
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MoS2/Ni3S2 nanorod arrays well-aligned on Ni foam: a 3D hierarchical efficient bifunctional catalytic electrode for overall water splitting†
Nan Zhang *a,
Junyu Leib,
Jianpeng Xiea,
Haiyan Huanga and
Ying Yu*a
*a,
Junyu Leib,
Jianpeng Xiea,
Haiyan Huanga and
Ying Yu*a
aState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing, P. R. China 102249. E-mail: zhangnan@cup.edu.cn; yybjpudhx@foxmail.com; Tel: +86-010-89739132
bPetrochemical Research Institute, PetroChina, Beijing, P. R. China 100195
First published on 29th September 2017
Abstract
Fabricating earth-abundant bifunctional water splitting electrocatalysts with high efficiencies to substitute Pt and IrO2 is in great demand for the development of clean energy conversion technologies. Via a simple one-step hydrothermal procedure, MoS2/Ni3S2 nanorod arrays well-aligned on Ni foam (MoS2/Ni3S2/NF) as a novel three dimensional (3D) hierarchical bifunctional catalytic electrode for overall water splitting has been successfully fabricated in this work. Ni foam (NF) as both 3D substrate and Ni source realized the uniform, dense and nearly vertical in situ self-growth of MoS2/Ni3S2 nanorods on it. Each nanorod had a coated structure with large surface contact regions. Benefiting from the elaborately designed architecture, MoS2/Ni3S2/NF exhibited excellent catalytic activity and good stability for both the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in an alkaline electrolyte. To reach the catalytic current density of 10, 100, 200, and 300 mA cm−2, it only required 187, 274, 300, and 320 mV overpotential for HER and 217, 275, 313, and 335 mV overpotential for OER respectively. Moreover, by employing MoS2/Ni3S2/NF as both the cathode and anode, the water splitting electrolyzer achieved excellent durability. Besides, it only needed very low cell voltages of 1.467, 1.593, 1.640 and 1.661 V to reach the current density of 10, 100, 200 and 300 mA cm−2, respectively, better than the performance of a benchmark combination. Therefore, MoS2/Ni3S2/NF is a promising non-noble-based bifunctional electrocatalyst for overall water splitting.
1. Introduction
Facing the serious environmental pollution and the conflict between the increasing global energy demand and the consumption of limited fossil fuels, clean and sustainable energy and energy conversion technologies have attracted much attention in recent years.1–3 Electrochemically splitting water into high purity hydrogen is a clean and effective technique to convert electrical energy into chemical energy which can be stored for later use.4,5 Accelerating the two half electrochemical reactions, the hydrogen evolution reaction (HER) at the cathode and oxygen evolution reaction (OER) at the anode, which comprise the water splitting process, are crucial for the overall efficiency of water splitting.6–8 Therefore it is necessary to fabricate excellent electrocatalysts for HER and OER. Heretofore, the most efficient HER catalysts are Pt-based materials,9,10 and the most efficient OER electrocatalysts are RuO2 and IrO2 based catalysts.11,12 Accordingly, there are two challenges for the development of HER and OER electrocatalysts. Firstly, the scarce crustal abundance and the high cost of noble metal-based materials severely limit their large-scale utilization. Secondly, the outstanding OER catalysts usually employed in alkaline conditions have poor HER activity while the superior HER catalysts often used in acid conditions have only moderate activity in OER.13,14 Improper pH condition leads to reduced efficiency of the catalysts. Therefore, different catalysts work in separated electrolytes in traditional electrolyzers for overall water splitting. As a consequence, fabricating bifunctional electrocatalysts for both HER and OER which can be applied in the same electrolyte, could simplify the water splitting system and reduce costs. In summary, the syntheses and investigations of low cost earth-abundant bifunctional electrocatalysts with high efficiency and durability are beneficial to the development of water splitting technology.To realize this goal, kinds of earth-abundant bifunctional electrocatalysts have been fabricated, such as transition metals,15,16 transition metal oxides,17–19 transition metal phosphides,20–25 transition metal nitrides,26 transition metal dichalcogenides (TMDs),27–29 etc. However, more strategies are still being tried for the improvement of the electrocatalytic performance. One strategy is to hybridize different catalysts together into a hierarchical nanostructure to construct the coupling interface regions thus increase the synergistic electrocatalytic activity,30,31 such as TiN@Ni3N,32 MnOx/Ni3S2/NF,33 CuO/Co3O4.34 Secondly, since the catalytic performances of electrocatalysts are highly dependent on their morphologies and structures, designing and fabricating the well-arranged nanoarray structures with large surface area, abundant active sites and efficient electron transport is another important strategy. For example, NiCo2S4 nanowire arrays,35 CoP nanorod arrays22 and Ni3S2 nanosheet arrays27 which were all supported on Ni foam (NF) have achieved excellent bifunctional electrocatalytic performances. Thirdly, introducing three dimensional (3D) substrates endowed the self-supported structure of the catalytic electrode which is advantageous for the convenient operation and application in technological devises. 3D skeletons such as NF,36,37 copper foam,38 or carbon cloth39 have been employed as substrates of various catalysts. Considering these three strategies together, we have been motivated to fabricate a 3D hybrid bifunctional catalytic electrode with a well-arranged nanoarray structure.
The earth abundant and rich valence state material, Ni3S2 has attracted tremendous interests of researchers due to its high conductivity and specific configuration.40 Therefore Ni3S2 has been widely investigated in various electrochemical applications, including supercapacitors,41,42 Li-ion batteries43,44 and non-enzymatic glucose detection,42,45 etc. Moreover, Ni3S2 has been found to be electrocatalytic active for HER45–47 and OER48,49 in recent years, and shown to be the promising bifunctional catalyst for overall water splitting.27,50 However, for superior electrocatalytic performance of Ni3S2, smart coupling engineering of different materials (such as nitrogen-doped graphene,51 CdS52) with Ni3S2 has been proved to be an effective strategy very recently. As an excellent HER electrocatalyst, MoS2 could be employed as a component of the hybrid bifunctional catalyst for improved HER catalytic activity53–56 However, integrating MoS2 with Ni3S2 into hierarchical bifunctional electrocatalysts for direct water splitting has been reported by very few articles. Therefore, novel MoS2-based 3D nanoarray hybrid catalytic electrode should be fabricated to achieve the improved bifunctional catalytic performance for overall water splitting.
In this work, MoS2/Ni3S2 nanorod arrays well-aligned on NF (MoS2/Ni3S2/NF) as an efficient 3D hierarchical bifunctional catalytic electrode for overall water splitting has been successfully fabricated for the first time via a simple one-step hydrothermal procedure. NF as the 3D substrate realized the uniform, dense and nearly vertical in situ self-growth of MoS2/Ni3S2 nanorods on it, and formed a self-supported catalytic electrode. And each MoS2/Ni3S2 nanorod consisted of Ni3S2 nanorod wrapped in MoS2 nanosheet. Moreover, in the hydrothermal procedure, we have changed the molar ratio of Mo precursor and S precursor, and obtained MoS2/Ni3S2/NF with tailored architectures. In these electrodes, the optimal MoS2/Ni3S2/NF has obtained excellent electrocatalytic performances for both HER and OER in alkaline electrolyte, 1.0 M KOH. During the overall water splitting process, MoS2/Ni3S2/NF as both cathode and anode has exhibited better electrocatalytic activity and stability than the integrated performance of Pt/C and IrO2. Therefore, MoS2/Ni3S2/NF has achieved the satisfied catalytic activity as a bifunctional electrocatalyst for overall water splitting.
2. Experimental section
2.1 Materials
Pt/C (20 wt%) was purchased from Sigma-Aldrich, and Nafion solution (5 wt%) was bought from Alfa Aesar. Thiourea was obtained from Sinopharm Chemical Reagent Co., Ltd (China). Sodium molybdate and hydrochloric acid were bought from Beijing Chemical Reagent Company (China). IrO2 and Ni powder (NP) were bought from Xiya reagent (China). NF with a thickness of 0.15 cm and 110 ppi (pore per square inch) was purchased from Suzhou Jia Shide Foam Metal Co., Ltd (China). Unless otherwise stated, reagents were of analytical grade and used as received. The water used throughout all experiments was deionized water from a Millipore system (>18 MΩ cm).2.2 Preparation of electrocatalysts
A hydrothermal approach was used for synthesizing MoS2/Ni3S2/NF and Ni3S2/NF, as well as MoS2/Ni3S2 (NP) which employed NP as Ni source. NF was cleaned by sonication sequentially in acetone, HCl aqueous solution (0.5 M), and water for 10 min each. In a typical synthesis procedure, the cleaned NF (2.5 cm × 3 cm) was immersed in 30 mL aqueous solution containing 0.0726 g of sodium molybdate and 0.2284 g thiourea. Then the solution together with NF was transferred into a 50 mL Teflon-lined stainless autoclave and kept at 200 °C for 12 h. After the autoclave naturally cooled at room temperature, MoS2/Ni3S2/NF was taken out from the autoclave and rinsed thoroughly with water and ethanol successively for several times and dried in an oven at 60 °C for 10 h. The loading weight of MoS2/Ni3S2 on NF was determined to be ∼7.6 mg cm−2 via a high-precision microbalance. In addition, Ni3S2/NF was synthesized using the same method and parameters without the addition of sodium molybdate.In control experiments, different amount of sodium molybdate was employed in the reaction system as 0.0362, 0.1452 and 0.2178 g respectively. As the feed ratio in the parenthesis stands for the molar ratio of Mo precursor and S precursor, MoS2/Ni3S2/NF obtained from the control experiments was respectively designated as MoS2/Ni3S2/NF (Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S = 0.5:10), MoS2/Ni3S2/NF (Mo:S = 2:10) and MoS2/Ni3S2/NF (Mo:S = 3:10). MoS2/Ni3S2/NF with the feed ratio of Mo:S = 1:10 prepared from the typical synthesis procedure was succinctly expressed as MoS2/Ni3S2/NF.
:S = 0.5:10), MoS2/Ni3S2/NF (Mo:S = 2:10) and MoS2/Ni3S2/NF (Mo:S = 3:10). MoS2/Ni3S2/NF with the feed ratio of Mo:S = 1:10 prepared from the typical synthesis procedure was succinctly expressed as MoS2/Ni3S2/NF.
MoS2/Ni3S2 (NP) was fabricated by the similar procedures except using NP as a substitute for NF. 30 mL aqueous solution containing 0.0726 g sodium molybdate, 0.2284 g thiourea and 0.2113 g NP were evenly dispersed, then transferred into the autoclave and kept at 200 °C for 12 h. The product was collected by centrifugation and washed by water and ethanol successively for three times. After drying in an oven at 60 °C for 10 h, MoS2/Ni3S2 (NP) powder was obtained. In this product, there was no NF as the 3D skeleton or Ni precursor. NP worked as Ni source for the synthesis of Ni3S2 in MoS2/Ni3S2 (NP).
The physical loading of Pt/C, IrO2 and MoS2/Ni3S2 (NP) on NF were operated as the following procedures. The electrocatalyst ink was respectively prepared by dispersing 7.6 mg electrocatalyst in 700 μL ethanol and 300 μL water containing 20 μL Nafion solution and treated by sonication until a homogeneous dispersion was obtained. Then the electrocatalyst ink was uniformly coated on the working area (1 cm × 1 cm) of NF and dried in an oven at 60 °C for 10 h. In this manuscript, Pt/C, IrO2 and MoS2/Ni3S2 (NP) overlaid on NF are referred to simply as Pt/C, IrO2 and MoS2/Ni3S2 (NP) respectively.
2.3 Characterization of the electrocatalysts
Scanning electron microscope (SEM) images, their corresponding energy-dispersive X-ray (EDX) spectra and EDX elemental mapping images were obtained on a Quanta 200 FEI scanning electron microscope. Transmission electron microscopy (TEM) images, high-resolution transmission electron microscopy (HRTEM) images and EDX elemental mapping images were carried out on an FEI TECNAI G2 F20 STWIN transmission electron microscopy operating at 200 kV. The X-ray diffraction (XRD) measurements were recorded in the range of 10–90° (2θ) on a D8 Focus diffractometer (Bruker) with Cu Kα radiation operated at 40 kV and 30 mA, which were applied to investigate the crystallographic structure of the as-fabricated products. X-ray photoelectron spectroscopy (XPS) analyses were carried out on a Thermo Scientific ESCALAB 250Xi X-ray photoelectron spectrometer with Al Kα X-ray radiation as the X-ray source for excitation.2.4 Electrochemical tests
All the electrochemical tests were conducted in the electrolyte of 1.0 M KOH. The HER and OER electrochemical experiments were performed with a three-electrode system. The system consisted of MoS2/Ni3S2/NF with an effective working area of 1 cm × 1 cm as the working electrode, Hg/HgO electrode as the reference electrode, and a graphite rod as the counter electrode. The overall water splitting experiments were operated in the electrolyzer with a two-electrode system composed of MoS2/Ni3S2/NF as both the cathode and the anode.Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were measured with the scan rate of 5 mV s−1. In the investigation of electrochemical double-layer capacitances, CV was measured in the potential ranging from 0.822 to 0.922 V with scan rates of 20, 40, 60, 80, 100, 120, 140, 160, 180, and 200 mV s−1, respectively. During the investigation of stability, CV was performed for 1000 cycles with a scan rate of 100 mV s−1 in the potential ranging from −0.303 to 0.097 V and 1.057 to 1.457 V respectively. The amperometric i–t curves were obtained at the static overpotentials which could obtain the current density of 10 mA cm−2. All the above data were iR corrected via the resistance test. Electrochemical impedance spectroscopy (EIS) measurements were carried out with frequencies ranging from 100 kHz to 0.01 Hz. The measurements above-mentioned were all conducted after bubbling nitrogen for 10 min at room temperature and without activation progress by a CHI 660e electrochemical analyzer (CH Instruments, Inc., Shanghai).
Hg/HgO electrode as the reference electrode in all the three-electrode system was calibrated according to the method previously reported.57 With platinum rotating disk electrode (0.126 cm2) as working electrode, platinum rod as counter electrode and Hg/HgO electrode as reference electrode in 1.0 M KOH solution saturated high purity hydrogen, CV was cycled from −1.0 to −0.8 V with scan rate of 1 mV s−1. The cross-over point at which the hydrogen evolution current changes into the hydrogen oxidation currents was taken as the thermodynamic (zero) potential for the hydrogen electrode reactions. As shown in Fig. S1,† the zero current point lies at −0.922 V versus Hg/HgO resulting in the correlation: Evs. RHE = Evs. Hg/HgO + 0.922 V. Therefore, all the potential values referenced to a reversible hydrogen electrode (RHE) measured were calibrated by the above equation.
3. Results and discussion
3.1 Material synthesis and characterization
MoS2/Ni3S2/NF was synthesized by a simple one-step hydrothermal procedure with NF as Ni source, sodium molybdate as Mo source and thiourea as S source. As the photograph shown in Fig. 1a, compared with the silver colored NF, the black color of MoS2/Ni3S2/NF indicates the successful growth of MoS2/Ni3S2 on NF. It should be ascribed to the black color of MoS2 since Ni3S2/NF shows to be brown. The bended MoS2/Ni3S2/NF in Fig. 1b illustrates the flexibility of this catalytic electrode since no structural damage happened on it after being stretched. The architecture of MoS2/Ni3S2/NF is shown by its SEM images. Compared with bare NF (Fig. S2†), MoS2/Ni3S2/NF had a rougher surface. NF as the substrate was densely and uniformly covered by MoS2/Ni3S2 nanorods, forming a 3D hierarchical self-supported catalytic electrode (Fig. 1c and d). Higher magnification SEM image (Fig. 1e) reveals the micron grade length MoS2/Ni3S2 nanorods with average diameter of ∼150 nm nearly vertically aligned on NF. However, when NP was employed as a substitute for NF, the prepared MoS2/Ni3S2 (NP) appeared to be irregular particles (Fig. S3a and b†) which consisted of nanorods and pieces (Fig. S3c†). It suggests that NF was necessary for the in situ directional self-growth of the 1D MoS2/Ni3S2 nanorod arrays. The rational reason may be that NF as Ni source promoted unidirectional transport of nickel ions from its surface toward the solution during the formation of MoS2/Ni3S2.27 In general, NF provided a 3D skeleton and large surface area for the growth of the catalyst. It realized the unique well-arranged nanorod array hierarchical nanostructure of the 3D catalytic electrode, MoS2/Ni3S2/NF. Furthermore, no obvious changes in the morphology of MoS2/Ni3S2/NF after ultrasonication treatment for 30 min as shown in Fig. S4† proves the good mechanical stability of MoS2/Ni3S2/NF. It could be attribute to the direct growth of MoS2/Ni3S2 nanorods from the substrate since NF worked as Ni source. Besides, the elemental composition of MoS2/Ni3S2 is confirmed to be only Mo, S and Ni by the EDX spectrum of MoS2/Ni3S2 nanorods (Fig. S5†). Moreover, the molar content of MoS2 in MoS2/Ni3S2 nanorods was determined to be about 18.2% through the quantitative values measured by EDX as shown in Table S1.† As the elemental mapping images shown in Fig. 1f and S6,† Mo, Ni and S elements have uniformly distributed not only on MoS2/Ni3S2 nanorods, but also on the whole 3D MoS2/Ni3S2/NF catalytic electrode. It demonstrated the uniform growth of the catalytic material MoS2/Ni3S2 on Ni foam substrate. On the other hand, Ni3S2/NF synthesized without the addition of Mo precursor was also investigated in this work. As shown in Fig. S7,† dwarf Ni3S2 nanobulges spread all over NF, with the elemental composition demonstrated by its EDX spectrum (Fig. S7d†). Compared with Ni3S2/NF, it should be emphasized that the addition of Mo precursor determined the 1D nanorod array structure of MoS2/Ni3S2 on NF with high specific surface area. Consequently, the introduction of Mo precursor played a crucial role in tailoring the morphology of our electrocatalyst.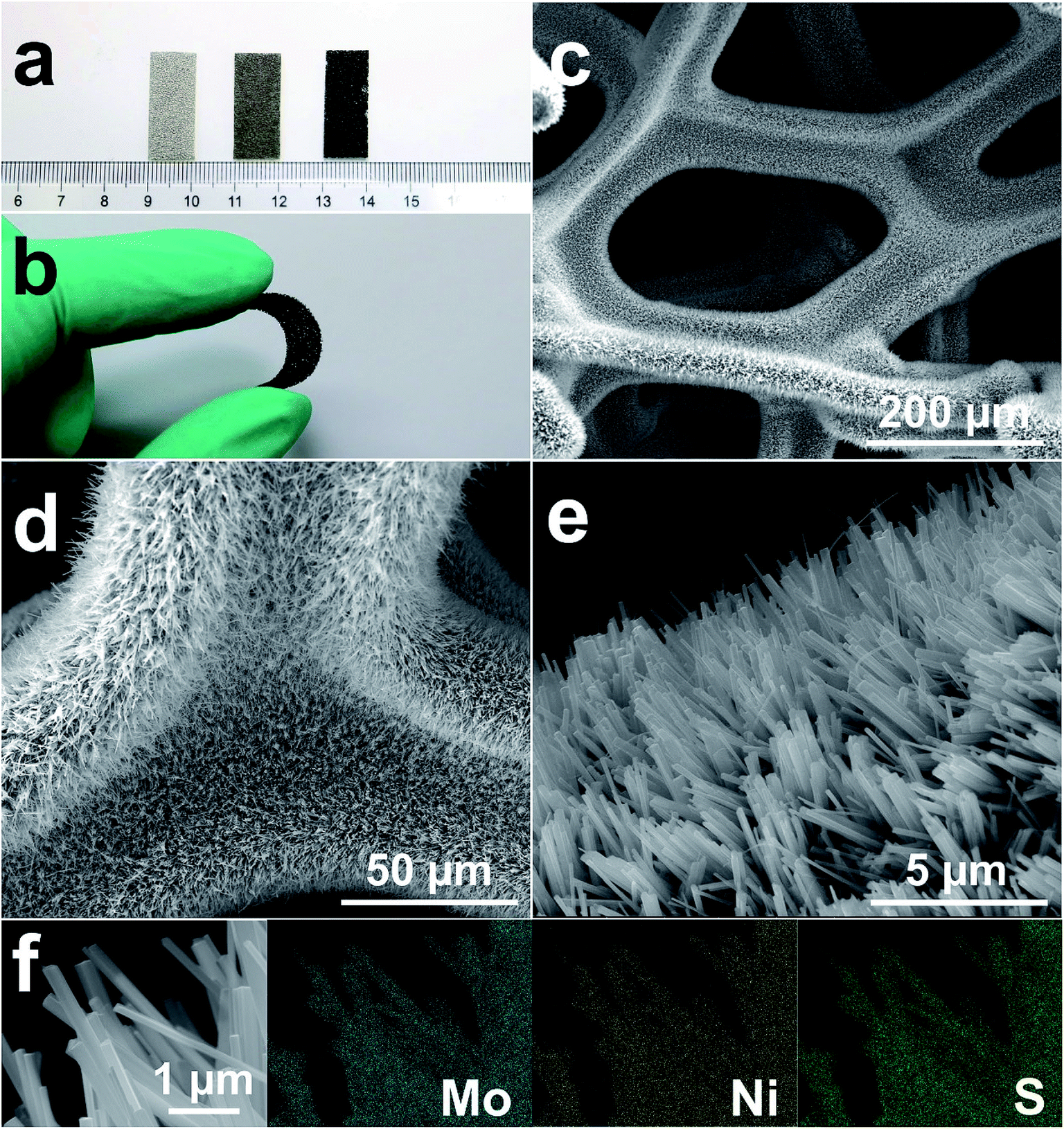 | ||
| Fig. 1 (a) The photograph of NF (left), Ni3S2/NF (middle), MoS2/Ni3S2/NF (right); (b) the photograph of the bending MoS2/Ni3S2/NF electrode; (c–e) the SEM images of MoS2/Ni3S2/NF at different magnifications; (f) SEM and the corresponding elemental mapping images of MoS2/Ni3S2 nanorods. | ||
Furthermore, TEM images of MoS2/Ni3S2 nanorod show individual nanorod obtained from MoS2/Ni3S2/NF by intense ultrasonication treatment. Through the different contrast between MoS2 and Ni3S2, Fig. 2a clearly shows the coating structure of MoS2/Ni3S2 as well as the interface between the outer MoS2 nanosheet and the inner Ni3S2 nanorod. MoS2 nanosheets were demonstrated to be accumulated into Ni3S2 nanorod and formed the edge-rich morphology (Fig. 2b). Fig. 2c exhibits the thin and crumpled MoS2 nanosheets and a more clearly view of the interface between the two phases. The HRTEM image in Fig. 2d displays the 0.62 nm interlayer distance which corresponds to the (002) facets of MoS2 and the a-few-layer structure of MoS2. As shown in Fig. 2e and f, the lattice distances of 0.40 nm and 0.28 nm were ascribed to the (101) and (110) facets of Ni3S2, respectively. To further inspect the two phases, elemental mapping images of the edge of a MoS2/Ni3S2 nanorod are shown in Fig. S8.† The comparison among Fig. S8c–e† shows that S elements and Mo elements have a wider distribution than Ni elements. Only S elements and Mo elements exist on the edge of MoS2/Ni3S2 nanorod without the appearance of Ni, which further demonstrated that the inner phase is Ni3S2 while the outer phase is MoS2. Overall, MoS2/Ni3S2 nanorod were demonstrated to be the coating structure with large surface contact regions between MoS2 and Ni3S2 phases, which may contributed to the synergistic effect for the electrocatalytic activity of MoS2/Ni3S2/NF.
 | ||
| Fig. 2 (a–c) TEM images and (d–f) HRTEM images of MoS2/Ni3S2 nanorod. | ||
The crystallographic structure of MoS2/Ni3S2/NF was further investigated by its XRD pattern shown in Fig. 3a. The strong peaks at 44.9°, 52.2°, and 76.7° are assigned to the metallic nickel of NF substrate as the small image above shown in Fig. 3a.27,58 Peaks at 22°, 31.4°, 38.3°, 50.3°, 55.5° were respectively ascribed to (101), (110), (021), and (122) facets of Ni3S2 referring to Ni3S2/NF's XRD pattern (the small image below shown in Fig. 3a).27,58 And the bulge at 13.6° corresponds to the (002) lattice plane of MoS2.55,56 As reported, the weak and broad (002) diffraction peak indicates the less stacking MoS2 ultrathin nanosheets,59,60 which is consistent with Fig. 2d.
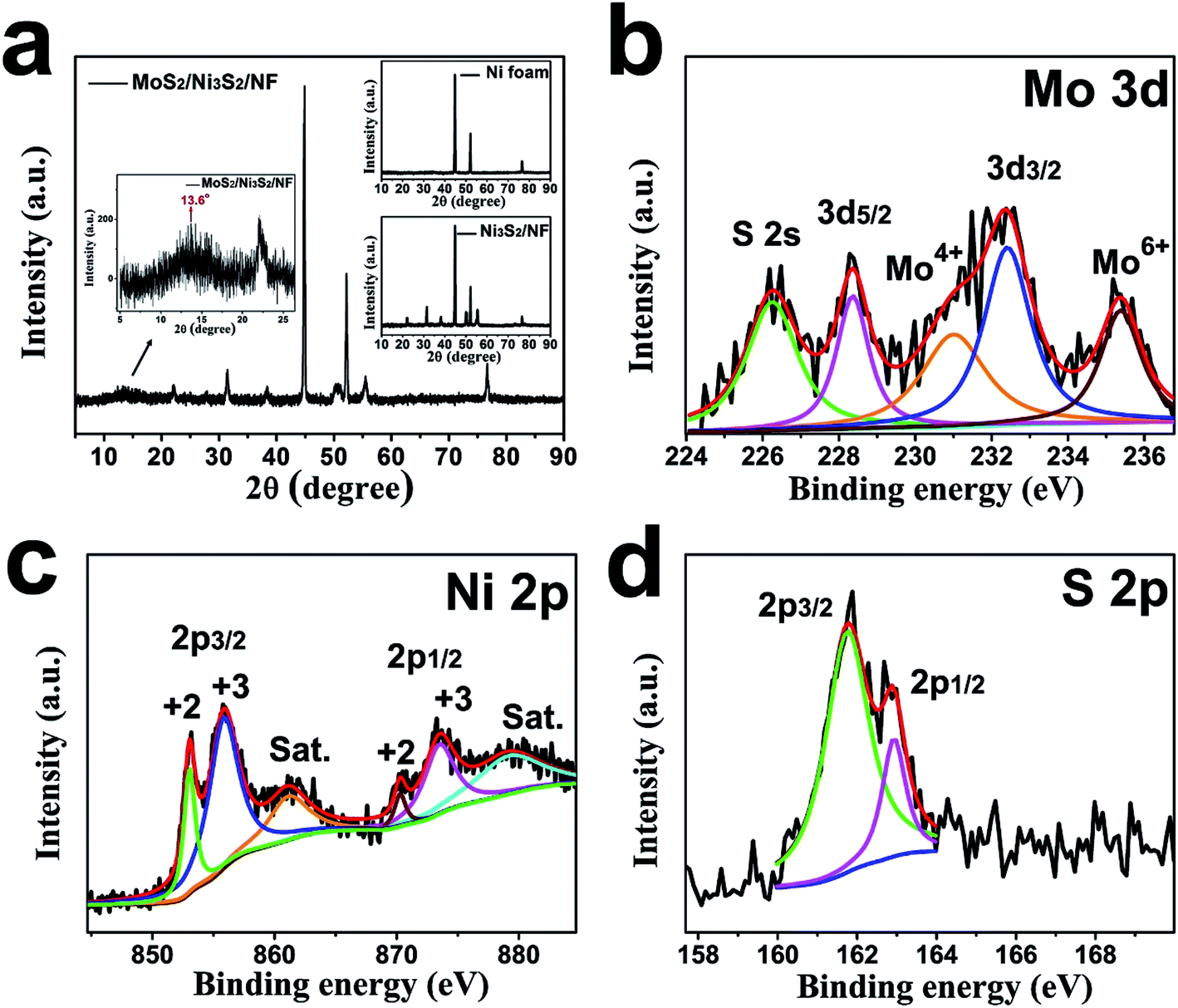 | ||
| Fig. 3 (a) The XRD patterns of MoS2/Ni3S2/NF with partial magnified, NF (the small image above) and Ni3S2/NF (the small image below); XPS spectra of (b) Mo 3d, (c) Ni 2p and (d) S 2p for MoS2/Ni3S2/NF (Sat. means shake-up satellites). | ||
The chemical valence states of the elements in MoS2/Ni3S2/NF were characterized by XPS. For Mo 3d shown in Fig. 3b, two significant peaks located at 228.4 eV and 232.4 eV were ascribed to Mo 3d5/2 and Mo 3d3/2 respectively, together with the binding energy at 231 eV for Mo4+ indicate the +4 oxidation state of Mo. Compared with pure MoS2, the Mo 3d peaks of MoS2/Ni3S2/NF exhibit a little shifts, which illustrates the existence of electronic interactions between MoS2 and Ni3S2.61,62 Besides the S 2s peak in Mo 3d spectrum, the peak at 235.4 eV corresponds to Mo6+ which often coexists with MoS2 as reported.63,64 The Ni 2p spectrum (Fig. 3c) was deconvoluted into two spin–orbit doublets and two shake-up satellites (identified as “Sat.”). The binding energies at 853.2 eV for Ni 2p3/2 and 870.3 eV for Ni 2p1/2 are spin–orbit characteristics of Ni2+, while the binding energies at 856.1 eV for Ni 2p3/2 and 873.7 eV for Ni 2p1/2 are spin–orbit characteristics of Ni3+.35 Moreover, in the S spectrum (Fig. 3d), the peaks at 161.8 eV and 163.0 eV respectively corresponding to S 2p3/2 and S 2p1/2 demonstrate the dominate state of S2−.
3.2 Hydrogen evolution and oxygen evolution activity
In this investigation, we varied the feeding amount of sodium molybdate to control different ratios of MoS2 in catalytic electrodes and tested their corresponding HER and OER performances. As shown in Fig. S9a,† the alignment of MoS2/Ni3S2 nanorods in MoS2/Ni3S2/NF (Mo:S = 0.5:10) with various diameters was sparse and disordered. When the molar ratio of Mo:S was 2:10, much thicker MoS2/Ni3S2 nanorods sparsely spread on NF and tilted in all directions thus attached with each other (Fig. S9c†). And for MoS2/Ni3S2/NF (Mo:S = 3:10), the much thicker and longer MoS2/Ni3S2 nanorods have grown on NF with more free space (Fig. S9d†). As shown in Fig. S10,† the best HER and OER activity has been achieved by MoS2/Ni3S2/NF (Mo:S = 1:10), which may be ascribed to its unique morphology. As shown in Fig. S9b,† compared with the MoS2/Ni3S2/NF of other Mo:S ratios, much slender MoS2/Ni3S2 nanorods with uniform diameter densely and nearly vertically aligned on NF. This well-arranged architecture not only achieved abundant active sites, but also realized the direct contact between the catalytic nanorods and the current collector, thus achieved high electron transfer efficiency. Hereafter, all the electrochemical studies of MoS2/Ni3S2/NF were conducted with the employment of MoS2/Ni3S2/NF (Mo:S = 1:10).
First, the HER electrocatalytic activity of MoS2/Ni3S2/NF was studied by LSV measurements in alkaline solution, 1.0 M KOH (Fig. 4). Herein, commercial Pt/C with negligible onset overpotential was tested as references. The onset overpotential of MoS2/Ni3S2/NF was demonstrated to be ∼50 mV (Fig. 4b), which was much smaller than those of Ni3S2/NF (126 mV), MoS2/Ni3S2 (NP) (179 mV) and NF (198 mV). As shown in Fig. 4a, compared with MoS2/Ni3S2 (NP), Ni3S2/NF and NF, further negative potential causes a much larger cathodic current density of MoS2/Ni3S2/NF. As a consequence, MoS2/Ni3S2/NF could afford the catalytic current density of 10, 100, 200 and 300 mA cm−2 at very low overpotential of 187, 274, 300 and 320 mV, respectively. Especially the small overpotential of 187 mV to reach 10 mA cm−2 is much lower than those of Ni3S2/NF (242 mV), MoS2/Ni3S2 (NP) (269 mV), and the reported non-precious catalysts including Co3O4@NCNTs/CP (380 mV), CoOx/CN (232 mV), Ni3S2/NF (223 mV), NiFe LDH/NF (210 mV), NiCo2S4 NW/NF (210 mV), Ni2P/NF (200 mV), and so on (see Table S2† for more details). Moreover, the Tafel plots in Fig. 4c were recorded with the linear regions which were fitted into the Tafel equation, yielding Tafel slopes of 90 mV dec−1 for MoS2/Ni3S2/NF, 87 mV dec−1 for Ni3S2/NF, 91 mV dec−1 for MoS2/Ni3S2 (NP) and 100 mV dec−1 for NF, respectively. On the basis of Tafel plots, we have obtained the exchange current density (j0), which is the most inherent measure of catalytic activity. The j0 of MoS2/Ni3S2/NF determined by extrapolation method was calculated to be 0.107 mA cm−2, much larger than those of Ni3S2/NF (0.0254 mA cm−2) and MoS2/Ni3S2 (NP) (0.013 mA cm−2). In contrast, the large j0 of MoS2/Ni3S2/NF implies the more favorable HER kinetics on it and higher HER catalytic activity.
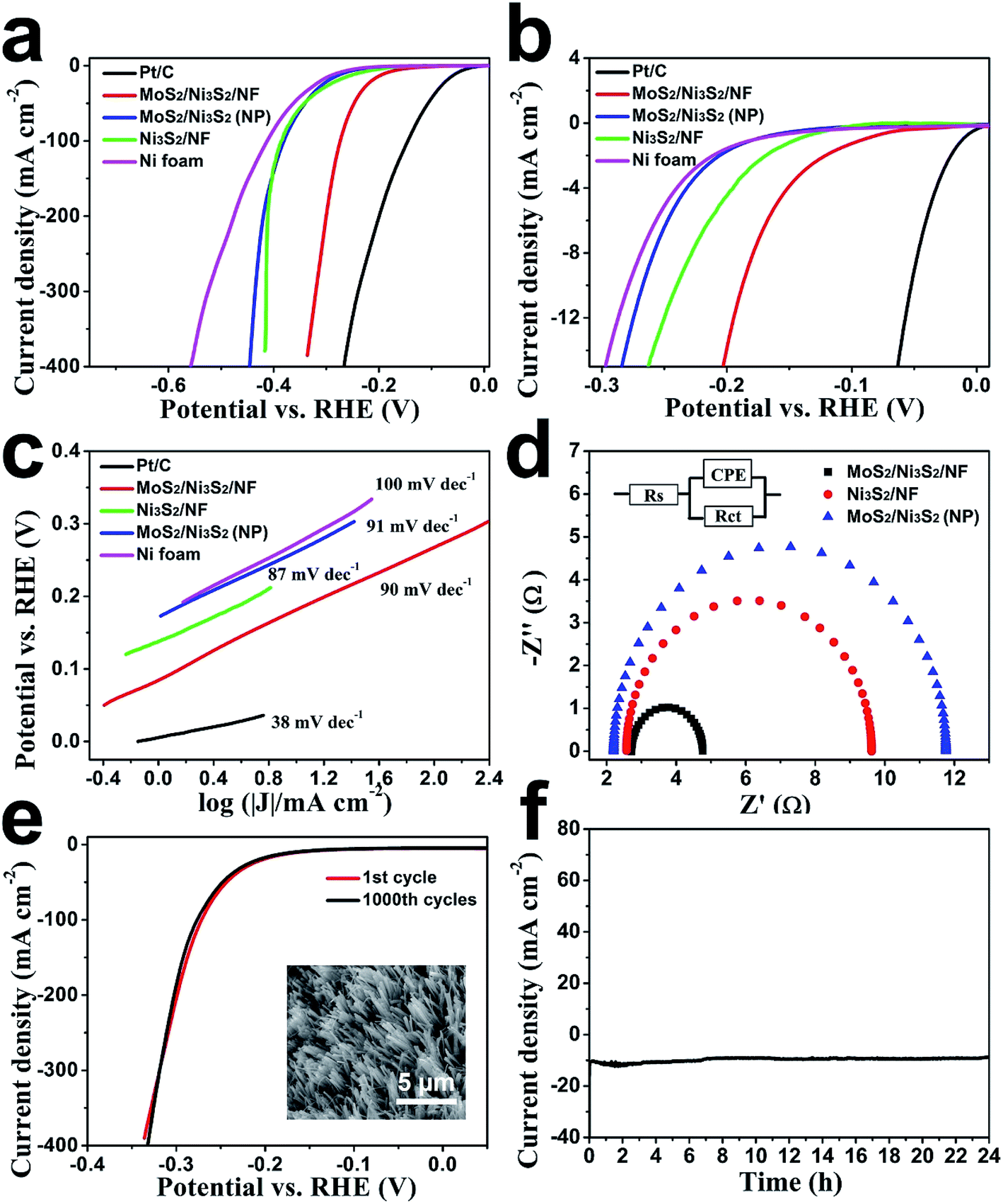 | ||
| Fig. 4 (a) LSV curves of Pt/C, MoS2/Ni3S2/NF, Ni3S2/NF, MoS2/Ni3S2 (NP) and NF in 1.0 M KOH, the (b) expanded region around the onsets of these polarization curves and the (c) corresponding Tafel plots of Pt/C, MoS2/Ni3S2/NF, Ni3S2/NF, MoS2/Ni3S2 (NP) and NF. (d) Nyquist plots of MoS2/Ni3S2/NF, MoS2/Ni3S2 (NP) and Ni3S2/NF measured at an overpotential of 275 mV. The inset circuit diagram shows the equivalent circuit model used to fit the experimental data. (e) Polarization curves for MoS2/Ni3S2/NF initially and after 1000 CV cycles were displayed. Inset: SEM image of MoS2/Ni3S2/NF after 1000 CV cycles (f) time-dependence of cathodic current density curve for MoS2/Ni3S2/NF. | ||
The electrode kinetics of HER catalyzed by the MoS2/Ni3S2/NF was investigated by the electrochemical impedance spectroscopy (EIS) as shown in Fig. 4d. In the electrochemical impedance spectrum, the charge transfer resistance (Rct) of MoS2/Ni3S2/NF (2.05 Ω), which is much smaller than those of Ni3S2/NF (7.05 Ω) and MoS2/Ni3S2 (NP) (9.56 Ω), indicates higher electron transfer efficiency thus higher hydrogen evolution efficiency. On one hand, it should be ascribed to the direct connection between NF and MoS2/Ni3S2, which notably accelerate the electron transport rate between the current collector and the active sites of electrocatalysts. On the other hand, the well-arranged MoS2/Ni3S2 nanorod arrays worked as numerous electric wires for efficient electron transfer from the current collector to the electrolyte. Moreover, the nearly vertical alignment of the catalyst best utilized the conductivity of the substrates.56,60,65 Consequently, electrons could be transported efficiently and rapidly from MoS2/Ni3S2/NF to the electrolyte, thus more hydrogen generated on the surface of this 3D electrode.
We further inspected the durability of the MoS2/Ni3S2/NF catalytic electrode. First, we employed CV measurement for this investigation. As shown in Fig. 4e, after 1000 CV cycles, MoS2/Ni3S2/NF electrode reveals a polarization curve similar to the initial one. The SEM image in the inset of Fig. 4e shows that MoS2/Ni3S2/NF maintains the original morphology after 1000 CV cycles. The durability of this electrode in a continuous HER process was also explored at the static potential of −0.183 V for 24 h. The slight degradation of the current density shown in Fig. 4f suggests the good stability of this catalytic electrode.
Subsequently, the OER electrocatalytic activity of MoS2/Ni3S2/NF was assessed in the same electrolyte (1.0 M KOH). As shown in Fig. 5a, the quasi-reversible oxidation (1.41 V) and reduction (1.33 V) peaks prior to OER catalytic current MoS2/Ni3S2/NF exhibited correspond to the redox reaction of the surface Ni species21 and expressed the transformation between NiII species and NiIII species as most Ni-based electrocatalysts have performed.35,40,66 To avoid the interference of the strong oxidative peak in the forward scan, we employed the cathodic sweep curve (reverse scan) in the corresponding CV curve for quantitative comparison. As shown in Fig. S11,† the onset overpotential of MoS2/Ni3S2/NF was approximately 209 mV, much smaller than those of IrO2 (251 mV), Ni3S2/NF (321 mV), MoS2/Ni3S2 (NP) (327 mV) and NF (381 mV). In addition, MoS2/Ni3S2/NF reached the catalytic current density of 10, 100, 200 and 300 mA cm−2 at the extremely low overpotentials of 217, 275, 313, and 335 mV respectively. Most importantly, the small overpotential of 217 mV to reach 10 mA cm−2 which is much lower than those of IrO2 (293 mV), Ni3S2/NF (378 mV), MoS2/Ni3S2 (NP) (415 mV), NF (545 mV), and many other earth-abundant bifunctional catalysts in Table S2,† demonstrates the higher OER activity of MoS2/Ni3S2/NF. Moreover, the kinetics of the catalysts were identified by the corresponding Tafel plots. As shown in Fig. 5b, the Tafel slope of MoS2/Ni3S2/NF is 38 mV dec−1, much smaller than those of IrO2 (72 mV dec−1), Ni3S2/NF (94 mV dec−1), MoS2/Ni3S2 (NP) (110 mV dec−1) and NF (148 mV dec−1). The small Tafel slope of MoS2/Ni3S2/NF suggests much more rapid reaction kinetics on this catalytic electrode and reveals the superior OER catalytic activity of MoS2/Ni3S2/NF.
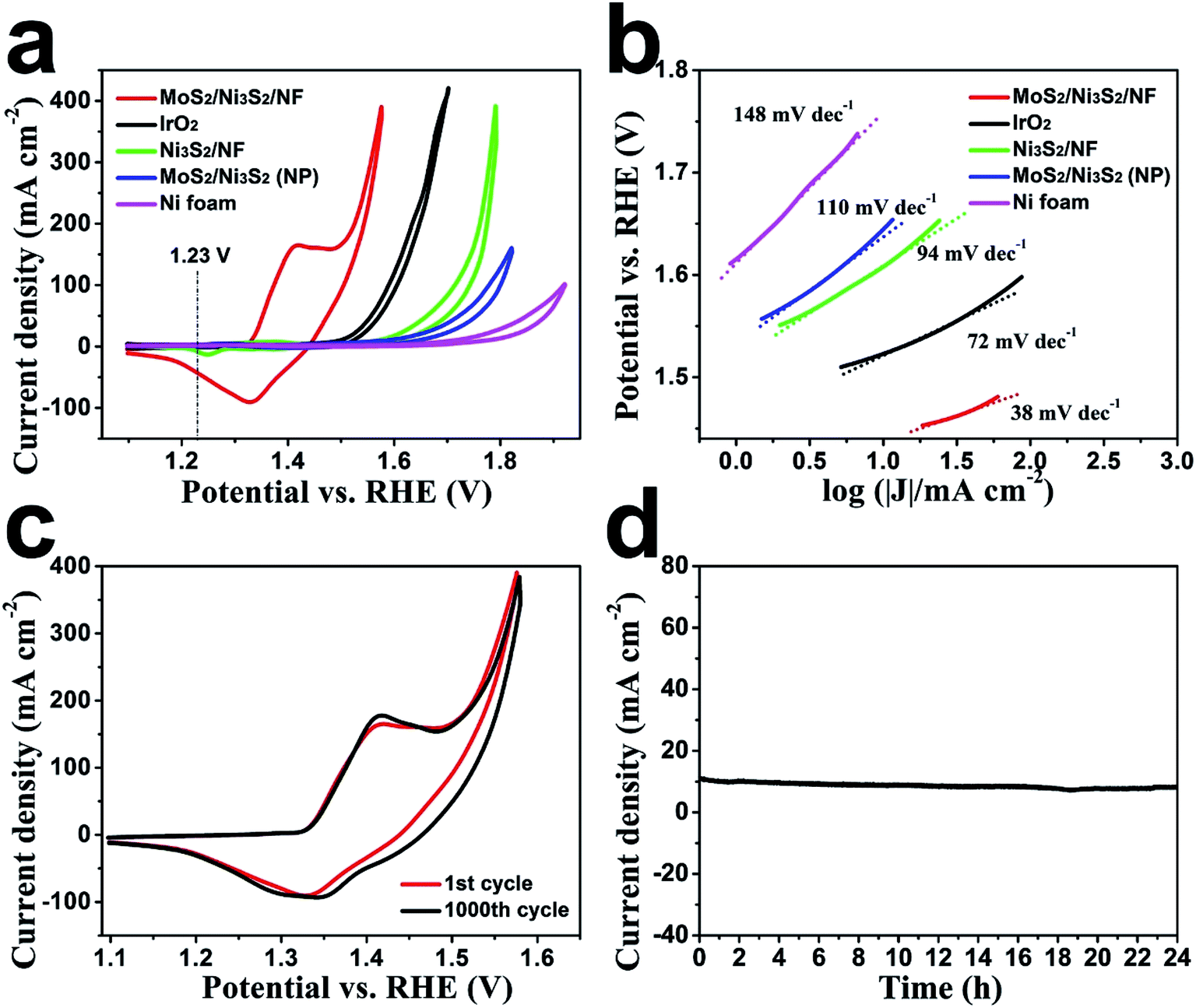 | ||
| Fig. 5 (a) Cyclic voltammograms of MoS2/Ni3S2/NF, IrO2, Ni3S2/NF, MoS2/Ni3S2 (NP) and NF in 1.0 M KOH; (b) the corresponding Tafel plots of MoS2/Ni3S2/NF, IrO2, Ni3S2/NF, MoS2/Ni3S2 (NP) and NF. (c) CV curves for MoS2/Ni3S2/NF initially and after 1000 CV cycles were displayed. (d) Time-dependence of anodic current density curve for MoS2/Ni3S2/NF. | ||
To comprehensively investigate the OER catalytic performance of MoS2/Ni3S2/NF electrode, its OER stability was further explored. As shown in Fig. 5c, only slight decays in current density have been observed after 1000 CV cycles. Additionally, as shown by the time-dependent current density curve in Fig. 5d, at the static potential of 1.437 V, slight degradation of the anodic current observed during 24 h has proved the long-term OER stability of MoS2/Ni3S2/NF.
Since the electron transfer efficiency discussed above (Fig. 4d) and the effective electrochemically active area are two essential factors for HER and OER catalytic activity, the effective electrochemically active area of MoS2/Ni3S2/NF was further estimated by employing CV measurements to measure the electrochemical double-layer capacitances (EDLCs, Cdl). As shown in Fig. 6, potential ranging from 0.822 to 0.922 V without Faradic current was selected to obtain the CV curves at various scan rates from 20 mV s−1 to 200 mV s−1, which meant that the current response in this region was attributed to the charging of the double layer. The halves of the positive and negative current density differences at the center of the scanning potential range are plotted versus the voltage scan rate in Fig. 6d, in which the slopes are the EDLCs. The Cdl of MoS2/Ni3S2/NF is 10.92 mF cm−2, which is about 13 times of the Ni3S2/NF's Cdl (0.85 mF cm−2) and 57 times of the MoS2/Ni3S2 (NP)'s Cdl (0.19 mF cm−2). The much larger Cdl of MoS2/Ni3S2/NF indicates the large effective electrochemical active area and abundant exposed active sites, which may be attributed to the elaborately designed architecture of MoS2/Ni3S2/NF. On one hand, the slender MoS2/Ni3S2 nanorod arrays uniformly, densely and nearly vertically grown on NF exposed numerous active sites. And the hierarchical 3D structure possessed sufficient space which benefited easy diffusion of the electrolyte into all well-exposed active sites thus notably increased the effective electrochemically active area. On the other hand, the covering structure of MoS2 ultrathin nanosheets wrapping Ni3S2 nanorods constructed large surface contact regions, then created more synergistic active sites for HER and OER.58,61,67
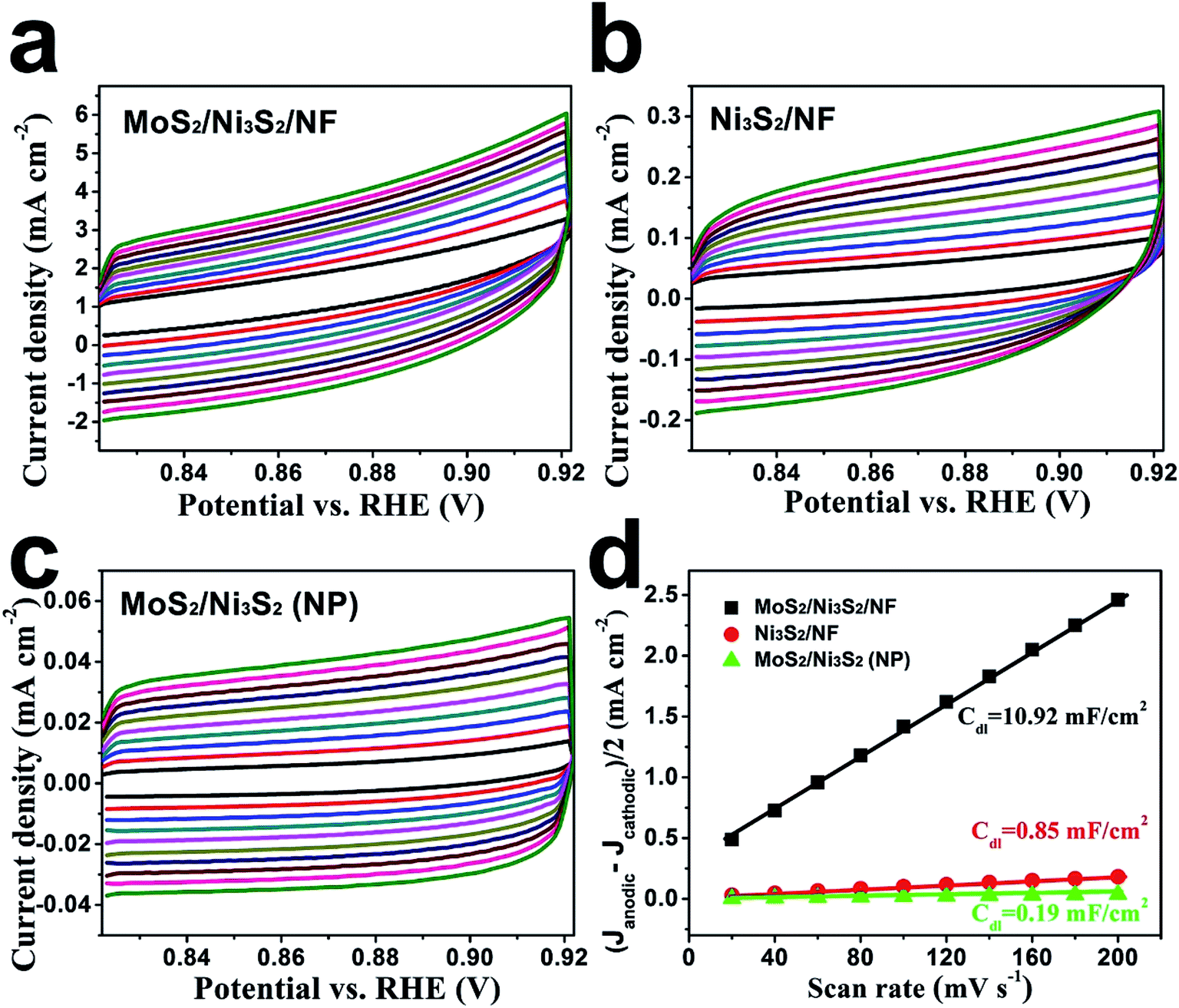 | ||
| Fig. 6 Cyclic voltammograms of (a) MoS2/Ni3S2/NF, (b) Ni3S2/NF and (c) MoS2/Ni3S2 (NP). The scan rates are 20, 40, 60, 80, 100, 120, 140, 160, 180, 200 mV s−1 respectively. (d) Linear fitting of the capacitive currents of the catalysts versus scan rates. | ||
3.3 Overall water splitting
Based on the aforementioned excellent HER and OER catalytic performance of MoS2/Ni3S2/NF, it is reasonably to anticipate that the MoS2/Ni3S2/NF can serve as a bifunctional 3D catalytic electrode for overall water splitting. To test this hypothesis, a water-splitting electrolyzer was constructed by employing MoS2/Ni3S2/NF as both the anode and the cathode (MoS2/Ni3S2/NF‖MoS2/Ni3S2/NF) with 1.0 M KOH as the electrolyte. As control experiments, we respectively prepared electrolyzers consisting of IrO2‖Pt/C, Ni3S2/NF‖Ni3S2/NF, MoS2/Ni3S2 (NP)‖MoS2/Ni3S2 (NP) and Ni foam‖Ni foam. As shown in Fig. 7a, the performance of MoS2/Ni3S2/NF catalyst couple is superior to those of others, and surpasses the benchmark HER and OER catalyst couple, IrO2‖Pt/C. For MoS2/Ni3S2/NF, it only needed low cell voltages of 1.467, 1.593, 1.640 and 1.661 V to afford the catalytic current density of 10, 100, 200 and 300 mA cm−2 respectively, lower than those of the bifunctional catalysts listed in Table S2,† which demonstrates its excellent overall water splitting catalytic activity. In addition, during the overall water splitting process, gas bubbles vigorously generated and released from both electrodes can be clearly observed in the electrolyzer (Fig. S12 and Video S1†).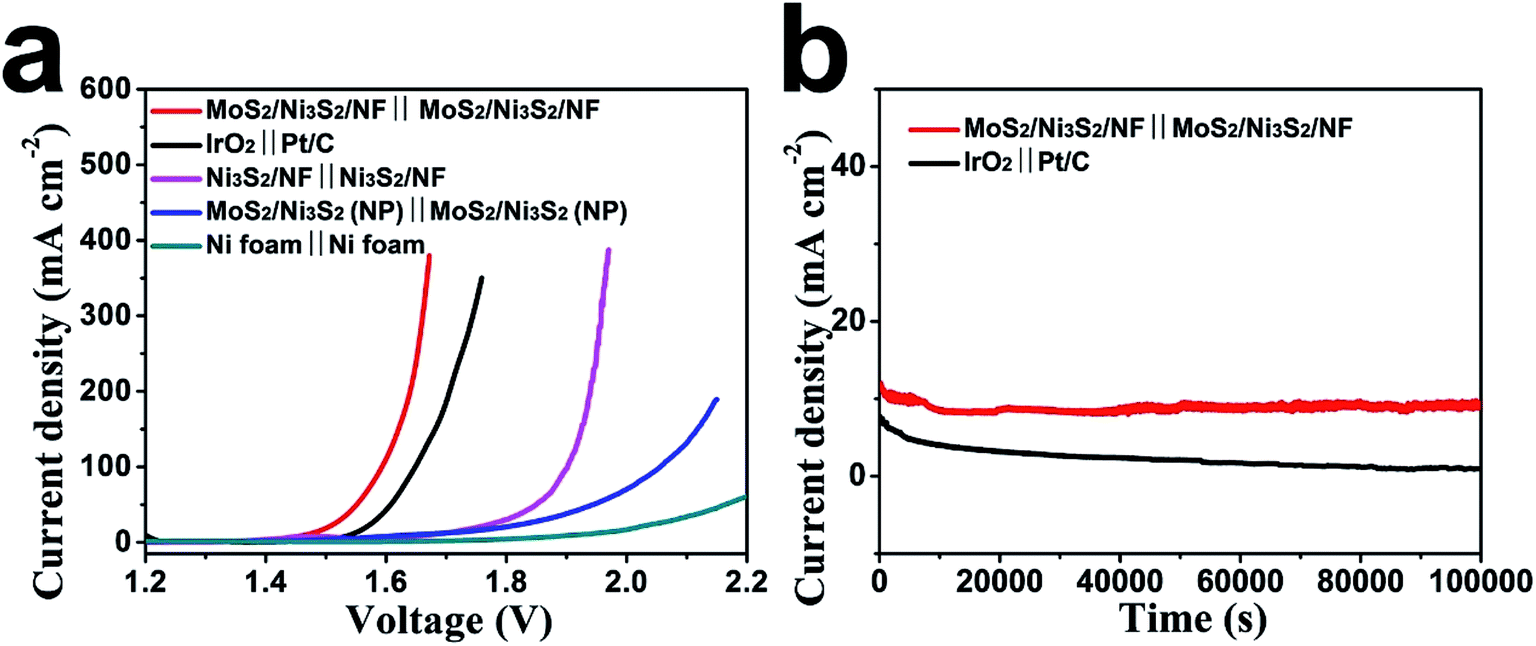 | ||
| Fig. 7 (a) Polarization curves of MoS2/Ni3S2/NF‖MoS2/Ni3S2/NF, IrO2‖Pt/C, Ni3S2/NF‖Ni3S2/NF, MoS2/Ni3S2 (NP)‖MoS2/Ni3S2 (NP) and Ni foam‖Ni foam in a two-electrode system. (b) Time-dependence of current density curve for MoS2/Ni3S2/NF‖MoS2/Ni3S2/NF and IrO2‖Pt/C in a two-electrode system. | ||
The stability of MoS2/Ni3S2/NF for overall water splitting was further explored as shown in Fig. 7b. The catalytic current density slightly decreased on MoS2/Ni3S2/NF‖MoS2/Ni3S2/NF at the static potential of 1.470 V within 10000 s. As a comparison, continuous and severe degradation of the current density occurred on IrO2‖Pt/C couple at the static potential of 1.530 V for this 10000 s electrolysis. It confirms the robust long-term stability of MoS2/Ni3S2/NF for overall water splitting.
4. Conclusion
In conclusion, we have successfully synthesized MoS2/Ni3S2/NF as a novel bifunctional catalytic electrode for overall water splitting by a simple one-step hydrothermal procedure. The uniform, dense and nearly vertical self-growth of MoS2/Ni3S2 nanorod arrays on the 3D substrate NF led to the large surface area and abundant exposed active sites. Moreover, the coating structure of MoS2/Ni3S2 has formed large surface contact regions between Ni3S2 and MoS2, which significantly increased the synergistic effect of the hybrid catalyst. In addition, NF as Ni source realized the in situ direct growth of MoS2/Ni3S2 nanorods from NF and resulted in enhanced electron transfer efficiency and stability. With the tailored architecture, the optimal MoS2/Ni3S2/NF acquired excellent catalytic activity and good durability for both HER and OER. It only needed 187, 274, 300, 320 mV overpotential to reach the catalytic current density of 10, 100, 200, 300 mA cm−2 respectively for HER, and 217, 275, 313, 335 mV overpotential to reach the current density of 10, 100, 200, 300 mA cm−2 respectively for OER. Most importantly, during the overall water splitting process, MoS2/Ni3S2/NF as catalytic electrodes for both cathode and anode approached 10, 100, 200 and 300 mA cm−2 at the low cell voltages of 1.467, 1.593, 1.640 and 1.661 V, respectively, along with good stability. The overall water splitting electrocatalytic performance of MoS2/Ni3S2/NF not only surpassed the benchmark OER and HER catalyst couple IrO2‖Pt/C, but also achieved better performance compared with many other non-noble-based bifunctional electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Science Foundation of China University of Petroleum, Beijing (No. 2462016YJRC022).References
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Z. Ma, X. Yuan, L. Li, Z.-F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 CAS.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeisser, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed.
- H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi, X. Meng, K. Wu and J. Ye, ACS Nano, 2016, 10, 684–694 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- L. C. Seitz, Z. Chen, A. J. Forman, B. A. Pinaud, J. D. Benck and T. F. Jaramillo, Chemsuschem, 2014, 7, 1372–1385 CrossRef CAS PubMed.
- L. F. Petrik, Z. G. Godongwana and E. I. Iwuoha, J. Power Sources, 2008, 185, 838–845 CrossRef CAS.
- N. Zhang, W. Ma, F. Jia, T. Wu, D. Han and L. Niu, Int. J. Hydrogen Energy, 2016, 41, 3811–3819 CrossRef CAS.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- L. An, L. Huang, P. Zhou, J. Yin, H. Liu and P. Xi, Adv. Funct. Mater., 2015, 25, 6814–6822 CrossRef CAS.
- W. Xu, Z. Lu, P. Wan, Y. Kuang and X. Sun, Small, 2016, 12, 2492–2498 CrossRef CAS PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Graetzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- J. Tian, N. Cheng, Q. Liu, X. Sun, Y. He and A. M. Asiri, J. Mater. Chem. A, 2015, 3, 20056–20059 CAS.
- X. Yu, T. Hua, X. Liu, Z. Yan, P. Xu and P. Du, ACS Appl. Mater. Interfaces, 2014, 6, 15395–15402 CAS.
- T. Sharifi, E. Gracia-Espino, X. Jia, R. Sandstrom and T. Wagberg, ACS Appl. Mater. Interfaces, 2015, 7, 28148–28155 CAS.
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785–3790 CrossRef CAS PubMed.
- C.-C. Hou, S. Cao, W.-F. Fu and Y. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 28412–28419 CAS.
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, ACS Catal., 2016, 6, 714–721 CrossRef CAS.
- Y.-P. Zhu, Y.-P. Liu, T.-Z. Ren and Z.-Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337–7347 CrossRef CAS.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- J. Li, J. Li, X. Zhou, Z. Xia, W. Gao, Y. Ma and Y. Qu, ACS Appl. Mater. Inter., 2016, 8, 10826–10834 CrossRef CAS PubMed.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067–4077 CrossRef CAS.
- Y. Wang, C. Xie, D. Liu, X. Huang, J. Huo and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18652–18657 CAS.
- L.-L. Feng, G. Yu, Y. Wu, G.-D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- W. Zhu, X. Yue, W. Zhang, S. Yu, Y. Zhang, J. Wang and J. Wang, Chem. Commun., 2016, 52, 1486–1489 RSC.
- I. H. Kwak, H. S. Im, D. M. Jang, Y. W. Kim, K. Park, Y. R. Lim, E. H. Cha and J. Park, ACS Appl. Mater. Interfaces, 2016, 8, 5327–5334 CAS.
- B. T. Sneed, A. P. Young, D. Jalalpoor, M. C. Golden, S. Mao, Y. Jiang, Y. Wang and C.-K. Tsung, ACS Nano, 2014, 8, 7239–7250 CrossRef CAS PubMed.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 CAS.
- Q. Zhang, Y. Wang, Y. Wang, A. M. Al-Enizi, A. A. Elzatahry and G. Zheng, J. Mater. Chem. A, 2016, 4, 5713–5718 CAS.
- Y. Wu, G.-D. Li, Y. Liu, L. Yang, X. Lian, T. Asefa and X. Zou, Adv. Funct. Mater., 2016, 26, 4839–4847 CrossRef CAS.
- X. Li, G. Guan, X. Du, J. Cao, X. Hao, X. Ma, A. D. Jagadale and A. Abudula, Chem. Commun., 2015, 51, 15012–15014 RSC.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 4661–4672 CrossRef CAS.
- G.-F. Chen, T. Y. Ma, Z.-Q. Liu, N. Li, Y.-Z. Su, K. Davey and S.-Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314–3323 CrossRef CAS.
- X. Xu, P. Du, Z. Chen and M. Huang, J. Mater. Chem. A, 2016, 4, 10933–10939 CAS.
- Q. Liu, S. Gu and C. M. Li, J. Power Sources, 2015, 299, 342–346 CrossRef CAS.
- D. Liu, Q. Lu, Y. Luo, X. Sun and A. M. Asiri, Nanoscale, 2015, 7, 15122–15126 RSC.
- P. Chen, T. Zhou, M. Zhang, Y. Tong, C. Zhong, N. Zhang, L. Zhang, C. Wu and Y. Xie, Adv. Mater., 2017, 29, 1701584 CrossRef PubMed.
- K. Krishnamoorthy, G. K. Veerasubramani, S. Radhakrishnan and S. J. Kim, Chem. Eng. J., 2014, 251, 116–122 CrossRef CAS.
- H. Huo, Y. Zhao and C. Xu, J. Mater. Chem. A, 2014, 2, 15111–15117 CAS.
- C.-W. Su, J.-M. Li, W. Yang and J.-M. Guo, J. Phys. Chem. C, 2014, 118, 767–773 CAS.
- D. Li, X. Li, X. Hou, X. Sun, B. Liu and D. He, Chem. Commun., 2014, 50, 9361–9364 RSC.
- T.-W. Lin, C.-J. Liu and C.-S. Dai, Appl. Catal., B, 2014, 154, 213–220 CrossRef.
- C. Tang, Z. Pu, Q. Liu, A. M. Asiri, Y. Luo and X. Sun, Int. J. Hydrogen Energy, 2015, 40, 4727–4732 CrossRef CAS.
- N. Jiang, Q. Tang, M. Sheng, B. You, D.-e. Jiang and Y. Sun, Catal. Sci. Technol., 2016, 6, 1077–1084 CAS.
- W. Zhou, X.-J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 CAS.
- X. Shang, X. Li, W.-H. Hu, B. Dong, Y.-R. Liu, G.-Q. Han, Y.-M. Chai, Y.-Q. Liu and C.-G. Liu, Appl. Surf. Sci., 2016, 378, 15–21 CrossRef CAS.
- C. Ouyang, X. Wang, C. Wang, X. Zhang, J. Wu, Z. Ma, S. Dou and S. Wang, Electrochim. Acta, 2015, 174, 297–301 CrossRef CAS.
- J.-J. Lv, J. Zhao, H. Fang, L.-P. Jiang, L.-L. Li, J. Ma and J.-J. Zhu, Small, 2017, 13, 1700264 CrossRef PubMed.
- S. Qu, J. Huang, J. Yu, G. Chen, W. Hu, M. Yin, R. Zhang, S. Chu and C. Li, ACS Appl. Mater. Interfaces, 2017, 9, 29660–29668 CAS.
- S. Trasatti, J. Electroanal. Chem., 1971, 33, 351–378 CrossRef CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- N. Zhang, W. Ma, T. Wu, H. Wang, D. Han and L. Niu, Electrochim. Acta, 2015, 180, 155–163 CrossRef CAS.
- N. Zhang, S. Gan, T. Wu, W. Ma, D. Han and L. Niu, ACS Appl. Mater. Interfaces, 2015, 7, 12193–12202 CAS.
- J. Yu, G. Chen, J. Sunarso, Y. Zhu, R. Ran, Z. Zhu, W. Zhou and Z. Shao, Adv. Sci., 2016, 3, e1603206 Search PubMed.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6701–6706 Search PubMed.
- C.-B. Ma, X. Qi, B. Chen, S. Bao, Z. Yin, X.-J. Wu, Z. Luo, J. Wei, H.-L. Zhang and H. Zhang, Nanoscale, 2014, 6, 5624–5629 RSC.
- Z. H. Deng, L. Li, W. Ding, K. Xiong and Z. D. Wei, Chem. Commun., 2015, 51, 1893–1896 RSC.
- H. Zhu, J. Zhang, R. Yanzhang, M. Du, Q. Wang, G. Gao, J. Wu, G. Wu, M. Zhang, B. Liu, J. Yao and X. Zhang, Adv. Mater., 2015, 27, 4752–4759 CrossRef CAS PubMed.
- H. Zhu, F. Lyu, M. Du, M. Zhang, Q. Wane, J. Yao and B. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 22126–22137 CAS.
- Y. Yan, X. Ge, Z. Liu, J.-Y. Wang, J.-M. Lee and X. Wang, Nanoscale, 2013, 5, 7768–7771 RSC.
- X. Xia, Z. Zheng, Y. Zhang, X. Zhao and C. Wang, Int. J. Hydrogen Energy, 2014, 39, 9638–9650 CrossRef CAS.
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- M. Kuang and G. Zheng, Small, 2016, 12, 5656–5675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07667c |
| This journal is © The Royal Society of Chemistry 2017 |