow larger and larger. When a larger PETG content was added, a higher CO2 solution in PS/PETG blends was obtained. A great degree of super-saturation would form during the cell growth stage and a larger foam expansion ratio was obtained. Nevertheless, the low-melt-strength PETG phase would affect the strength of a cell wall. Thick PETG particles dispersed in the PS phase resulted in a fragile bubble wall and then bubble coalescence and rupture would easily occur.
As a consequence, the degree of super-saturation dramatically decreased and slowly reached a balanced state, as presented in Fig. 7(d). The bubbles solidified and final foam morphologies formed. The PS/PETG blend foams had higher expansion ratios than that of PS. Moreover, smaller cell size and higher cell density were obtained in PS/PETG blend foams. In PS/PETG blend foams, sparse PETG particles dispersed in the PS phase had strong cell walls and a closed-cell morphology was presented, as seen in Fig. 5(b) and (c). Conversely, obvious open-cell morphology was observed in the PS/PETG blend foams which had higher PETG content additions, as seen in Fig. 5(d) and (e). Therefore, with CO2 as a blowing agent, the foamability of PS was highly improved and the cell morphology could also be tuned, to a certain degree, by the addition of PETG.
4. Conclusion
In this paper, the morphologies of PS/PETG blends were initially investigated. It was found that the interfaces between PS and PETG were clearly observed since PS and PETG were immiscible. The PETG phase dispersed homogeneously in the PS matrix and maintained an almost unchanged scale with additional content increasing from 10 wt% to 25 wt%. Afterwards, the rheology of PS/PETG blends was characterized and the values were little changed in comparison with PS. Furthermore, the melt strengths of PS, PETG, and PS/PETG blends were also measured. Results showed that PS had a much higher melt strength than that of PETG and the addition of PETG in the PS matrix would weaken the melt strength of all the blends. Finally, the foamability of PS and PS/PETG blends was investigated and the mechanism was discussed. During the foaming process, addition of the PETG phase not only offered the many interfaces, which facilitated foaming nucleation, but also increased the CO2 solution concentration for all the blends. This resulted in a foam with higher expansion ratio and uniform cell structure in comparison with that of PS. The foam morphology and expansion ratio could be tuned by the added PETG content. However, the PETG content should be controlled to under a certain degree, or a serious open-cell phenomenon will happen.Acknowledgements
Financial supports from Young Backbone Project of Beijing Academy of Science and Technology and Innovative Team Project of Beijing Academy of Science and Technology are gratefully acknowledged.References
- K. Wang, Y. Pang, F. Wu, W. Zhai and W. Zheng, J. Supercrit. Fluids, 2016, 110, 65 CrossRef CAS.
- M. Sauceau, J. Fages, A. Common, C. Nikitine and E. Rodier, Prog. Polym. Sci., 2011, 36, 749 CrossRef CAS.
- C. Zhang, B. Zhu, D. Li and L. Lee, Polymer, 2012, 53, 2435 CrossRef CAS.
- C. Vo, F. Bunge, J. Duffy and L. Hood, Cell. Polym., 2011, 3, 137 Search PubMed.
- Y. Long, F. Sun, C. Liu and X. Xie, RSC Adv., 2016, 6, 23726 RSC.
- J. Yang, Y. Sang, F. Chen, Z. Fei and M. Zhong, J. Supercrit. Fluids, 2012, 62, 197 CrossRef CAS.
- C. Cutierrez, J. Rodriguez, I. Gracia, A. Lucas and M. Garcia, J. Supercrit. Fluids, 2013, 76, 126 CrossRef.
- R. Sharudin and M. Ohshima, J. Appl. Polym. Sci., 2013, 38104, 2245 CrossRef.
- Y. Wang, Y. Zhang, Y. Liu, L. Zhang, S. Ren, J. Lu, X. Wang and N. Fan, J. Pet. Sci. Eng., 2017, 154, 234 CrossRef CAS.
- J. Reglero, P. Viot and M. Dumon, J. Cell. Plast., 2011, 47, 535 CrossRef CAS.
- X. Liao, H. Zhang, Y. Wang, L. Wu and G. Li, RSC Adv., 2014, 4, 45109 RSC.
- L. Chen, D. Rende, L. Schadler and R. Ozisik, J. Mater. Chem. A, 2013, 1, 3837 CAS.
- D. Tomasko, A. Burley, L. Feng, S. Yeh, K. Miyazono, S. Kumar, I. Kusaka and K. Koelling, J. Supercrit. Fluids, 2009, 47, 493 CrossRef CAS.
- R. Gendron and M. Champagne, J. Cell. Plast., 2006, 42, 127 CrossRef.
- K. Arora, A. Lesser and T. Mccarthy, Macromolecules, 1998, 31, 4614 CrossRef CAS.
- J. Yang, L. Huang, Y. Zhou, Z. Chen and M. Zhong, J. Supercrit. Fluids, 2013, 82, 13 CrossRef CAS.
- A. Jose, M. Jean, P. Matthieu and D. Michel, J. Supercrit. Fluids, 2011, 57, 87 CrossRef.
- X. Wang, W. Wang, B. Liu, Z. Du and X. Peng, J. Cell. Plast., 2016, 52, 595 CrossRef.
- K. Wang, Y. Pang, F. Wu, W. Zhai and W. Zheng, J. Supercrit. Fluids, 2016, 110, 65 CrossRef CAS.
- B. Zhu, W. Zha, J. Yang, C. Zhang and L. Lee, Polymer, 2010, 51, 2177 CrossRef CAS.
- J. Pinto, M. Dumon and M. Rodriguez-Perez, Recent Developments in Polymer Macro, Micro and Nano Blends, Elsevier, Duxford, 2017, vol. 9, pp. 237–288 Search PubMed.
- R. Sharudin and M. Ohshima, J. Appl. Polym. Sci., 2013, 128, 2245 CrossRef CAS.
- D. Kohlhoff, A. Nabil and M. Ohshima, Polym. Adv. Technol., 2012, 23, 1350 CrossRef CAS.
- G. Zhong, K. Wang, L. Zhang, Z. Li, H. Fong and L. Zhu, Polymer, 2011, 52, 5397 CrossRef CAS.
- R. Liao, W. Yu and C. Zhou, Polymer, 2010, 51, 568 CrossRef CAS.
- S. Lee, J. Cell. Plast., 2010, 46, 321 CrossRef CAS.
- W. Zhai, H. Wang, J. Yu, J. Dong and J. He, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1641 CrossRef CAS.
- T. Otsuka, K. Taki and M. Ohshima, Macromol. Mater. Eng., 2008, 293, 78 CrossRef CAS.
- K. Taki, K. Nitta, S. Kihara and M. Ohshima, J. Appl. Polym. Sci., 2005, 97, 1899 CrossRef CAS.
- J. Ruiz, M. Pedros, J. Tallon and M. Dumon, J. Supercrit. Fluids, 2011, 58, 168 CrossRef CAS.
- C. Yu, D. Shi, J. Wang, H. Shi, T. Jiang, Y. Yang, G. Hu and R. Li, Mater. Des., 2016, 107, 171 CrossRef CAS.
- M. Luna and G. Filippone, Eur. Polym. J., 2016, 79, 198 CrossRef.
- C. Fan, C. Wan, F. Gao, C. Huang, Z. Xi, Z. Xu, L. Zhao and T. Liu, J. Cell. Plast., 2015, 52, 277 CrossRef.
- R. Dupaix and M. Boyce, Polymer, 2005, 46, 4827 CrossRef CAS.
- W. Jiang, R. Bao, W. Yang, Z. Liu, B. Xie and M. Yang, Mater. Des., 2014, 59, 524 CrossRef CAS.
- X. Zhang, B. Li, K. Wang, Q. Zhang and Q. Fu, Polymer, 2009, 50, 4737 CrossRef CAS.
- D. Saheb and J. Jog, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2439 CrossRef CAS.
- A. Rizvi, A. Tabatabaei, M. Barzegari, S. Mahmood and C. Park, Polymer, 2013, 54, 4645 CrossRef CAS.
- Y. Handa, B. Wong and Z. Zhang, Polym. Eng. Sci., 1999, 39, 55–61 Search PubMed.
- P. Guo, Y. Xu, M. Lu and S. Zhang, Ind. Eng. Chem. Res., 2015, 54, 217 CrossRef CAS.
- W. Kong, J. Bao, J. Wang, G. Hu, Y. Xu and L. Zhao, Polymer, 2016, 90, 331 CrossRef CAS.
- H. Huang and H. Xu, Polym. Adv. Technol., 2011, 22, 822 CrossRef CAS.
- X. Wang, W. Liu, H. Zhou, B. Liu, H. Li, Z. Du and C. Zhang, Polymer, 2013, 54, 5839 CrossRef CAS.
- S. Patricia, Y. Marcio and R. Nicole, Polymer, 2005, 46, 2610 CrossRef.
- N. Tokita, Rubber Chem. Technol., 1977, 50, 292 CrossRef CAS.
- Y. Sung, M. Han, J. Hyun, W. Kim and H. Lee, Polymer, 2003, 44, 1681 CrossRef CAS.
- P. Macaubas and N. Demarquette, Polymer, 2001, 42, 2543 CrossRef CAS.
- J. Colton and N. Suh, Polym. Eng. Sci., 1997, 27, 485 Search PubMed.
- P. Gong and M. Ohshima, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1173 CrossRef CAS.
- M. Maiti and R. Jasra, Ind. Eng. Chem. Res., 2012, 51, 10607 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |