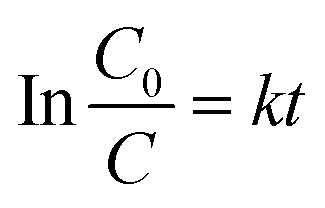DOI:
10.1039/C7RA07441G
(Paper)
RSC Adv., 2017,
7, 42774-42782
Rational fabrication of a graphitic-C3N4/Sr2KNb5O15 nanorod composite with enhanced visible-light photoactivity for degradation of methylene blue and hydrogen production†
Received
6th July 2017
, Accepted 31st August 2017
First published on 5th September 2017
Abstract
A g-C3N4/Sr2KNb5O15 nanorod composite photocatalyst was simply prepared by direct growth of graphitic C3N4 on one-dimensional Sr2KNb5O15 nanorods and evaluated by degradation of methylene blue (MB) and water splitting for H2 production under visible light irradiation. By coupling g-C3N4 and Sr2KNb5O15 nanorods, the nanocomposite with an optimal 77 wt% g-C3N4 exhibited 6.2 times higher activity for photodegradation than the bare g-C3N4 and the increase in H2 production rate from an aqueous methanolic solution reached up to 12.1 fold after appropriate photodeposition of 0.03 wt% Rh cocatalysts. Furthermore, a comparative study on the preparation methods shows the photodegradation rate of the nanocomposite prepared by the direct growth method was found to be 3.4 times higher than those formed just by physical mixing. The results demonstrate the importance of the formation of proper nano-interfaces in the nanocomposite. Combined with theoretical prediction of the band structures, a possible mechanism is thus proposed including the formation of proper interfaces between g-C3N4 and Sr2KNb5O15 by direct growth approachs promoting spatial separation of photoinduced electron–hole pairs.
1. Introduction
The search for efficient heterogeneous photocatalysts has attracted considerable interest over the past decades due to their potential applications in environmental clean-up and solar energy conversion.1–3 Among the various semiconductor-based photocatalytic materials, titania (TiO2) is recognized as the most promising candidate for eliminating organic pollutants because of its low cost, high abundance in the Earth's crust, nontoxicity, and chemical stability.4,5 However, the low quantum efficiency of TiO2 photocatalysts, due to the absorption of only ultraviolet (UV) light (band gap of ∼3.2 eV), and fast recombination of photogenerated electron–hole pairs limits their practical application.
In recent years, doping of metal cations (W, V, Fe, and Cr, etc.)6–8 or nonmetal anions (C, N, B, S and F, etc.)9–13 was extensively researched in order to develop visible light-driven photocatalysts, but unfortunately it usually gives rise to accelerated charge recombination, and interruption of the surface structures.14,15 In order to boost solar energy efficiency, dye-sensitized solar cells have also been explored to enhance the light absorption in the visible region.16,17 Currently among all the strategies delicate construction of hetero- or homo-junction semiconductor photocatalyst composite structures is thought to be the most promising approach for extending light absorption and improving charge separation.18–20 Up to now, several semiconductor couples have been demonstrated to efficiently promote charge separation by the interfaces between the combined semiconductors, especially employing TiO2 as one part.21–27
As a metal-free semiconductor, polymeric graphitic carbon nitride (g-C3N4), firstly reported by Wang et al. in 2009; it can enable both half reactions of water splitting in the presence of sacrificial reagents under visible light irradiation.28,29 The small band gap (ca. 2.70 eV) capable of visible light absorption, a quite negative conduction band minimum (−1.12 eV vs. NHE) and the high thermal and chemical stability render g-C3N4 as one of the most promising candidates for the fabrication of heterostructured composites.30 By coupling of g-C3N4 with another narrow band-gap semiconductor, such as N–TiO2,31 N–H2Ta2O6,32 N–In2TiO5,33 TaON,34 BiPO4,35 Bi2WO6,36 and α-Fe2O3,37 etc., the formation of heterojunctions has been demonstrated to promote the separation of photogenerated charge carriers thus improving the photocatalytic efficiency.38,39 On the other hand, owing to the delocalized π-electrons from the conjugated π system of g-C3N4,40 the combination of g-C3N4 with wide band gap semiconductors has been also developed. Successful examples include TiO2,41,42 ZnO,43 titanate,14,44 and Zn2GeO4.45 In the previous work,46 we have reported that the tetragonal tungsten bronze-type Ta substituted Sr2KNb5O15 nanorod photocatalysts possess a unique tunnel structure with a built-in electric field along [001] direction and exhibit relatively high activity for photocatalytic water splitting under UV irradiation. The Sr2KNb5O15 with a minimum band gap of ∼3.2 eV shows a great potential as an alternative photocatalyst to TiO2. However, the challenge in photocatalytic efficiency still remains and more efficient photocatalytic structures need to be developed.19
Inspired by this, here we report, for the first time, a g-C3N4/Sr2KNb5O15 nanocomposite photocatalyst simply prepared by direct growth of g-C3N4 on Sr2KNb5O15 nanorods. Under visible-light irradiation, the photocatalytic properties were significantly enhanced by coupling of g-C3N4 with Sr2KNb5O15 for the degradation of methylene blue (MB) and photocatalytic hydrogen production. The highest photocatalytic activity was observed in the g-C3N4/Sr2KNb5O15 nanocomposite with optimum g-C3N4 content of 77 wt%. The proposed mechanism was associated with the role of g-C3N4 as a visible light absorber unit, and the function of the inner Sr2KNb5O15 nanorods as an electron separator, which is capable of adopting photoinduced electrons from the g-C3N4, i.e. the photoinduced electrons from the conduction band of g-C3N4 are injected into the Sr2KNb5O15, while the photoinduced holes remain in the g-C3N4, thus promoting spatial separation of photoinduced electron–hole pairs. Furthermore, the study of Rh metal as co-catalysts for improving photocatalytic H2 production points out that in the composite materials system the proper use of noble metal co-catalysts is an important issue. The resulting inhibition effect at high dosage of Rh may produce, affecting the interfacial charge transfer between g-C3N4 and Sr2KNb5O15.
2. Experimental
Synthesis of photocatalysts
All reagents used in this study were used without further purification. The Sr2KNb5O15 nanorods were prepared by molten salt method, as reported in our previous work.46 Stoichiometric amounts of precursors SrCO3 (>99.99%, Aldrich), K2CO3 (>99.9%, Alfa Aesar) and Nb2O5 (99.99%, Alfa Aesar) with potassium chloride (KCl: 99.5%, Honeywell Riedel-de Haën) at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were heated at 850 °C in air for 2 h. The g-C3N4 was prepared by directly heating melamine (C3H6N6, Alfa Aesar, >99%) to 520 °C for 4 h.47 The direct growth method was used for preparation of g-C3N4/Sr2KNb5O15 nanocomposite photocatalysts. Briefly, an appropriate weight ratio of melamine and Sr2KNb5O15 was added into ethanol. The bottle was placed in an ultrasonic bath for 30 min for complete dispersion and then stirred in a fume hood. After drying at 80 °C overnight, the resultant mixture was heated to 520 °C in a covered porcelain crucible for 4 h. According to this method, different weight ratios of g-C3N4/Sr2KNb5O15 samples with g-C3N4 contents of 24 wt%, 55 wt%, 77 wt% and 99 wt% were synthesized, and denoted as 24CNNb, 55CNNb, 77CNNb and 99CNNb, respectively. For comparison, a physical mixture of the pre-synthesized g-C3N4 and Sr2KNb5O15 was also prepared without heat treatment (denoted as M77CNNb).
:2 were heated at 850 °C in air for 2 h. The g-C3N4 was prepared by directly heating melamine (C3H6N6, Alfa Aesar, >99%) to 520 °C for 4 h.47 The direct growth method was used for preparation of g-C3N4/Sr2KNb5O15 nanocomposite photocatalysts. Briefly, an appropriate weight ratio of melamine and Sr2KNb5O15 was added into ethanol. The bottle was placed in an ultrasonic bath for 30 min for complete dispersion and then stirred in a fume hood. After drying at 80 °C overnight, the resultant mixture was heated to 520 °C in a covered porcelain crucible for 4 h. According to this method, different weight ratios of g-C3N4/Sr2KNb5O15 samples with g-C3N4 contents of 24 wt%, 55 wt%, 77 wt% and 99 wt% were synthesized, and denoted as 24CNNb, 55CNNb, 77CNNb and 99CNNb, respectively. For comparison, a physical mixture of the pre-synthesized g-C3N4 and Sr2KNb5O15 was also prepared without heat treatment (denoted as M77CNNb).
Characterization
The X-ray diffraction patterns of all samples were recorded with a PANalytical MPD diffractometer using Cu-Kα radiation (λ = 0.1541 nm), and the data were collected from 10° to 60° (2θ). Static N2 physisorption measurements were carried out at −196 °C using a Quantachrome Autosorb-1MP system. All samples were degassed at 200 °C for 6 h before the measurements. Thermogravimetric analyses were conducted with a TG/DSC NETZSCH STA 409 PC instrument from room temperature to 1000 °C at a heating rate of 5 °C min−1 under nitrogen flow of 300 mL min−1. UV-Vis diffuse reflectance spectra were measured using MgO as a reference on a Cary 4000 UV/Vis Varian spectrophotometer. Band gap energies were calculated by analysis of the Tauc-plots resulting from Kubelka–Munk transformation of diffuse reflectance spectra. The transmission electron microscopy and high-resolution transmission electron microscopy characterization of the materials were carried out using a Philips/FEI Tecnai F20 S-TWIN electron microscopy instrument operating at 200 kV. X-ray photoelectron spectroscopy (XPS) measurements were carried out in an ultra-high vacuum (UHV) set-up equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV), operated at 14.5 kV and 35 mA, and a high resolution Gammadata-Scienta SES 2002 analyzer. The base pressure in the measurement chamber was maintained at about 5 × 10−10 mbar. The measurements were carried out in the fixed transmission mode with pass energy of 200 eV resulting in an overall energy resolution of 0.25 eV. A flood gun was applied to compensate the charging effects. The Casa XPS software with a Gaussian–Lorentzian product function and Shirley background subtraction was used for peak deconvolution. To obtain a stable fit, single contributions of N 1s and C 1s signal deconvolution were constrained to have their full width at half maximum (FWHM) within the same range. The photoluminescence (PL) spectra were recorded at room temperature by using a Horiba LabRam HR spectrometer with the 325 nm line of He–Cd laser as excitation source.
Photocatalytic evaluation
Photodegradation of methylene blue. Methylene blue (MB) was adopted as the typically tested hazardous organic model pollutant to evaluate the photocatalytic performance of as-prepared samples. The photocatalytic reaction system consisted of a 300 W Xe arc lamp as a light source (Newport Corporation), a GG400 filter (providing the visible light of different wavelength, >400 nm) and a Schott KG3 IR filter (preventing from thermal catalytic effect). All experiments were conducted at room temperature in air. In a typical run, 50 mg of catalysts was added into 100 mL of 10.0 mg L−1 MB solution. Prior to irradiation, the suspension was magnetically stirred in the dark for 40 min to ensure the adsorption–desorption equilibrium of MB on the surface of the photocatalyst. Under irradiation approximately 4 mL suspension was collected at given time intervals and separated through a syringe driven filter unit (Millex, Millipore Corp., USA). The concentration change of MB was detected by measuring its characteristic absorption peak at 665 nm on a Cary 4000 UV/Vis Varian spectrophotometer, a Perkin-Elmer Lambda 650 UV-Vis spectrometer was employed during the recovery experiments.
Photocatalytic H2 production. The photocatalytic H2 production was conducted in an air-tight reactor and a 500 W mid-pressure Hg arc lamp equipped with a cooling water filter at 15 °C and a 420 nm cut-off filter was used as a visible light irradiation source. The evolved H2 was determined by gas chromatography (GC7900, Techcomp Ltd., Beijing, China) equipped with a MS-5A column and a thermal conductivity detector. High-purity N2 (N2, 5.0 quality) was used as carrier. In a typical run, 30 mg of the powders were added in 30 mL of 10 vol% aqueous methanolic solution and then dispersed by ultrasonication for 10 min. The Rh metal was stepwise deposited by in situ photoreduction using hexachlororhodate(III) (Na3RhCl6, Sigma Aldrich). Prior to irradiation, the whole system was purged with high-purity N2 to remove air completely.
3. Results and discussion
Physicochemical properties
Estimation of the g-C3N4 content in the g-C3N4/Sr2KNb5O15 composite samples was carried out by thermogravimetric analysis (TG, see Fig. S1†). The g-C3N4 content in the composite samples was more or less the same as the corresponding values calculated from balance measurement (see Table 1), and all TG weight losses had similar profiles within the experimental error. For example, the content of g-C3N4 in the 77CNNb sample can be calculated to be approximately 80 wt% from the TG curve. The slight weight loss being observed before 300 °C corresponded to desorption of water. The major weight loss started at about 540 °C, and finished at about 730 °C, which can be attributed to the decomposition of g-C3N4. After 730 °C, no significant mass change was observed and accordingly the mass residue of the Sr2KNb5O15 material can be determined. Moreover, the exposed surface areas of these nanocomposite samples were determined by the Brunauer–Emmett–Teller (BET) method from nitrogen adsorption–desorption measurements. As shown in Table 1, a slight decrease was observed in the g-C3N4/Sr2KNb5O15 composite samples with g-C3N4 content higher than 55 wt%, probably due to the influence of constituent g-C3N4 with relative low BET surface area.
Table 1 Physicochemical characterization of g-C3N4/Sr2KNb5O15 nanocomposite samples
| Sample |
C3N4a (wt%) |
SBETb (m2 g−1) |
Egc (eV) |
| Estimated by TG. Measured by the BET method. Determined from the extrapolated straight line portion of the Tauc plot at (F(R) hv)1/2 = 0 (see Fig. 3 below). |
| Sr2KNb5O15 |
0 |
19.1 |
3.21 |
| 24CNNb |
30% |
17.1 |
3.22/2.64 |
| 55CNNb |
54% |
19.6 |
2.66 |
| 77CNNb |
80% |
14.6 |
2.66 |
| 99CNNb |
98% |
13.2 |
2.65 |
| g-C3N4 |
100% |
11.2 |
2.66 |
Crystal structure
The X-ray powder diffraction (XRD) patterns of the g-C3N4/Sr2KNb5O15 nanocomposite samples with different content of g-C3N4 are shown in Fig. 1. The samples were well-crystallized and no significant peak shift can be observed. Fig. S2† illustrates in a schematic diagram the crystal structures of Sr2KNb5O15 and g-C3N4. The bare Sr2KNb5O15 can be indexed to tetragonal tungsten bronze structure (JCPDS No. 34-0108), which consists of a framework of corner-shared NbO6 octahedral and three cationic tunnels A1, A2, and A3 along [001] direction (A1 and A2 sites are partially occupied by Sr and K atom and partially vacant, and A3 sites are vacant). The XRD pattern of bare g-C3N4 shows two distinct peaks at 13.2° and 27.4°, which can be indexed as the (100) and (002) diffraction planes, respectively, showing the construction of graphitic planes from tri-s-triazine units were connected by planar amino groups.48 The small-angle diffraction peak at 13.2° gives evidence to the (100) interplanar structural packing of tri-triazine units. The strong peak at 27.4° is the characteristic (002) interplanar stacking peak, corresponding to the interlayer distance of aromatic systems. The results indicate the formation of well-built g-C3N4 layer structure, in good agreement with the literature.15 For the g-C3N4/Sr2KNb5O15 nanocomposite samples, it was hard to differentiate the overlapping diffraction peaks of g-C3N4 at 27.4° and Sr2KNb5O15 at 27.7° as long as the content of g-C3N4 was lower than 55 wt%. With the increased content of g-C3N4, the stronger intensity of the diffraction peak appearing at around 27.5° in the 77CNNb and 99CNNb samples mostly result from their superimposition effects. As shown in Fig. 1, similar XRD patterns were observed for the nanocomposite samples being prepared by direct growth (77CNNb) or by physically mixing (M77CNNb). In both cases no structural interaction between g-C3N4 and Sr2KNb5O15 is observed.
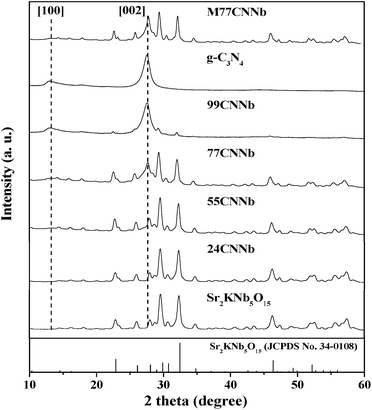 |
| | Fig. 1 XRD patterns of the g-C3N4/Sr2KNb5O15 nanocomposite samples. | |
Microstructure
Fig. 2 shows the representative transmission electron microscopy (TEM) images of bare Sr2KNb5O15 (a), bare g-C3N4 (b), 77CNNb nanocomposite (c), and the high resolution transmission electron microscopy (HRTEM) image of 77CNNb nanocomposite (d), respectively. The bare Sr2KNb5O15 sample exhibited nanorod morphology with diameters in the range of 100–400 nm (Fig. 2a) and lengths up to a few microns (Fig. 2b), while the bare C3N4 particles are irregular aggregated. As compared with bare Sr2KNb5O15 and g-C3N4, it can be clearly seen (Fig. 2c) that most of the Sr2KNb5O15 nanorod surfaces were intimately surrounded by g-C3N4 in the nanocomposite with high g-C3N4 content (77CNNb), indicating that this coverage might favor the formation of sufficient interfaces. In order to further examine the structure, HRTEM analysis was employed. The HRTEM images of the 77CNNb nanocomposite (Fig. 2d and S3†) confirm the expected interplanar spacing of 0.32 nm of the [211] crystal planes of Sr2KNb5O15 as well as the coverage of the Sr2KNb5O15 nanorod by g-C3N4. Presumably, the photo-induced charge transfer via the interfaces was spatially smooth which was fundamental for improving the photocatalytic activity, as will be discussed in detail below.
 |
| | Fig. 2 Representative TEM images of bare Sr2KNb5O15 (a) bare g-C3N4 (b) 77CNNb nanocomposite (c), and HRTEM image of the 77CNNb nanocomposite (d), respectively. | |
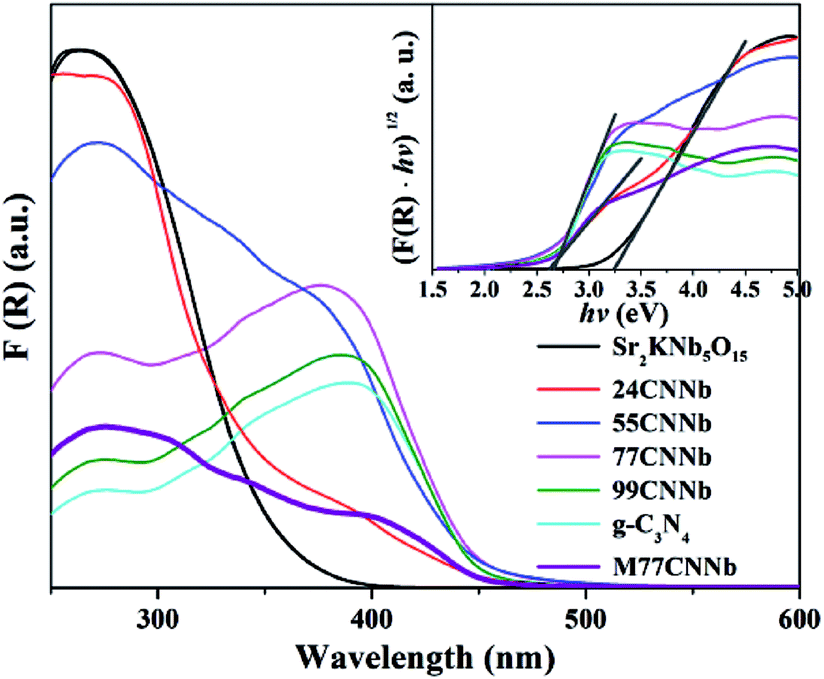
Optical property
Fig. 3 shows Kubelka–Munk transformed UV-Vis diffuse reflection spectra (DRS) and Tauc plots (inset) of the g-C3N4/Sr2KNb5O15 nanocomposites. The band gaps of bare Sr2KNb5O15 and g-C3N4 can be estimated to be about 3.21 eV and 2.66 eV, respectively. A shoulder in the absorption was clearly observed for the g-C3N4/Sr2KNb5O15 nanocomposites with low g-C3N4 content (24CNNb) and the mixed M77CNNb sample. The two different band gaps as estimated from the Tauc plots can be assigned to the absorption bands of their components. Along with increasing g-C3N4 content the intense absorbance of the Sr2KNb5O15 component disappeared in the UV region and their absorption edges become almost similar to that of bare g-C3N4, implying the Sr2KNb5O15 nanorods were sufficiently covered by a thick g-C3N4 layer.
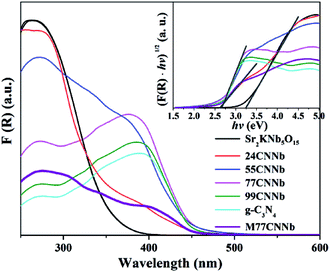 |
| | Fig. 3 Kubelka–Munk transformed UV-Vis diffuse reflection spectra and Tauc plots (inset) of the g-C3N4/Sr2KNb5O15 nanocomposite samples. | |
Surface analysis
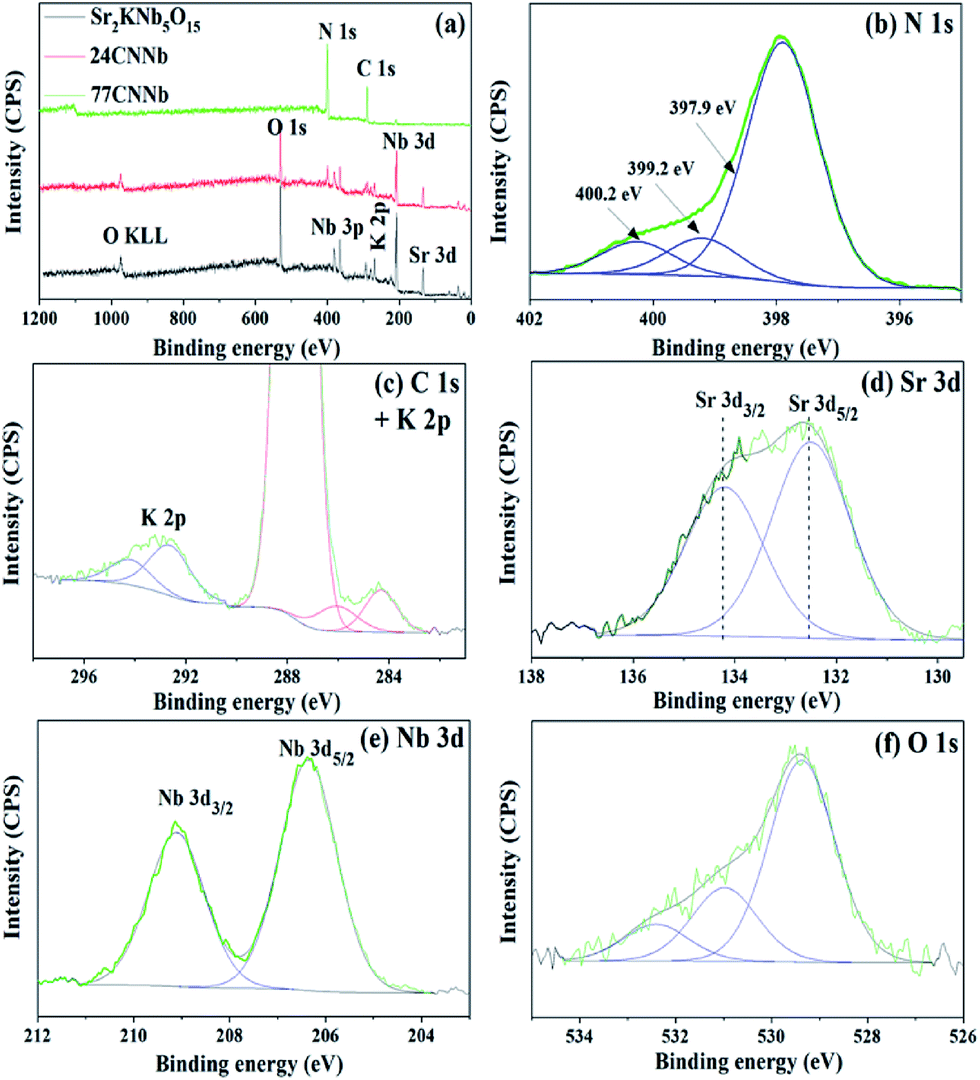
The X-ray photoelectron spectroscopy (XPS) was employed to analyze the oxidation state and the chemical environment of constituent elements on the surface of the bare Sr2KNb5O15, 24CNNb and 77CNNb samples. As can be seen from the survey spectra in Fig. 4a, the main constituent elements on the surface of bare Sr2KNb5O15 sample are Sr, K, Nb, O with sharp photoelectron peaks and low amount of adsorbed C in the form of carbonates. Compared with the XPS spectrum of bare Sr2KNb5O15, there are no significant changes in binding energy of these constituent elements in the XPS spectra of 24CNNb and 77CNNb nanocomposites (see Fig. S4†). However, the observed content of Sr, K, Nb and O elements significantly decrease while at the same time, the signals of C 1s and N 1s are obviously stronger in intensity. As seen in Table 2, with the increasing g-C3N4 content from 24CNNb to 77CNNb samples, the calculated atom concentration of N element increases by a factor of about 2.67, in good agreement with the increased amount of g-C3N4. However, the atom concentration of Sr and Nb elements decreased by a factor of ca. 12.7 and 16.6, respectively, indicating a large amount of g-C3N4 covering the surface of Sr2KNb5O15 nanorods, which is consistent with the TEM observation.
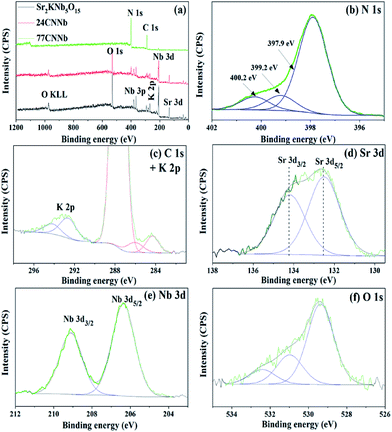 |
| | Fig. 4 Survey XPS spectra of bare Sr2KNb5O15, 24CNNb and 77CNNb nanocomposite samples, respectively (a); and high resolution XPS spectra of N 1s (b), C 1s and K 2p (c), Sr 3d (d), Nb 3d (e) and O 1s (f) of the 77CNNb nanocomposite sample, respectively. | |
Table 2 Atom concentration (at%) for N, Sr and Nb derived from XPS spectra
| Sample |
N (at%) |
Sr (at%) |
Nb (at%) |
| Sr2KNb5O15 |
0 |
11.97 |
21.80 |
| 24CNNb |
19.78 |
7.77 |
14.46 |
| 77CNNb |
52.79 |
0.61 |
0.87 |
The high resolution N 1s, C 1s, K 2p, Sr 3d, Nb 3d and O 1s XPS spectra of the 77CNNb nanocomposite sample are shown in Fig. 4b–f, respectively. The N 1s XPS spectrum (Fig. 4b) can be deconvoluted into three peaks at 397.9, 399.2, and 400.2 eV, which can be ascribed to C–N–C, N-(C)3 and C–N–H groups of g-C3N4.49,50 The C 1s core level XPS spectrum in Fig. 4c can be fitted into three peaks at 284.3, 286.1 and 287.7 eV, respectively. The XPS peak with binding energy of 284.3 eV can be assigned to the saturated hydrocarbons including adventitious carbon. The peaks at 286.1 and 287.7 eV can be ascribed to the C–N–C and the C–(N)3 group, respectively.50,51 Therefore, the above results confirm the presence of graphite-like sp2-bonded graphitic carbon nitride. The XPS spectra of K 2p (Fig. 4c), Sr 3d (Fig. 4d) and Nb 3d (Fig. 4e) can be fitted well into two peaks corresponding to their angular momentum of electron. The K 2p3/2 and K 2p1/2 peaks are observed at 292.7 and 294.3 eV, respectively.52 The Sr 3d5/2 and Sr 3d3/2 peaks are located at 132.5 and 134.2 eV, respectively.53 The Nb 3d peaks at the binding energies of 206.4 and 290.1 eV can be attributed to Nb 3d3/2 and 3d5/2 levels, respectively.54,55 Therefore, the results indicate that only one chemical state for each element of K, Sr, and Nb exists in the nanocomposite, i.e., chemical state of K+, Sr2+, and Nb5+. Furthermore, Fig. 4f shows that the asymmetric O 1s spectrum can be deconvoluted into three peaks at 529.3, 531.0 and 532.4 eV, respectively. The main peak positioned at a low binding energy of 529.3 eV can be attributed to lattice oxygen in the form of O2−.56 While the other minor and broad peaks centered at 531.0 and 532.4 eV are usually assigned to the surface adsorbed oxygen in form of CO32−, H2O and O2.57–59
Photocatalytic degradation of methylene blue
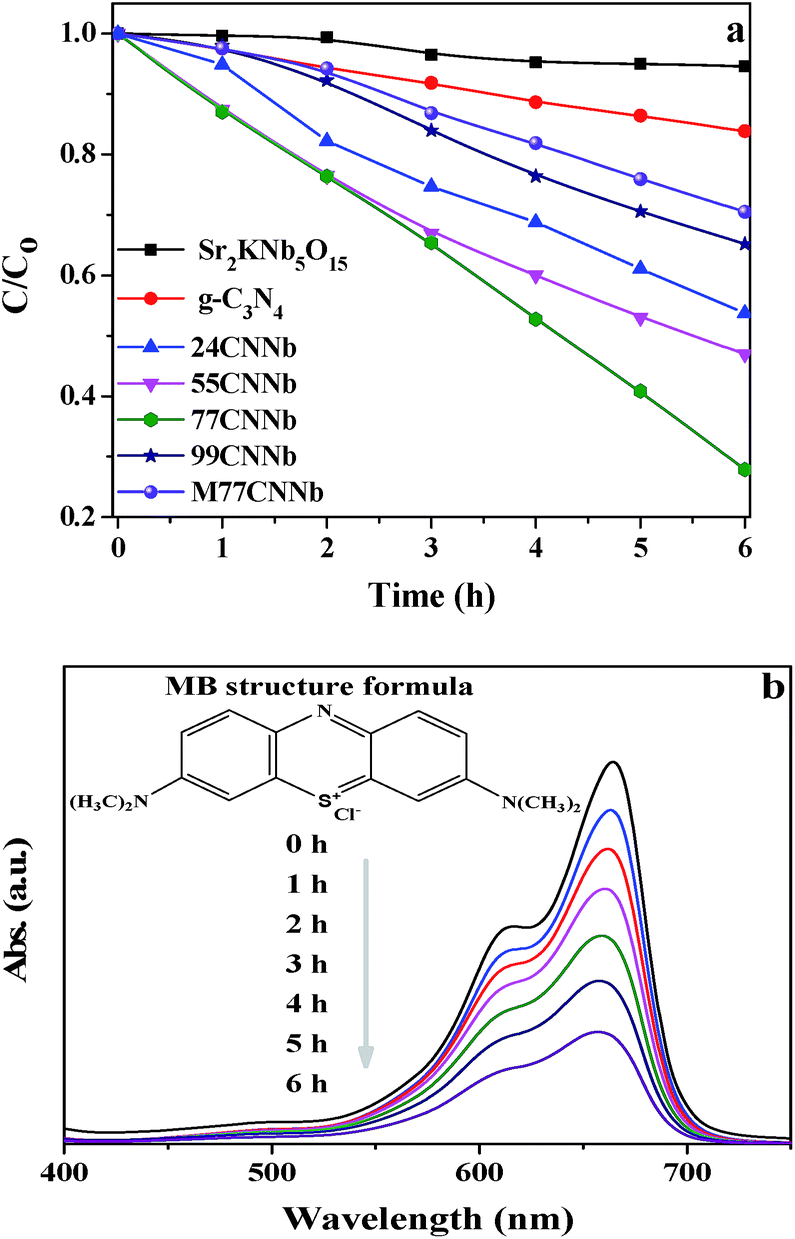
Fig. S5† shows the wavelength distribution of the irradiation light with a GG400 cutoff filter employed in the photocatalytic degradation of methylene blue (MB) experiments. The photocatalytic abilities of the g-C3N4/Sr2KNb5O15 nanocomposites are presented in Fig. 5a. It is noted that the adsorption of MB on the as-synthesized photocatalysts was negligible after the adsorption–desorption equilibrium was reached in the dark. After irradiation for 6 h, the bare Sr2KNb5O15 nanorods exhibit almost no activity, due to their relatively large band gap of 3.21 eV, which is only capable of absorbing shortwave ultraviolet light. Meanwhile, it also reflects that the self-sensitized degradation of MB over Sr2KNb5O15 did not exist in the system and the self-decomposition of MB can be also regarded as negligible. Only 16.2% MB was photodegraded over bare g-C3N4 under the same conditions, showing the poor activity of g-C3N4. By coupling of g-C3N4 with Sr2KNb5O15, the photocatalytic activities were significantly improved and reached the maximum in the 77CNNb sample and then decreased in 99CNNb sample. The results indicate the cooperative effect between the C3N4 and Sr2KNb5O15 components. The relatively high g-C3N4 content in the nanocomposites is very essential to ensure sufficient surface coverage of the Sr2KNb5O15 nanorods, which presumably plays a crucial role in formation of proper nano-interfaces, thereby leading to the significant improvement of the photocatalytic abilities. Fig. 5b illustrates the variations in characteristic absorbance band of MB at 665 nm, which was assigned to the absorption of the conjugated π-system.60 It can be clearly seen that the absorbance of MB obviously decreased with the increasing irradiation time over the 77CNNb nanocomposite.
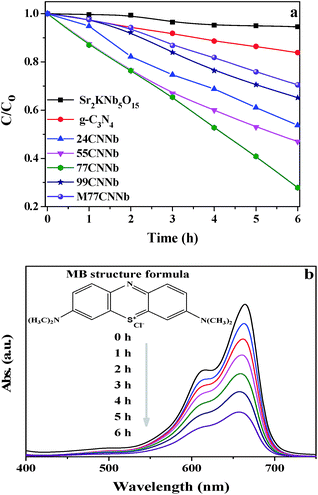 |
| | Fig. 5 (a) Photodegradation of MB under visible light (λ > 400 nm) irradiation over g-C3N4/Sr2KNb5O15 nanocomposite samples. The M77CNNb nanocomposite, bare Sr2KNb5O15 and g-C3N4 were also tested for comparison. (b) Absorption changes of MB solution over the 77CNNb nanocomposite sample during photocatalytic process (inset is the MB structural formula). | |
In order to determine the photocatalytic reaction rate, first-order kinetics was confirmed by plotting ln(C0/C) against the illumination time. The kinetic equation can be expressed as follows:
| |
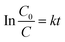 | (1) |
where
C0 and
C are the concentrations of MB solution at times 0 and
t in
eqn (1), the rate constant of
k (h
−1) is equal to the corresponding slope of the fitting line, and
t is the illumination time (h). The MB photodegradation on each sample follows the first-order kinetics model, as show in Fig. S6.
† The resulting rate constants of g-C
3N
4/Sr
2KNb
5O
15 nanocomposites of different content of g-C
3N
4, M77CNNb nanocomposite, bare Sr
2KNb
5O
15 and g-C
3N
4 are illustrated in
Fig. 6. The photooxidation rate constants of g-C
3N
4/Sr
2KNb
5O
15 nanocomposites increased with increasing content of the constituent g-C
3N
4. The highest photooxidation rate constant of 77CNNb nanocomposite was estimated to be
ca. 0.183 h
−1, which was about 6.2 times higher than that of the bare g-C
3N
4 (0.029 h
−1) and 3.4 times that of the M77CNNb nanocomposite (0.053 h
−1). It is worthy to note that the photooxidation rate constant of 99CNNb (0.067 h
−1) is still higher than that of bare g-C
3N
4 and M77CNNb nanocomposite. The results indicate that the g-C
3N
4/Sr
2KNb
5O
15 nanocomposite prepared by direct grown method was greatly beneficial for the formation of the intimate contact between the g-C
3N
4 and Sr
2KNb
5O
15, thus contributing to improve the electron transfer
via proper interfaces. In contrast, an improper weight ratio of g-C
3N
4/Sr
2KNb
5O
15 leads to the loss of favorable interfaces between g-C
3N
4 and Sr
2KNb
5O
15.
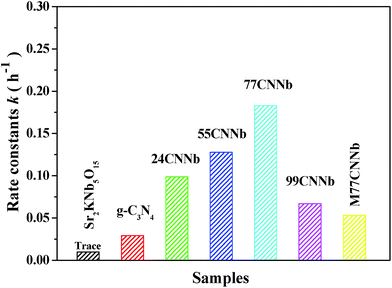 |
| | Fig. 6 First-order rate constant for the photodegradation of MB under visible light irradiation (λ > 400 nm) over the g-C3N4/Sr2KNb5O15 nanocomposite photocatalysts with different C3N4 content, M77CNNb nanocomposite, bare Sr2KNb5O15 and g-C3N4. | |
Recyclability
The recyclability of the photocatalyst is an important parameter of the photocatalytic process for practical application. After each cycle, fresh MB solution was used for next photocatalytic experiment, and taking into account the loss of photocatalyst during the sampling process, the photocatalyst was collected from previous parallel experiments followed by washing and drying. As shown in Fig. S7a,† the 77CNNb nanocomposite shows a slight decrease in degradation efficiency after three cycles, which might be due to the trace amount of MB and the degradation byproducts adsorbed on the surface even after intensive washing. As seen from the XRD patterns of the 77CNNb sample before and after the photoreactions (Fig. S7b†), no significant change was observed. Thus it can be concluded that g-C3N4/Sr2KNb5O15 could be recognized as a stable photocatalyst for decomposing organic contaminant pollutants exposed to visible light irradiation.61–63
Photoluminescence spectra
Photoluminescence analysis was conducted to understand the electron–hole pair recombination properties of prototypical g-C3N4 and 77CNNb nanocomposite samples. As shown in Fig. S8,† the intensity of the broad emission peak appearing at around 440 nm from g-C3N4 was obviously higher than that from 77CNNb. The decreased peak intensity in the 77CNNb nanocomposite can be explained by the reduction of the recombination rate of electron–hole pairs, which is consistent with the photocatalytic MB degradation results.
Photocatalytic H2 production from water splitting
As shown in Fig. 7, under visible light irradiation (λ > 420 nm), the 77CNNb nanocomposite, pristine g-C3N4 and Sr2KNb5O15 samples were further investigated for photocatalytic H2 production from water splitting. All the measurements were reproducibly conducted at least three times with deviations less than 5%. No H2 evolution was observed for the pristine Sr2KNb5O15. The presence of methanol as an electron donor and then stepwise photodecomposition of Rh metal as cocatalyst significantly improved the average H2 production rates of both the g-C3N4 and the 77CNNb nanocomposite. However, surprisingly, with stepwise photodecomposition of 0.03 wt% Rh cocatalysts, the enhancement of the g-C3N4 was obviously larger than for 77CNNb, as indicated by arrows in Fig. 7. BET surface areas and optical band gaps can be easily excluded as the dominant factor because, as mentioned above, the 77CNNb nanocomposite has a larger BET area than g-C3N4 and both of them have a similar band gap. Thus this trend can only be ascribed to an inhibition effect of the deposited Rh, and the large potential difference between Rh and g-C3N4, which were probably the most influential factors on the changes of enhancement extent (as will be discussed in detail below).
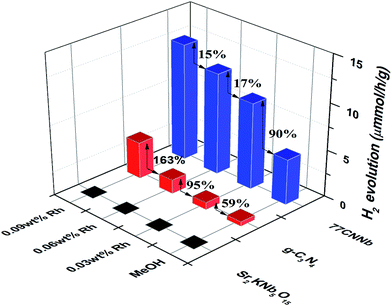 |
| | Fig. 7 Time course of photocatalytic H2 evolution over the 77CNNb nanocomposite, pristine g-C3N4 and Sr2KNb5O15 samples (reaction conditions: catalyst, 30 mg; 30 mL of 10 vol% methanolic solution). | |
Proposed photocatalytic mechanism
According to the report by Scaife for oxides not containing partly filled d-levels,64 the following equation can be used to approximately determine the flat band potential:where Vfb and Eg represent a flat band potential and a band gap, respectively. The conduction band (CB) and valance band (VB) edge potentials of Sr2KNb5O15 were roughly estimated to be about −0.27 and 2.94 V, respectively. Thus under visible light irradiation, the wide band-gap Sr2KNb5O15 is photoinactive. On the other hand, Wang, et al.28 reported that by density functional theory (DFT) calculations, the LUMO and HOMO energy levels of g-C3N4 are estimated at −1.12 and 1.57 V, respectively. As seen in Fig. 8, effective spatial charge separation between g-C3N4 and Sr2KNb5O15 is proposed to account for the enhanced visible-light photocatalytic activity, i.e., the g-C3N4 behaves as visible light absorber, and with the aid of smooth nano-interfaces, the cooperative Sr2KNb5O15 nanorods act as an electron separator, which is capable of adopting photoexcited electrons from g-C3N4. The electron transfer process and redistribution behavior of photogenerated holes in the side of g-C3N4 and photogenerated electrons in the side of Sr2KNb5O15 greatly reduce the possibilities of electron–hole pairs recombination and thus enhance the photodegradation rate. Moreover, in order to understand the effect of Rh metal for interfacial charge transfer process during photocatalytic H2 production reaction, the work function of Rh metal (∼4.98 eV) was provided (Fig. 8).65 As a result of the high work function value of Rh metal, Schottky barriers at the interfaces were formed between Rh metal with g-C3N4 and Sr2KNb5O15, respectively, thus significantly enhancing the electron transfer efficiencies of the g-C3N4 and the 77CNNb nanocomposite for photocatalytic H2 production. In case of the nanocomposite two electron transfer channels from g-C3N4 and Sr2KNb5O15 to Rh, respectively, will be built up. And considering the formed coverage structure of g-C3N4/Sr2KNb5O15 and large potential difference between g-C3N4 and Rh metal, it can be reasonably inferred that Rh metal co-catalysts would be photoreduced preferentially on surface active sites of g-C3N4, rather than Sr2KNb5O15. Therefore, with the increased amount of the deposited Rh on g-C3N4, a large amount of the Schottky barriers was formed between Rh with g-C3N4. Consequently, the electron transfer between Sr2KNb5O15 with g-C3N4 and Rh metal became weaker and was competitively inhibited. That is probably the primary reason why the enhancement extent on H2 production rate was decreased in 77CNNb nanocomposite. Note, the study also points out that in the composite materials system the proper use of noble metal as co-catalysts is an important issue for improving photocatalytic performance.
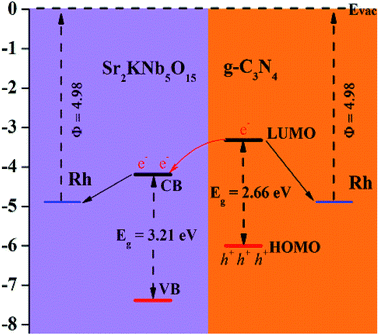 |
| | Fig. 8 Proposed mechanism of g-C3N4/Sr2KNb5O15 nanorod nanocomposites. | |
4. Conclusions
In summary, a novel g-C3N4/Sr2KNb5O15 nanocomposite photocatalyst was simply prepared by directly grown graphitic C3N4 on Sr2KNb5O15 nanorods at 520 °C for 4 h. The structure properties, surface morphology and element chemical states of the as-synthesized samples have been investigated. Compared to bare Sr2KNb5O15, and g-C3N4, the photocatalytic property was significantly enhanced by coupling of g-C3N4 with Sr2KNb5O15 for degradation of methylene blue (MB) and photocatalytic H2 production from aqueous methanolic solution with stepwise in situ photodepositon of Rh. The results demonstrated that g-C3N4 acts as a visible-light-harvesting unit, and the inner Sr2KNb5O15 nanorods is capable of adopting photoexcited electrons from g-C3N4, thus greatly enhancing spatial charge separation via the formed nano-interfaces.
By a comparative investigation of the direct growth method with physical mixing method of pre-synthesized g-C3N4 and Sr2KNb5O15, the photodegradation rate of the former is found to be about 3.4 times higher than of the latter, indicating how important the formation of proper nano-interfaces is. The work may provide further insight for rational construction of C3N4-based composite and offer an alternative strategy to develop visible-light-driven photocatalysts for the potential applications of water treatment and other environmental remediation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We greatly appreciate Prof. Dr M. Muhler (RUB, Germany) for his support, Dr T. Reinecke (RUB, Germany) for XRD measurements and Dr W. H. Zhang (RUB, Germany) for the TG measurements. This work was financially supported by the National Natural Science Foundation of China (Grant Number: 51402193, 51572173, 51602197 and 11402149), Shanghai Eastern Scholar Program (Grant Number: QD2016014) and Shanghai Pujiang Talent Program (Grant Number: 16PJ1407700).
Notes and references
- A. Mills and S. LeHunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- S. K. Lee and A. Mills, Platinum Met. Rev., 2003, 47, 61–72 CAS.
- F. Wang, T. A. Shifa, X. Zhan, Y. Huang, K. Liu, Z. Cheng, C. Jiang and J. He, Nanoscale, 2015, 7, 19764–19788 RSC.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- H. Yamashita, M. Harada, J. Misaka, M. Takeuchi, K. Ikeue and M. Anpo, J. Photochem. Photobiol., A, 2002, 148, 257–261 CrossRef CAS.
- N. Negishi, M. Fujino, H. Yamashita, M. A. Fox and M. Anpo, Langmuir, 1994, 10, 1772–1776 CrossRef CAS.
- W. Y. Choi, A. Termin and M. R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1994, 33, 1091–1092 CrossRef.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908–4911 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454 CrossRef CAS.
- X. Zong, C. H. Sun, Z. G. Chen, A. Mukherji, H. Wu, J. Zou, S. C. Smith, G. Q. Lu and L. Z. Wang, Chem. Commun., 2011, 47, 6293–6295 RSC.
- R. Marschall and L. Wang, Catal. Today, 2014, 225, 111–135 CrossRef CAS.
- X. Xu, G. Liu, C. Randorn and J. T. S. Irvine, Int. J. Hydrogen Energy, 2011, 36, 13501–13507 CrossRef CAS.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- U. B. Cappel, S. M. Feldt, J. Schoneboom, A. Hagfeldt and G. Boschloo, J. Am. Chem. Soc., 2010, 132, 9096–9101 CrossRef CAS PubMed.
- P. Wang, P. Chen, A. Kostka, R. Marschall and M. Wark, Chem. Mater., 2013, 25, 4739–4745 CrossRef CAS.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- Z. Cheng, F. Wang, T. A. Shifa, K. Liu, Y. Huang, Q. Liu, C. Jiang and J. He, Appl. Phys. Lett., 2016, 109, 053905 CrossRef.
- K. Vinodgopal, I. Bedja and P. V. Kamat, Chem. Mater., 1996, 8, 2180–2187 CrossRef CAS.
- Y. Bessekhouad, D. Robert and J.-V. Weber, Catal. Today, 2005, 101, 315–321 CrossRef CAS.
- Z. Bian, J. Zhu, S. Wang, Y. Cao, X. Qian and H. Li, J. Phys. Chem., 2008, 112, 6258–6262 CAS.
- K. Li, B. Chai, T. Y. Peng, J. Mao and L. Zan, ACS Catal., 2013, 3, 170–177 CrossRef CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- H. J. Yan and H. X. Yang, J. Alloys Compd., 2011, 509, L26–L29 CrossRef CAS.
- P. A. Sant and P. V. Kamat, Phys. Chem. Chem. Phys., 2002, 4, 198–203 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe, K. Domen, Y. D. Hou, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- Y. Wu and M. Wark, New Concepts in Photocatalysis, RSC Publishing, London, 2016, ch. 6, pp. 129–161 Search PubMed.
- N. Yang, G. Q. Li, W. L. Wang, X. L. Yang and W. F. Zhang, J. Phys. Chem. Solids, 2011, 72, 1319–1324 CrossRef CAS.
- Q. Y. Li, B. Yue, H. Iwai, T. Kako and J. H. Ye, J. Phys. Chem. C, 2010, 114, 4100–4105 CAS.
- Y. Liu, G. Chen, C. Zhou, Y. D. Hu, D. G. Fu, J. Liu and Q. Wang, J. Hazard. Mater., 2011, 190, 75–80 CrossRef CAS PubMed.
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC.
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS.
- L. Ge, C. C. Han and J. Liu, Appl. Catal., B, 2011, 108, 100–107 CrossRef.
- X. She, J. Wu, H. Xu, J. Zhong, Y. Wang, Y. Song, K. Nie, Y. Liu, Y. Yang and M. T. F. Rodrigues, Adv. Energy Mater., 2017, 1700025 CrossRef.
- J. Fu, Y. L. Tian, B. B. Chang, F. N. Xi and X. P. Dong, J. Mater. Chem., 2012, 22, 21159–21166 RSC.
- D. Jiang, L. Chen, J. Zhu, M. Chen, W. Shi and J. Xie, Dalton Trans., 2013, 42, 15726–15734 RSC.
- B. Chai, T. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC.
- Q. Y. Li, L. L. Zong, Y. Y. Xing, X. D. Wang, L. G. Yu and J. J. Yang, Sci. Adv. Mater., 2013, 5, 1316–1322 CrossRef CAS.
- H. J. Yan and H. X. Yang, J. Alloys Compd., 2011, 509, 26–29 CrossRef.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC.
- Y. Zeng, Y. Wang, J. Chen, Y. Jiang, M. Kiani, B. Li and R. Wang, Ceram. Int., 2016, 42, 12297–12305 CrossRef CAS.
- L. Sun, Y. Qi, C. J. Jia, Z. Jin and W. Fan, Nanoscale, 2014, 6, 2649–2659 RSC.
- P. Wang, L. Schwertmann, R. Marschall and M. Wark, J. Mater. Chem. A, 2014, 2, 8815–8822 CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- M. Xiong, L. Chen, Q. Yuan, J. He, S. L. Luo, C. T. Au and S. F. Yin, Carbon, 2015, 86, 217–224 CrossRef CAS.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083 RSC.
- B. Chai, T. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC.
- M. Kaba, N. Raklaoui, M. F. Guimon and A. Mas, J. Appl. Polym. Sci., 2005, 97, 2088–2096 CrossRef CAS.
- H. Konno, K. Sasaki, M. Tsunekawa, T. Takamori and R. Furuichi, Jpn. Anal., 1991, 40, 609–616 CrossRef CAS.
- S. Koba, R. Nagata, M. Onishi, Y. Ozono, K. Shibata, S. Higo, I. Kawano, T. Kushida, Y. Kim and T. Ogushi, Int. J. Mod. Phys. B, 1998, 12, 1755–1762 CrossRef CAS.
- F. Garbassi, J. Bart and G. Petrini, J. Electron Spectrosc. Relat. Phenom., 1981, 22, 95–107 CrossRef CAS.
- T. Xia, Q. Li, J. Meng and X. Cao, Mater. Chem. Phys., 2008, 111, 335–340 CrossRef CAS.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- R. Cole, N. Brooks and P. Weightman, Phys. Rev. Lett., 1997, 78, 3777 CrossRef CAS.
- N. Wakiya, K. Kuroyanagi, Y. Xuan, K. Shinozaki and N. Mizutani, Thin Solid Films, 2000, 372, 156–162 CrossRef CAS.
- Y. Zhang-Steenwinkel, J. Beckers and A. Bliek, Appl. Catal., A, 2002, 235, 79–92 CrossRef CAS.
- P. Qu, J. C. Zhao, T. Shen and H. Hidaka, J. Mol. Catal. A: Chem., 1998, 129, 257–268 CrossRef CAS.
- H. Cheng, B. Huang, Y. Dai, X. Qin and X. Zhang, Langmuir, 2010, 26, 6618–6624 CrossRef CAS PubMed.
- S. Y. Chai, Y. J. Kim, M. H. Jung, A. K. Chakraborty, D. Jung and W. I. Lee, J. Catal., 2009, 262, 144–149 CrossRef CAS.
- Y. Zang and R. Farnood, Appl. Catal., B, 2008, 79, 334–340 CrossRef CAS.
- D. E. Scaife, Sol. Energy, 1980, 25, 41–54 CrossRef CAS.
- W. M. Haynes, CRC handbook of chemistry and physics CD-ROM version, 90th edn, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The crystallographic structure, TG curves, XPS data, TEM images, UV-Vis spectra, first-order kinetics model, recycle measurements and PL spectra. See DOI: 10.1039/c7ra07441g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Ilya Sinevc,
Feng Suna,
Huijun Lia,
Ding Wanga,
Qian Li
*ab,
Ilya Sinevc,
Feng Suna,
Huijun Lia,
Ding Wanga,
Qian Li