
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hollow Pt–Ag nanoparticles by oxygen-assisted acid etching as electrocatalysts for the oxygen reduction reaction†
Shutang Chena,
Sravan Thotaa,
Gurpreet Singhb,
Túlio J. Aímolab,
Christopher Koenigsmann *b and
Jing Zhao*ac
*b and
Jing Zhao*ac
aDepartment of Chemistry, University of Connecticut, Storrs, Connecticut 06269, USA. E-mail: jing.zhao@uconn.edu; Fax: +1-860-4862981; Tel: +1-860-4862443
bDepartment of Chemistry, Fordham University, Bronx, New York 10458, USA. E-mail: ckoenigsmann@fordham.edu; Fax: +1-718-8174432; Tel: +1-718-8174439
cInstitute of Materials Science, University of Connecticut, Storrs, Connecticut 06269, USA
First published on 4th October 2017
Abstract
Hollow bimetallic nanostructures have recently shown promising performance for the oxygen reduction reaction (ORR) in fuel cells. In this work, we report the synthesis of hollow Pt–Ag nanoparticles of varying sizes by O2-assisted acid etching of Ag@Pt core@shell nanostructures at room temperature. With a Pt shell less than 6 nm thick, the O2 dissolved in acetic acid could oxidize the Ag core. Subsequently, silver oxide was dissolved in acetic acid and turned into Ag+ ions. During this process, the Ag atoms diffused into the lattice of the Pt shell, and Ag@Pt core@shell nanostructures evolved into hollow Pt–Ag alloy nanoparticles. The as-synthesized hollow Pt–Ag nanocatalysts maintained specific ORR activities that were enhanced beyond the specific activity for commercial carbon supported Pt nanoparticles. The 5.8 nm hollow Pt–Ag nanoparticles displayed the highest activity of 1.12 mA cm−2. Over the course of an accelerated durability test, the 5.8 nm nanoparticles retained 95% and 87% of their initial electrochemical surface area and specific ORR activity. The enhanced activity and durability can be ascribed to the high surface area of the porous structure and the new d-band center due to the hollow morphology and Pt–Ag alloy formation. This work demonstrates a simple strategy for fabricating small porous nanoparticles, which can be potentially used as electrocatalysts in PEM fuel cells.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are among the most promising technologies to power electric vehicles, because they can directly convert chemical energy into electricity at efficiency higher than internal combustion engines with no pollutant emission.1–4 In a typical PEMFC, carbon supported Pt nanoparticles (NPs) are used to catalyze the cathode reaction, i.e. oxygen reduction reaction (ORR).5,6 Although Pt/C has reasonable catalytic performance towards the ORR, the high cost and poor durability of Pt catalysts hinders the large scale applications of PEMFCs. An effective method to lower the cost of catalyst is to alloy Pt with transition metals, such as Ag, Cu, Ni, and Co.7–11 Alloying can also modify the electronic structure of Pt, which further enhances the catalytic activity and durability.12 By tuning the size, morphology, and surface structure of the Pt-based alloy nanostructures, their catalytic performance can be enhanced towards ORR, methanol oxidation reaction (MOR), and formic acid oxidation reaction.13–17 Among the various Pt-alloy electro-catalysts, Pt–Ag is particularly interesting because Pt–Ag alloy is less susceptible to dissolution in weak acidic electrolytes compared to Pt–Cu, Pt–Ni and Pt–Co.18–25Pt-Based hollow NPs have significantly enhanced catalytic activity and durability toward ORR compared to their solid counterpart, because their hollow interiors and porous walls increase the specific surface area that participates in the catalytic reaction.26–40 The most common synthetic strategy to fabricate hollow Pt–Ag nanocatalysts is based on the galvanic replacement between a Pt precursor and sacrificial Ag templates, pioneered by the Xia group.24,41–43 Their recent study has demonstrated that Pt–Ag alloy nanocages have great catalytic performance for ORR.24 Alternatively, our group reported that thermal annealing of Ag@Pt core@shell NPs would induce a structural reconstruction in the solid NPs and convert them into hollow Pt–Ag NPs.44 But the thermal treatment requires high temperature, which is not desirable in catalyst fabrication. And the catalytic activity and durability of the hollow NPs produced by thermal annealing was not satisfactory for effectively catalyzing ORR and still needs to be improved.
In many previous reports,45–48 Pt-based nanocatalysts are treated with acetic acid prior to electrochemical measurements, in order to exchange the long surface ligand used in the synthesis with the relatively short acetate ligand.35,49 Inspired by this idea, we employ for the first time an acetic acid etching to transform Ag@Pt core@shell NPs into hollow nanostructures with varying shell thickness. Although Ag does not directly react with acetic acid, Ag can dissolve in acetic acid in the presence of oxygen.50,51 The presence of oxygen and also the thickness of Pt shell are critical for the formation of the hollow nanostructures. Under proper conditions, Ag atoms in the Ag@Pt NPs can be oxidized to Ag+ ions. During this process, some of the silver atoms diffuse into the lattice of the Pt shell producing a hollow NP with a Pt–Ag alloy shell i.e. hollow Pt–Ag NPs (Scheme 1).
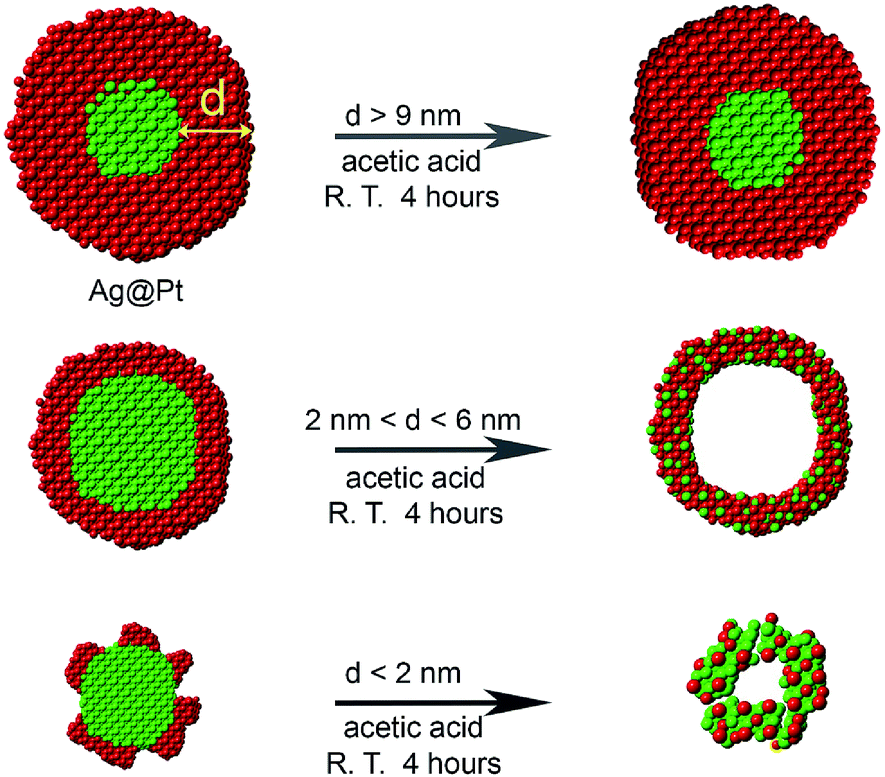 | ||
| Scheme 1 Synthesis of Pt–Ag hollow NPs from Ag@Pt core@shell nanostructures via an oxygen etching process: 4Ag + O2 + 4H+ → 4Ag+ + 2H2O. | ||
The advantage of this process is that it can be performed in solution at room temperature, which is a major advance over our previous method41 that required an extended heating protocol. Since the NPs are treated in acetic acid, the hollow NPs naturally have short acetic acid as the ligands, which can be selectively removed by an electrochemical treatment to activate the particles as catalysts for ORR. Moreover, the size and composition of the hollow nanocatalysts can be adjusted by etching different sized Ag@Pt NPs. We also demonstrate that these acid-etched particles have superior catalytic durability.
Among the different sized hollow catalysts we examined in this study, 5.8 nm hollow Pt–Ag NPs showed the highest specific ORR activity of 1.12 mA cm−2, almost four times of that of the commercial Pt/C catalysts (0.28 mA cm−2). After 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles of an accelerated durability test, the electrochemically active surface area (ESA) and specific activity of the porous Pt–Ag NPs decreased only slightly, demonstrating the high durability of these catalysts.
000 cycles of an accelerated durability test, the electrochemically active surface area (ESA) and specific activity of the porous Pt–Ag NPs decreased only slightly, demonstrating the high durability of these catalysts.
2. Experimental section
2.1 Chemicals
Silver nitrate (AgNO3, 99%), dihydrogen hexachloroplatinate hexahydrate (H2PtCl6·6H2O, 99.9%), oleylamine (OLA, 70%), and hexadecylamine (HDA, 90%) were purchased from Sigma Aldrich. Acetic acid (CH3COOH, ACS purity) was purchased from Alfa. Perchloric acid (HClO4, Optima Grade) was purchased from Fisher Scientific and was diluted with ultrapure water (18.2 MΩ cm) water produced by a Millipore DirectQ5 UV system. Ethanol (200 proof, ACS/USP grade) and isopropyl alcohol (Optima Grade, anhydrous) were purchased from Pharmco-Aaper and Fisher Scientific, respectively. A Nafion solution with a concentration of 0.025% was prepared by diluting a 5% stock solution (Aldrich) in absolute ethanol. Activated carbon black was purchased from Orion. HiSPEC 3000 carbon supported Pt nanoparticles (Pt NP/C) with a 20% precious metal loading (lot: Y16B043) were purchased from Alfa Aesar and served as a commercial standard for ORR catalysts. All chemicals were used as received.2.2 Synthesis of Ag@Pt core@shell NPs
The Ag@Pt core@shell NPs of different sizes were obtained following the previous protocol with slight modification.44 Here, we describe the synthesis of Ag@Pt core@shell NPs with a diameter of 6 nm. Briefly, HDA (40 mmol, 10.73 g), and H2PtCl6·6H2O (0.1 mmol, 51.3 mg) were loaded into a 50 mL three-neck flask. After removal of oxygen in the solution by continuous nitrogen flow, the reaction mixture was heated to 200 °C with a heating rate of 20 °C min−1. AgNO3 (51.0 mg, 0.3 mmol) in 1.0 mL of OLA was quickly injected into the reaction flask after the reaction solution turned gray. And the reaction was allowed to proceed for 20 min at 200 °C. The reaction flask was cooled to 100 °C and ethanol was added, followed by centrifugation for 30 s at 2500 rpm and the precipitate was collected and redispersed in toluene. For the synthesis of 12 nm and 22 nm Ag@Pt NPs, the reaction conditions were kept the same as the above, except that the amount of Ag precursors was decreased to 17.0 mg and 3.4 mg, respectively.2.3 Synthesis of hollow Pt–Ag NPs
Ag@Pt core@shell NPs (10 mg), and 6.0 mL of 25% (volume ratio) acetic acid in water were added to a 10.0 mL vial equipped with a magnetic stir bar. The mixed solution was sonicated for 5 min, and it turned black. The solution was then stirred for 4 h at room temperature. The products were precipitated out by adding ethanol to the solution, and separated by centrifugation at 5000 rpm for 2 min. The final products were re-dispersed in water.2.4 Catalysts preparation
Carbon black (16 mg) was added to 10 mL of a 1:1 mixture of water and ethanol (Vwater:Vethanol = 1:1), and sonicated for 1 h. Subsequently, 4.9 mg of 5.8 nm hollow Pt–Ag nanocatalysts were added to the carbon black solution and the mixed dispersion was stirred for 5 h at room temperature. Pt–Ag nanocatalysts on carbon support (20 wt% Pt) were then collected via centrifugation at 6000 rpm for 20 min and the precipitate was washed twice with ethanol. For the loading of 12.5 nm and 22.4 nm hollow Pt–Ag NPs, the same procedure was applied, except that the amount of catalysts was 4.6 mg and 4.3 mg, respectively.
2.5 Electrochemical measurement
The as-prepared carbon supported hollow Pt–Ag NP catalysts were dispersed into absolute ethanol with a concentration of 2 mg mL−1 forming catalyst inks. The commercial Pt NP/C were dispersed into a 25% isopropyl alcohol in ultrapure water mixture with a concentration of 2 mg mL−1. Prior to electrochemical measurements, glassy carbon rotating disk electrodes (GC-RDE, Pine Instruments, 5 mm) were polished to a mirror finish with aluminum oxide paste. A 5 μL aliquot of the respective catalyst ink was dropped onto the surface of the electrode and allowed to dry in air. The catalyst modified electrodes were sealed with one 5 μL drop of a 0.025% Nafion solution, which was allowed to dry in air. The as-prepared electrodes were then immersed in ultrapure water to remove residual impurities.Electrochemical measurements were performed with a WaveDriver 20 bipotentiostat coupled with an MSR rotating disk electrode apparatus supplied by Pine Instruments. A custom-made electrochemical cell was assembled with a platinum counter electrode (Alfa Aesar, 1.5 cm2) and an Ag/AgCl reference electrode (3 M Cl−) housed in a double junction chamber (BASi instruments). Electrochemical measurements were performed in a 0.1 M HClO4 electrolyte prepared by diluting the concentrated acid in ultrapure water. All potentials are reported with respect to the reversible hydrogen electrode (RHE), unless otherwise specified.
Cyclic voltammetry was performed in de-oxygenated 0.1 M HClO4 with a scan rate of 20 mV s−1. The electrochemically accessible surface area (ESA) was determined from the integration of the hydrogen adsorption peaks in the cyclic voltammogram, utilizing the conversion factor, 0.21 mC cm−2. The capacitive contribution from the carbon support was removed during the integration of the hydrogen adsorption peaks. In this case, the surface of the catalyst may possess a small fraction of silver atoms, which do not contribute to the hydrogen adsorption charge. But we expect this effect to be minimal since the CV results show that there was no detectable oxidation signal from Ag surface sites.
The ORR activity was measured utilizing the thin-layer rotating disk electrode technique.52 Polarization curves were obtained with a scan rate of 10 mV s−1 in oxygen-saturated 0.1 M HClO4 electrolyte with a rotation rate of 1600 rpm. The current in the polarization curve is normalized to the geometric surface area of 0.196 cm2 for the 0.5 mm diameter glassy carbon electrode. The kinetic current (Ik) was calculated at 0.9 V, utilizing the Koutecky–Levich equation, and was normalized to the ESA to provide an area-normalized kinetic current density (Jk, specific activity). Specific activities are reported as the average value of the activity measured from three separately prepared electrodes. The durability of the catalysts was examined utilizing a method outlined by the U.S. Department of Energy adapted for testing ORR catalysts under half-cell conditions.53 The potential is cycled between 0.6 V and 1.0 V at a rate of 50 mV s−1 in a solution of 0.1 M HClO4 left open to the atmosphere. Cyclic voltammograms and polarization curves are obtained in increments of 5000 cycles to determine the surface area and catalytic activity.
2.6 Instrumentation
JEOL 2010 microscope was used to acquire the low resolution TEM images of the NPs with an accelerating voltage of 200 kV. High-resolution TEM (HRTEM) images, high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images, and energy-dispersive X-ray (EDX) mapping were obtained using FEI-Talos microscope at an accelerating voltage of 200 kV. X-ray diffraction (XRD) patterns of the NPs were obtained using a Rigaku Ultima IV Power X-ray Diffractometer with Cu Kα radiation operated at a tube voltage of 40 kV and current of 44 mA. X-ray photoelectron spectrometer (XPS) data were collected on a Thermo Scientific ESCALAB 250Xi XPS. A powder sample was attached to a silica substrate and loaded in the XPS chamber. Calibration of the binding energy was carried out by calibrating the binding energy of the C 1s peak to 284.76 eV.3. Results and discussion
The synthesis of the electrocatalytically active hollow Pt–Ag NPs involves two steps (Scheme 1). In the first step, Ag@Pt core@shell NPs were synthesized by a seed-mediated co-reduction method we have recently developed.44 And then, the Ag core was removed from the core@shell NPs by chemical etching in the presence of O2. Ag@Pt core@shell NPs of three different sizes and Pt shell thickness were synthesized by controlling the molar ratio of Pt/Ag precursors. For the Pt/Ag feeding ratio of 5:2 and 1:1, Ag@Pt core@shell NPs of 22.4 and 12.5 nm were obtained, as determined from the TEM images in Fig. 1. EDX mapping analysis shows that Ag is present in the core and Pt is localized in the shell. The core@shell structure is further confirmed by EDX line scans (Fig. S1†). The Ag trace has one peak at the center of the NP, while the Pt trace has two side peaks. In these two types of core@shell NPs, the Pt shell is complete, and the corresponding Pt shell thickness is 5.6 nm and 2.9 nm for the 22.4 nm and 12.5 nm NPs, respectively (measured from EDX mapping images as shown in Fig. S2†). When the feeding molar ratio of Pt/Ag was reduced to 1:3, Pt grew in islands on the Ag core and thus formed an incomplete Pt shell (Fig. 1F). From these results, we found that as the amount of Ag precursor increased in the reaction, the average size and shell thickness of core@shell NPs decreased. Correspondingly, the atomic ratio of Pt to Ag in the core@shell NPs decreases from 77:23, 54:46, to 28:72, for the 22.4 nm, 12.5 nm, and 5.8 nm NPs (Fig. S3†), respectively.
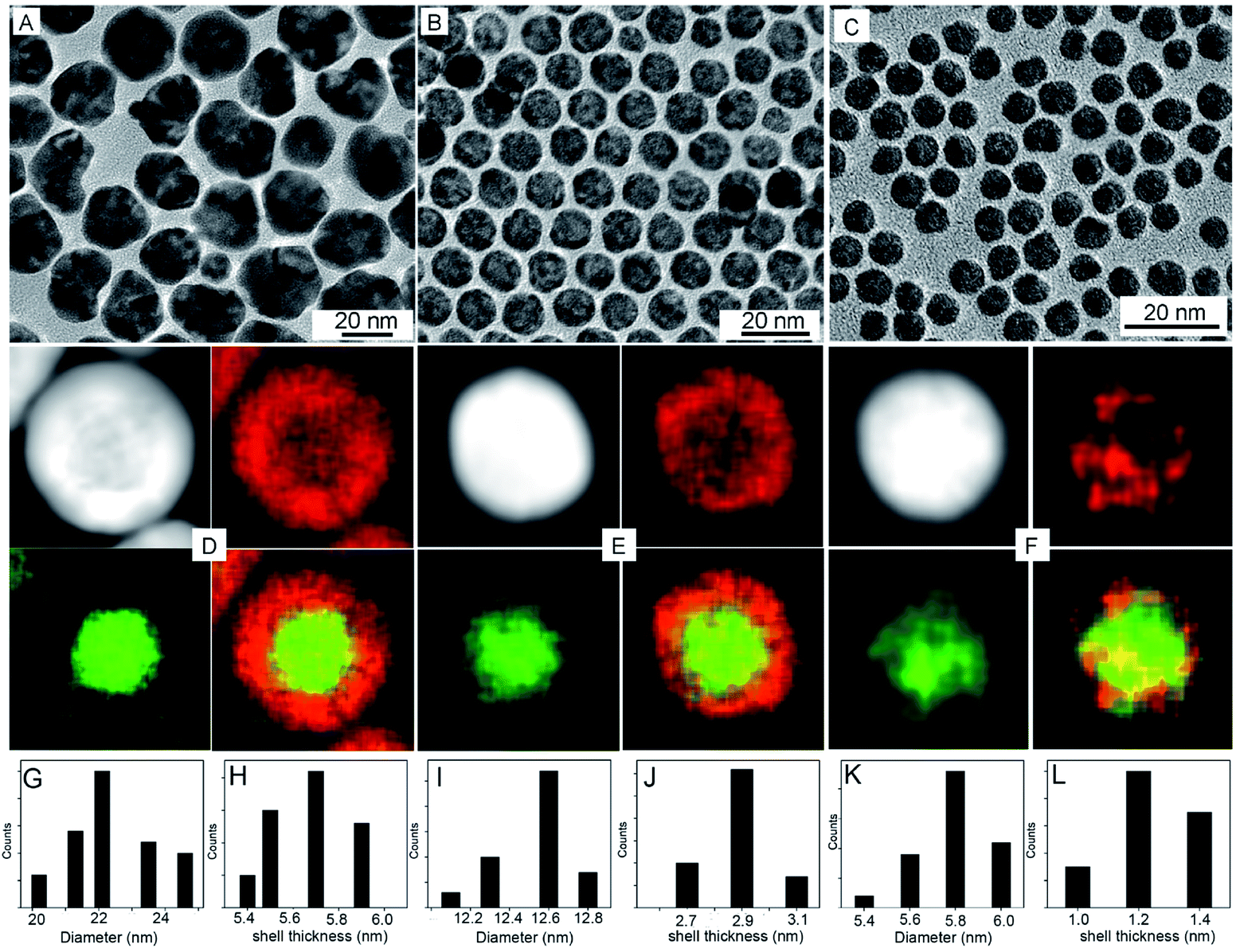 | ||
| Fig. 1 TEM, HAADF-STEM, and EDX elemental mapping images of Pt@Ag core@shell NPs with different sizes, (A and D) 22.4 nm, (B and E) 12.5 nm, (C and F) 5.8 nm, respectively. (G–L) Their corresponding diameter and shell size distribution patterns. Red represents Pt component and green represents Ag component in the mapping images. | ||
Utilizing pre-synthesized Ag@Pt core@shell NPs as precursors, hollow Pt–Ag NPs were generated by simply dispersing the NPs in acetic acid solution for 4 h at room temperature. It is worth noting that the presence of oxygen in the solution is critical to the formation of hollow NPs. Although Ag does not usually react with acetic acid at room temperature, Ag can be oxidized to silver oxide by oxygen. Silver oxide will then react with acetic acid and dissolve in the media.47,48 This chemical etching process is described in eqn (1).
| 4Ag + O2 + 4H+ → 4Ag+ + 2H2O | (1) |
The Ag atoms can be oxidized to Ag+ ions and thus dissolve from the Ag@Pt core@shell NPs into solution. When this occurs, Ag+ ions can be detected in the supernatant of the sample after centrifugation. Specifically, Cl− ions were added to the supernatant, which resulted in the formation of a visible, white insoluble precipitate of silver chloride.36 Moreover, two experiments were performed with 12.4 nm Ag@Pt core@shell NPs: one in air and a control in a deoxygenated solution prepared by bubbling argon. All the other reaction conditions were kept constant. The solution containing oxygen from the air produced hollow Pt–Ag structures. In contrast, the Pt@Ag core@shell structures remained unchanged in the deoxygenated solution (Fig. S4†). This result confirmed that the oxygen is indeed crucial to the structure evolution of the core@shell structures into hollow structures.
Since Pt is inert in acetic acid under ambient conditions, the Pt shell in the Ag@Pt core@shell NPs is expected to either significant slow the rate of oxidation of the Ag core or prevent oxidation altogether. On the basis of our collective results, we find that the thickness of the Pt shell is the primary factor that determines whether the Ag core can be etched by acetic acid.
To examine the effect of the Pt shell thickness on the formation of hollow NPs, Pt@Ag core@shell NPs with varying shell thickness were treated with acetic acid in air. Ag@Pt core@shell NPs with relatively thick Pt shells of 9.1 nm did not undergo etching when treated with acetic acid (Fig. S5†). On the other hand, when the Pt shell thickness was reduced to 5.6 nm (the overall size of the NPs is 22.4 nm), completely hollow Pt–Ag NPs were formed after the reaction, as shown in Fig. 2A. The light contrast in the center of Pt–Ag NPs indicated that the Pt–Ag NP became hollow after etching, as also confirmed by HAADF-STEM image (Fig. 2D). EDX mapping images showed that both Pt and Ag were homogeneously distributed throughout the hollow NP. Similar hollow nanostructures were obtained after etching the Ag@Pt core@shell NPs with 2.9 nm shell (Fig. 2B and E). When the etching reaction was performed using the Ag@Pt core@shell NPs with an incomplete Pt shell, small porous Pt–Ag NPs were produced, as shown in Fig. 2C and F. This observation suggests that the relatively thick Pt shell can protect the Ag core from oxidizing in acidic media.
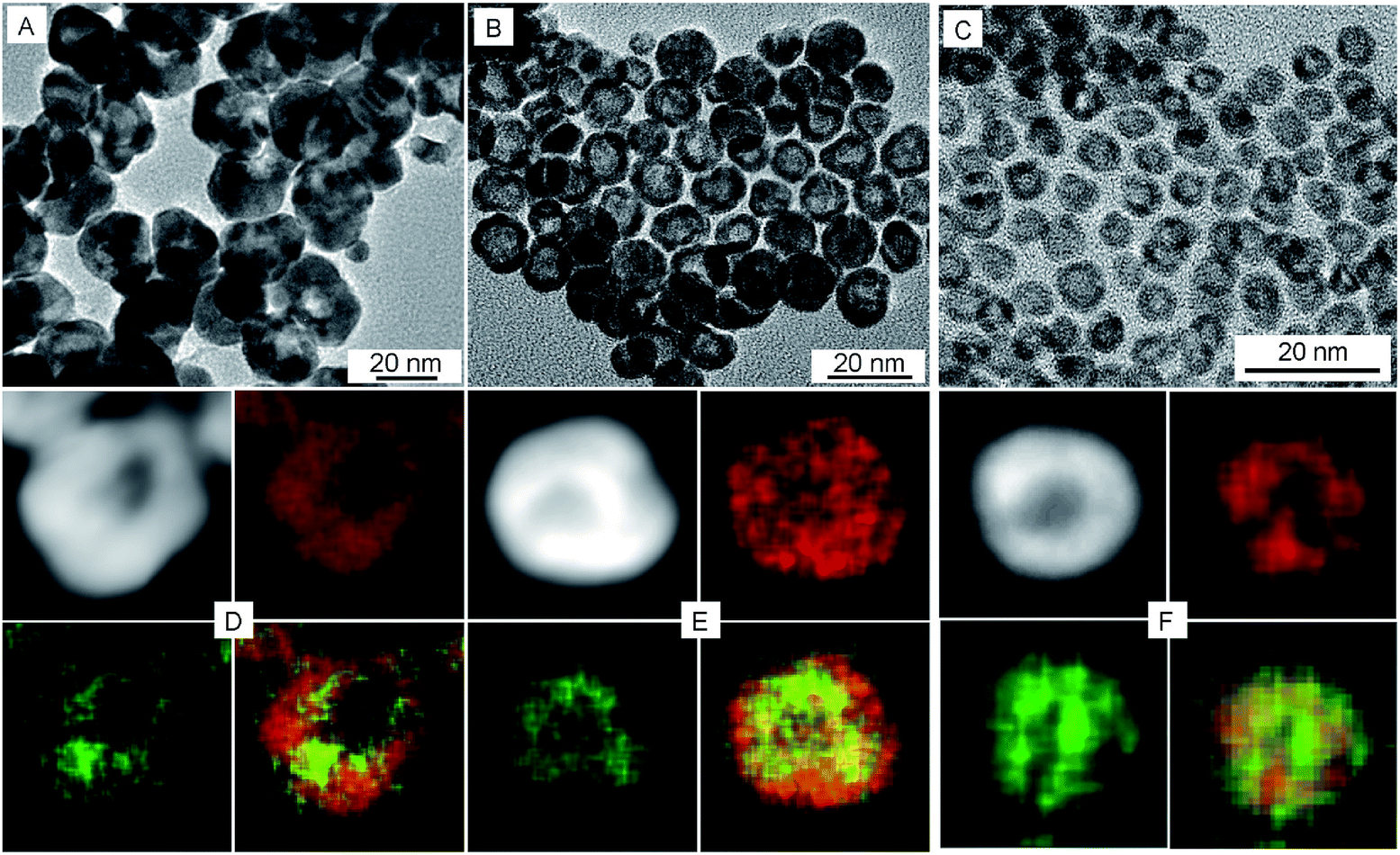 | ||
| Fig. 2 TEM, HAADF-STEM, and EDX elemental mapping images of Pt–Ag hollow NPs with different sizes, (A and D) 22.4 nm, (B and E) 12.5 nm, (C and F) 5.8 nm, respectively. Red represents Pt component and green represents Ag component in the mapping images. | ||
A closer look at the hollow NPs by HRTEM shows that they are spherical in shape, with holes on the surfaces and an internal cavity in the center (Fig. 3). The lattice fringes with spacing of 0.228 nm are between that of the Pt (111) planes and Ag (111) planes, which suggests the formation of Pt–Ag alloy. The atom ratios as determined by EDX measurements of Pt and Ag in three sized hollow NPs are 93:7, 88:12, and 82:18 (Fig. S6†), respectively. The overall atomic fraction of Pt/Ag in the hollow Pt–Ag NPs increased compared to that of the Pt@Ag core@shell NPs, because most of the Ag atoms were dissolved from the original structures into solution. However, the atomic fraction of Pt/Ag decreased, e.g. from 94:6 to 88:12 for the 12.4 nm NPs, if we compared the wall of the hollow NPs and the shell of the Ag@Pt NPs according to the EDX analysis (Fig. S7†). The increase in the Ag content of the shell indicates that some Ag atoms diffused into the Pt shell during the etching process.41 The diffusion occurred because Ag atoms have much smaller radii than Pt atoms, and therefore they can easily form an alloy within the Pt host lattice. The combined effects of the hollow structure and the formation of a Pt–Ag alloy results in a favourable change in the energy of the weighted center of the Pt d-band as determined by XPS measurements,25 which is predicted to increase their activity for electrocatalysis.51
 | ||
| Fig. 3 HRTEM images of 5.8 nm Pt–Ag porous NPs. The circles denote the pores on the particle surface. | ||
Fig. 4A shows the powder X-ray diffraction (XRD) analysis of the crystal structure and composition for all the three different sized hollow NPs. The four peaks in the XRD patterns were indexed to the (111), (200), (220), and (311) diffraction of a face-centered-cubic (fcc) structure. The peak positions were located in between those of pure Ag and pure Pt metals, due to the formation of bimetallic Pt–Ag hollow NPs. The diffraction from the index facets of hollow NPs was shifted to lower 2θ angles compared to the pure Pt NPs, which was indicative of a lattice expansion compared to pure Pt. The expansion of the lattice relative to pure Pt can be attributed to the larger lattice constant of silver (4.08 Å) relative to that of Pt (3.92 Å). This has been observed previously in hollow Pt–Ag nanocubes and NPs.29,36
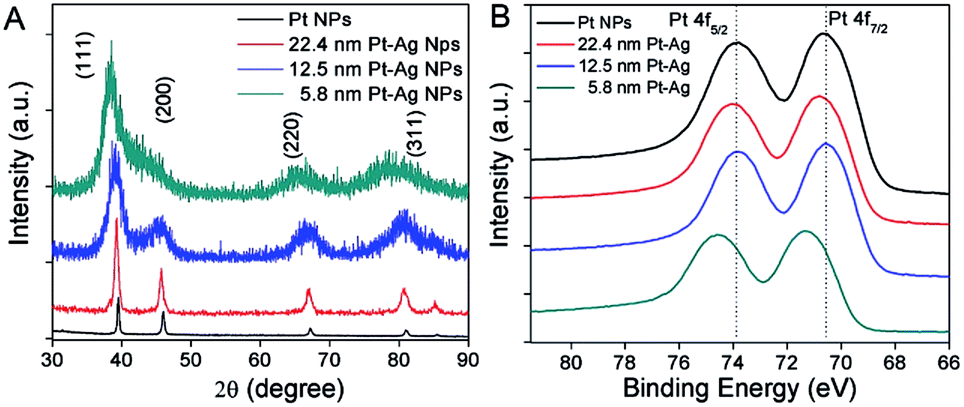 | ||
| Fig. 4 XRD patterns (A) and XPS spectra (B) of hollow Pt–Ag NPs with varying sizes. | ||
The electronic properties of hollow Pt–Ag NPs were further examined by X-ray photoelectron spectroscopy (XPS). Fig. 4B shows that the peaks attributed to Pt 4f5/2 and Pt 4f7/2 in XPS were well resolved. When compared with the Pt NPs (black curve in Fig. 4B), the binding energy of Pt 4f in the hollow Pt–Ag NPs increased by 0.2 eV for 22.4 nm and 0.7 eV for 5.8 nm, respectively. Given that the reference peak (C 1s, 284.5 eV) had no shift, the change of binding energy is likely due to the alloying effect and lattice strain.7,51,54 Because the electronegativity of Ag is less than that of Pt, Ag will donate electrons to Pt, resulting in negative shifting bonding energy of Pt 4f from hollow Pt–Ag NPs via a ligand effect.51 In this case, we observed a positive shift of the binding energy of Pt 4f from hollow Pt–Ag NPs, comparing to Pt NPs. Positive shifts in binding energy have been observed previously and are attributed to the competing effects of geometric strain in the Pt lattice due to the Ag atoms in the alloy and the ligand effect.33,55
For NP based electrocatalysts, their performance is highly dependent on the surface ligands. Long chain ligands are often used in the synthesis to stabilize the structure and control the morphology of the NPs.56 However, the strongly adsorbed ligands are detrimental to catalytic performance, since they block the active sites on the surface of the catalyst.34,44,57 In our method, acetic acid not only removes Ag atoms from the Ag@Pt core@shell NPs creating the hollow structure, but also exchanges with and removes the hexadecylamine molecules from the NP surface (Fig. S8†). The short and weakly adsorbed acetic acid allows the molecules to easily access the catalytic surfaces, therefore, improves the catalytic activity of the NPs.
The hollow Pt–Ag NPs of 22.4 nm, 12.4 nm and 5.8 nm were loaded on carbon black (Fig. S9†), and their electrochemical properties were examined by cyclic voltammetry (CV) carried out in 0.1 M HClO4. Initially, the surface of the NPs was activated electrochemically by cycling the potential to 1.3 V, in order to oxidize and desorb the acetic acid. The electrochemical activation process resulted in a significant irreversible oxidation wave between 1.0 and 1.3 V, consistent with the oxidation of adsorbed organic species. The intensity of the oxidation wave decreased during the first 50 cycles inevitably producing a stable CV. During the activation process, the hydrogen adsorption and oxide features of the CV increased in intensity and stabilized after 50–75 cycles, suggesting that the capping ligands were fully removed. As a control, we performed the same activation protocol on the commercial Pt NP/C catalysts. Cycling the commercial particles to 1.3 V for 100 cycles had essentially no effect on either the CV or the catalytic activity. On this basis, we conclude that the effect is primarily isolated to the removal of organic surface species in the Pt–Ag NPs and not to surface roughening effects or the formation of new active sites.
After activation, the CVs of the hollow Pt–Ag NPs (Fig. 5A) display the reversible hydrogen adsorption/desorption (Hads) in the region of 0 to 0.4 V and the onset of oxide formation at 0.7–0.8 V, which are characteristic of nanostructured Pt-based catalysts.52,58,59 The feature associated with the oxidation of Ag surface sites at ∼0.5–0.7 V was not observed in our catalysts suggesting that surface is predominantly Pt with relatively few silver sites.60,61 Interestingly the position of the oxide reduction peaks (Fig. 5A) in the CVs for the 22.4 nm, 12.5 nm, and 5.8 nm hollow Pt–Ag NPs were localized at ∼800 mV. On average, these values are shifted by ∼35 mV relative to the peak for commercial Pt NPs/C (Fig. S10†) localized at 765 mV. This suggests that the adsorption energy of oxygen to the surface is weaker in the hollow Pt–Ag NPs.
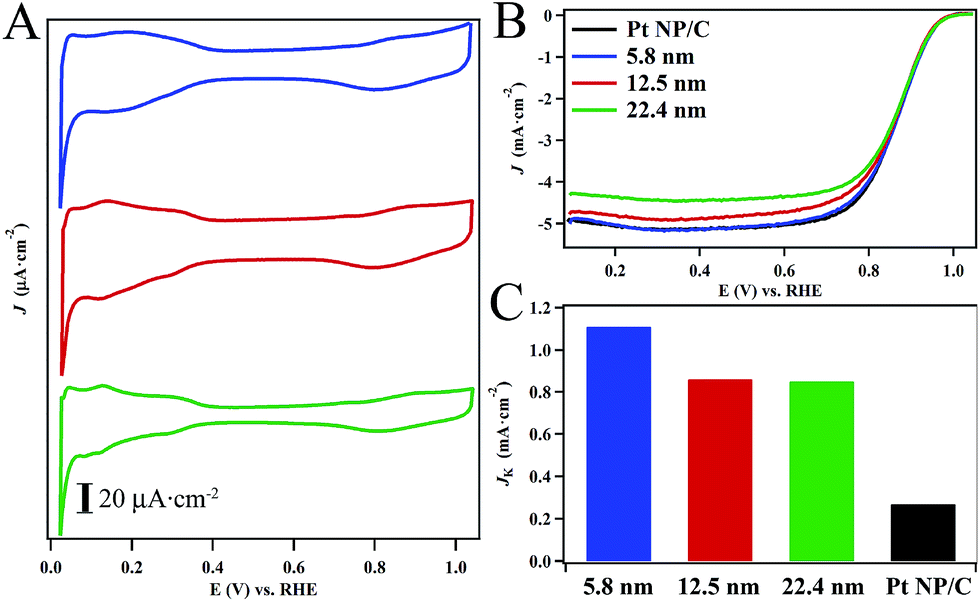 | ||
| Fig. 5 Electrochemical characterization of the hollow Pt–Ag nanocatalysts in 0.1 M HClO4. Cyclic voltammograms (A) were obtained in deoxygenated electrolyte at a scan rate of 20 mV s−1. Polarization curves (B) of hollow Pt–Ag nanocatalysts obtained in oxygen saturated electrolyte at a rotation rate of 1600 rpm. Specific activity (Jk) normalized to the ESA of the various catalysts (C) calculated at 0.9 V vs. RHE. | ||
Fig. 5B depicts polarization curves obtained from the hollow Pt–Ag NPs in oxygen saturated electrolyte (c.f. ESI for details†). The specific activity measured at 0.9 V (Fig. 5C) normalized to the ESA of the 22.4 nm, 12.5 nm, and 5.8 nm hollow Pt–Ag NPs were determined to be 0.82 ± 0.03 mA cm−2, 0.83 ± 0.05 mA cm−2, and 1.11 ± 0.02 mA cm−2, respectively. The activity of the Pt–Ag hollow NPs is significantly higher than the activity of 0.28 mA cm−2 for the Pt NP/C. In a prior report, the specific activity of Pt–Ag nanocages with a Pt content of ∼6% was determined to be 0.66 mA cm−2 at 0.9 V.36 In another report, the specific activity of hollow Pt–Ag alloy particles and Pt–Ag core–shell particles was found to be 0.10 mA cm−2 and 0.12 mA cm−2, respectively in a sulfuric acid electrolyte.34 Thus, the specific activity of our hollow Pt–Ag NPs is comparable to or better than other recent examples of high quality hollow Pt–Ag nanostructures.
Rotating ring-disk electrode measurements (Fig. S11†) were performed to explore the mechanism of ORR on the 5.8 nm hollow Pt–Ag NPs. Fits of the Koutecky–Levich plot (Fig. S12†) revealed that the experimental value for the constant ‘B’ of 0.0955 mA s−1/2 was in excellent agreement with the theoretical value of 0.0914 mA s−1/2 calculated for the four-electron reduction of oxygen to water. Moreover, no peroxide was detected at the ring electrode in the kinetically controlled region of the polarization curve. Collectively, these results suggest that the four-electron reduction process predominates with the hollow Pt–Ag catalyst.
In prior reports, the enhanced activity in hollow Pt–Ag nanostructures relative to solid Pt1−xAgx alloy-type NPs and to Pt@Ag core@shell NPs was attributed to the electronic structure of the hybrid structure induced by the alloy-type composition and the hollow morphology.29,32,37 In both the alloy-type and core@shell type NPs, the tensile strain induced by the addition of Ag atoms resulted in an expanded Pt lattice, which resulted in a shift of the Pt 4f peaks to lower binding energies in the XPS spectrum.24,30,57 The shift to lower binding energies suggested that the d-band center was shifted to higher energies, which has been shown to correlate with lower ORR activities by Nørskov and co-workers.62
However, the position of the Pt 4f peaks for the hollow Pt–Ag NPs was comparable to or slightly higher than that of Pt. We attribute this effect to the hollow morphology of the particles. In a previous report, Adzic and co-workers measured the relative degree of expansion strain on the surface atoms of hollow and solid Pt NPs.35 Solid NPs are widely known to have high degrees of microstrain leading to significant expansion of the surface atoms relative to Pt (111). However, the degree of microstrain was much lower in the case of the hollow nanoparticles, leading to less surface expansion relative to Pt (111). DFT calculations revealed that the hollow morphology promotes contraction of the surface atoms, which mitigates the effect of surface expansion strain associated with small diameter solid NPs. Based on these prior results in elemental Pt NPs, we believe that the hollow morphology has a similar strain reducing effect in the Pt–Ag alloy-type hollow NPs where expansion strain is induced by the addition of silver atoms to the alloy. Thus, the hollow morphology is expected to lower the observed d-band center of hollow NPs relative to solid alloy-type and core–shell morphologies, which is consistent with our XPS findings.
Herein, the XPS results from the 22.4 nm and 12.5 nm hollow Pt–Ag NPs maintain comparable Pt 4f binding energies to that of Pt NP/C, which is consistent with prior results for 17 nm hollow Pt–Ag NPs.29 The specific activities of 0.82 mA cm−2 and 0.83 mA cm−2 for these particles are consistent with the XPS results, which predicts that the position of their d-band centers are nearly identical. Interestingly, the Pt 4f peak for the 5.8 nm Pt–Ag hollow NPs was shifted positively relatively to the Pt NP/C, which suggested that the d-band center was lower in the case of the smaller particles. The shift in the XPS peaks was accompanied by a corresponding increase in the specific activity of 1.35 fold to 1.11 mA cm−2.
One plausible explanation for the enhanced activity in the 5.8 nm hollow Pt–Ag NPs is the higher silver content in these particles. In a prior report, increasing silver content from 80% to 94% in hollow Pt–Ag nanocages led to an increase in the measured specific activity from ∼0.30 mA cm−2 to 0.66 mA cm−2.36 The increase in activity was accompanied by a shift in the Pt 4f XPS peaks to higher binding energies suggesting a significant composition-dependent effect on the electronic structure of the hollow NPs. A positive shift of the binding energy of Pt 4f from hollow Pt–Ag NPs arises from the electron transfer between Pt and Ag. Although pure Ag atoms do not have any unpaired electrons and any d-band vacancy, according to the Pauli exclusion principles, the electron transfer would lead to the increase of the number of unpaired electrons numbers of Ag in the Pt–Ag hollow catalysts.63 During ORR, active oxygen can preferential adsorb onto the metals with a number of unpaired electrons, which would further facilitate the ORR kinetics on these Pt–Ag hollow catalysts.64
The adsorption strength of oxygen species may also shed light on the effect of the silver content. The surface oxidation of Pt-based species occurs at potentials above 0.6–0.7 V leading to the characteristic, reversible oxidation wave in the CV.52 Initially, the surface is oxidized leading to the formation of adsorbed hydroxyl species, OH*, which are converted to adsorbed oxide species, O*, at higher potentials. The onset of the oxidation wave in the anodic sweep and the peak of the surface oxide reduction wave can be employed to determine the relative adsorption strength of surface oxide species, which depends upon a multitude of factors including catalyst composition.
The effect of composition on the adsorption strength of oxide species in Pt–Ag alloys was recently examined via a combined computational and experimental approach.65 The computations revealed the silver dopant contributes to significant changes in the adsorption energy of both O* and OH* species relative to pure platinum. Specifically, the adsorption energy of O* was reduced relative to pure platinum while the adsorption energy of OH* was increased. This effect can be attributed to the relative adsorption energies of these species on Ag (111) and Pt (111), wherein O* binds more strongly to Pt (111) and OH* binds more strongly Ag (111).66 In the case of Pt–Ag alloys, the stronger adsorption of OH* is likely a result of the combined electronic effects of introducing silver into the platinum lattice along with the structural ‘lattice expansion’ effect. We believe that the strain reducing qualities of the hollow morphology may act to counter the lattice expansion effect of the silver dopant leading to lower overall adsorption energies of O* and OH* intermediates. This is consistent with the 35 mV positive shift of the oxide reduction peak of the hollow Pt–Ag hollow NPs relative to the commercial Pt NPs, which suggests that the adsorption strength of oxide species is lower in the hollow Pt–Ag NPs.
In this case, the 5.8 nm hollow Pt–Ag NPs maintained the highest silver content of 18% relative to the silver content of 12% and 7% for the 12.5 nm and 22.4 nm NPs. However, despite having different silver contents, the 12.5 nm and 22.4 nm hollow Pt–Ag NPs maintained essentially identical ORR activities. Thus, the trend in activity is not entirely explained by the composition of the particles.
An alternative explanation is that there is a size-dependent effect that leads to higher activity in the 5.8 nm hollow Pt–Ag NPs. Specifically, the peak in Hads region associated with low-coordination atoms and defect sites at 0.13 V decreases in intensity as particle size is decreased from 22.4 nm to 5.8 nm.65–69 Lower defect site densities are advantageous for the electrocatalytic reduction of oxygen since defects strongly adsorb oxygen preventing effective ORR kinetics and also contribute to better catalyst durability.68–71 It is also plausible that the smaller size of the hollow NPs may enhance the beneficial structural effects of the hollow morphology, which was predicted in the calculations performed by Adzic and co-workers.35 However, additional computational and experimental investigation is necessary to fully explain the enhanced performance in the 5.8 nm NPs.
We selected the 5.8 nm NPs for ORR durability testing, since they evince the highest catalytic activity and maintain the lowest defect site density. Polarization curves (Fig. 6) and CVs (Fig. S13†) obtained over the course of the test reveal that there is a minimal change in the performance of the NPs. After 20000 cycles, the half-wave potential shifts by only 8 mV and the particles retain 95% of their electrochemical surface area. Under the same conditions, the half-wave potential of the commercial Pt NP/C (Fig. S14†) shifted by over 16 mV and retained only 61.3% of their initial electrochemical surface area. The performance of the commercial Pt NP/C were consistent with previous reports.4,72 TEM and mapping images obtained of the particles confirmed the hollow structures of the catalysts were maintained even after 20000 cycles. There is a slight size decrease of the hollow NPs after the durability test (Fig. S15†). EDX analysis indicates the molar ratio of Pt/Ag is 94:6 after 20000 cycles, as compared to 82:18 before the reaction. This is likely due to dissolution of Ag atoms from the NPs in acid media during the cycling (Fig. S15E†).
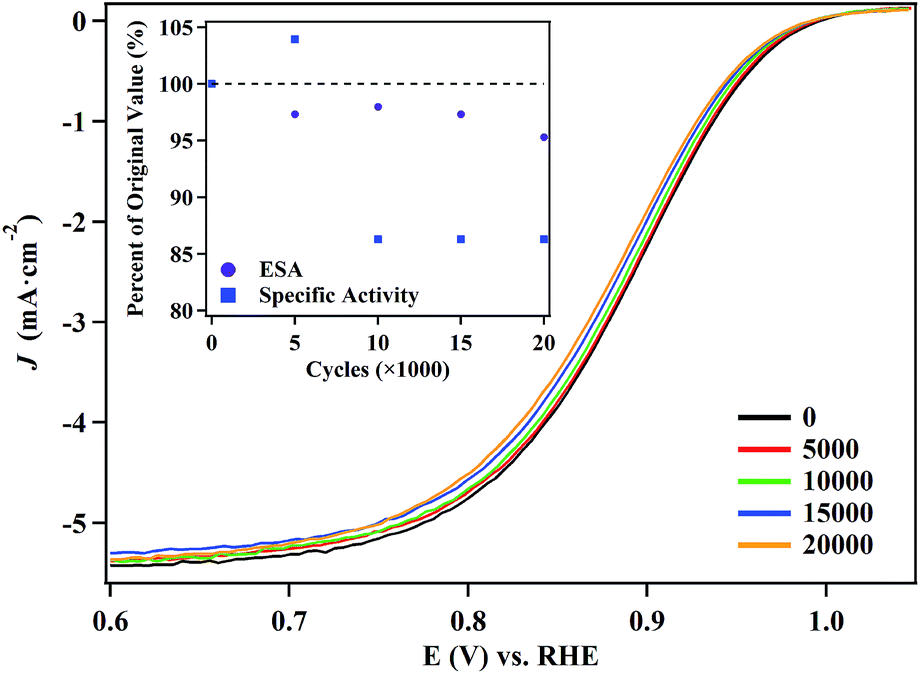 | ||
| Fig. 6 Polarization curves obtained from the 5.8 nm hollow Pt–Ag nanocatalysts over the course of a 20000 cycle accelerated durability test. The electrochemical surface area (ESA) and specific activity of the catalyst (inset) are shown as a function of the number of cycles. | ||
4. Conclusions
In conclusion, we have developed a simple synthetic method to fabricate hollow Pt–Ag NPs of varying sizes, and evaluated their catalytic activity. The hollow NPs were produced via O2-assisted etching of Ag@Pt core@shell NPs in acetic acid. The thickness of Pt shell and the presence of oxygen play important roles for forming the hollow nanostructures. Size- and composition-controlled hollow Pt–Ag NPs were obtained when the shell of Pt thickness was less than 6 nm. Hollow Pt–Ag nanocatalysts demonstrated enhanced ORR activity, higher than that of the commercial Pt/C. The 5.8 nm porous nanocatalysts had the highest specific activity of 1.12 mA cm−2, which was almost four times than that of the commercial Pt/C (0.28 mA cm−2). We believe that the hollow morphology and the silver content contribute to the increased activity in the Pt–Ag hollow NPs. In terms of the structural effects, the normally deleterious lattice expansion associated with adding silver into Pt–Ag alloys is mitigated by the hollow morphology, which has been shown to relieve the surface expansion strain in prior reports. In terms of the electronic effects, silver dopants into Pt–Ag alloys has been shown to weaken the interaction of certain oxygen adsorbates, which in this case may contribute to the increased ORR activity. In terms of durability, the ESCA and specific activity of the porous nanocatalysts only decreased very slightly, after 20000 cycles. We envision such simple strategy for designing small porous nanostructures could be extended to other bimetallic catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JZ thanks the American Chemical Society Petroleum Research Fund (PRF# 54004-DN15) for financial support. The TEM studies were performed using the facilities in the UConn/FEI Center for Advanced Microscopy and Materials Analysis (CAMMA). We thank Mr Muhammad G. Salim for the assistance in XPS measurements. TJA and GS would like to thank the Fordham College at Rose Hill for financial support through Undergraduate Research Grants. TJA would also like to thank the Brazilian Science Mobility Program for financial support.Notes and references
- T. Chen and V. O. Rodionov, ACS Catal., 2016, 6, 4025–4033 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- C. He, S. Desai, G. Brown and S. Bollepalli, Electrochem. Soc. Interface, 2005, 14, 41–44 CAS.
- N. M. Marković, T. J. Schmidt, V. Stamenković and P. N. Ross, Fuel Cells, 2001, 1, 105–116 CrossRef.
- Fuel Cell Catalysts, ed. M. T. M. Koper, Wiley Interscience, Hoboken, NJ, 2009 Search PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- N. S. Porter, H. Wu, Z. Quan and J. Fang, Acc. Chem. Res., 2013, 46, 1867–1877 CrossRef CAS PubMed.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- Y. Lu, S. Du and R. Steinberger-Wilckens, Appl. Catal., B, 2016, 199, 292–314 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- C. Koenigsmann, M. E. Scofield, H. Liu and S. S. Wong, J. Phys. Chem. Lett., 2012, 3, 3385–3398 CrossRef CAS.
- C. Koenigsmann and S. S. Wong, Energy Environ. Sci., 2011, 4, 1161–1176 CAS.
- E. Antolini and J. Perez, J. Mater. Sci., 2011, 46, 1–23 CrossRef.
- Y. Feng, G. Zhang, J. Ma and B. Xu, Phys. Chem. Chem. Phys., 2011, 7, 3863–3872 RSC.
- N. Zhang, F. Chen, X. Wu, Q. Wang, A. Qaseema and Z. Xia, J. Mater. Chem. A, 2017, 5, 7043–7054 CAS.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- B. Han, C. E. Carlton, J. Suntivich, Z. Xu and Y. Shao-Horn, J. Phys. Chem. C, 2015, 119, 3971–3978 CAS.
- M. Oezaslan, F. Hasché and P. Strasser, J. Electrochem. Soc., 2012, 159, B444–B454 CrossRef CAS.
- Y.-T. Pan, Y. Yan, Y.-T. Shao, J.-M. Zuo and H. Yang, Nano Lett., 2016, 16, 6599–6603 CrossRef CAS PubMed.
- X. Yang, L. T. Roling, M. Vara, A. O. Elnabawy, M. Zhao, Z. D. Hood, S. Bao, M. Mavrikakis and Y. Xia, Nano Lett., 2016, 16, 6644–6649 CrossRef CAS PubMed.
- D. Zhao, Y.-H. Wang, B. Yan and B.-Q. Xu, J. Phys. Chem. C, 2009, 113, 1242–1250 CAS.
- L. Lu, G. Sun, S. Xi, H. Wang, H. Zhang, T. Wang and X. Zhou, Langmuir, 2003, 19, 3074–3077 CrossRef CAS.
- J. Cao, M. Guo, J. Wu, J. Xu, W. Wang and Z. Chen, J. Power Sources, 2015, 277, 155–160 CrossRef CAS.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- T. Fu, J. Fang, C. Wang and J. Zhao, J. Mater. Chem. A, 2016, 4, 8803–8811 CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl and Y. M. Wang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- Y. Kang, F. Li, S. Li, P. Ji, J. Zeng, J. Jiang and Y. Chen, Nano Res., 2016, 9, 3893–3902 CrossRef CAS.
- H. Liu and J. Yang, J. Mater. Chem. A, 2014, 2, 7075–7081 CAS.
- Z. Peng, J. Wu and H. Yang, Chem. Mater., 2009, 22, 1098–1106 CrossRef.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- J. X. Wang, C. Ma, Y. Choi, D. Su, Y. Zhu, P. Liu, R. Si, M. B. Vukmirovic, Y. Zhang and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 13551–13557 CrossRef CAS PubMed.
- J. A. Bordley and M. A. El-Sayed, J. Phys. Chem. C, 2016, 120, 14643–14651 CAS.
- M. Liu, F. Chi, J. Liu, Y. Song and F. Wang, RSC Adv., 2016, 6, 62327–62335 RSC.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C. Chen, R. Yu, Q. Zhang, L. Gu, B. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. Goddard, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- R. Chattot, T. Asset, J. Drnec, P. Bordet, J. Nelayah, L. Dubau and F. Maillard, Nano Lett., 2017, 17, 2447–2453 CrossRef CAS PubMed.
- J. Chen, B. Wiley, J. McLellan, Y. Xiong, Z.-Y. Li and Y. Xia, Nano Lett., 2005, 5, 2058–2062 CrossRef CAS PubMed.
- C.-L. Lee and C.-M. Tseng, J. Phys. Chem. C, 2008, 112, 13342–13345 CAS.
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed.
- S. Chen, S. Thota, X. Wang and J. Zhao, J. Mater. Chem. A, 2016, 4, 9038–9043 CAS.
- K. Lee, S. W. Kang, S.-U. Lee, K.-H. Park, Y. W. Lee and S. W. Han, ACS Appl. Mater. Interfaces, 2012, 4, 4208–4214 CAS.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- K.-H. Park, Y. W. Lee, S. W. Kang and S. W. Han, Chem.–Asian J., 2011, 6, 1515–1519 CrossRef CAS PubMed.
- C. Koenigsmann, E. Sutter, R. R. Adzic and S. S. Wong, J. Phys. Chem. C, 2012, 116, 15297–15306 CAS.
- A. T. N. Dao, D. M. Mott, K. Higashimine and S. Maenosono, Sensors, 2013, 13, 7813–7826 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321–6328 CrossRef CAS PubMed.
- US Department of Energy, Office of Energy Efficiency and Renewable Energy, Hydrogen, Fuel Cells and Infrastructure Technologies Program, 2010, pp. 1–34 Search PubMed.
- B. Hammer and J. K. Nørskov, in Advances in Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- M. Asano, R. Kawamura, R. Sasakawa, N. Todoroki and T. Wadayama, ACS Catal., 2016, 6, 5285–5289 CrossRef CAS.
- J. Wu, P. Li, Y.-T. F. Pan, S. Warren, X. Yin and H. Yang, Chem. Soc. Rev., 2012, 41, 8066–8074 RSC.
- C. Koenigsmann and S. S. Wong, Energy Environ. Sci., 2011, 4, 1161–1176 CAS.
- D. Strmcnik, D. Tripkovic, D. van der Vliet, V. Stamenkovic and N. M. Markovic, Electrochem. Commun., 2008, 10, 1602–1605 CrossRef CAS.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- D. Liu, M. Xie, C. Wang, L. Liao, L. Qiu, J. Ma, H. Huang, R. Long, J. Jiang and Y. Xiong, Nano Res., 2016, 9, 1590–1599 CrossRef CAS.
- K. Kim, K. L. Kim and K. S. Shin, J. Phys. Chem. C, 2011, 115, 23374–23380 CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111, 404–410 CAS.
- M. J. Eslamibidgoli, J. Huang, T. Kadyk, A. Malek and M. Eikerling, Nano Energy, 2016, 29, 334–361 CrossRef CAS.
- Y.-T. Liang, S.-P. Lin, C.-W. Liu, S.-R. Chung, T.-Y. Chen, J.-H. Wang and K.-W. Wang, Chem. Commun., 2015, 51, 6605–6608 RSC.
- S. C. Huang, C. H. Lin and J. H. Wang, J. Phys. Chem. C, 2010, 114, 9826–9834 CAS.
- N. M. Markovic and P. N. Ross, in Interfacial Electrochemistry: Theory, Experiment and Applications, ed. A. Wieckowski, Marcel Dekker, Inc, New York, 1999, vol. 1, pp. 821–841 Search PubMed.
- R. R. Adzic and J. X. Wang, J. Phys. Chem. B, 1998, 102, 8988–8993 CrossRef CAS.
- J. X. Wang, N. M. Markovic and R. R. Adzic, J. Phys. Chem. B, 2004, 108, 4127–4133 CrossRef CAS.
- J. X. Wang, J. Zhang and R. R. Adzic, J. Phys. Chem. A, 2007, 111, 12702–12710 CrossRef CAS PubMed.
- R. R. Adzic and J. X. Wang, J. Phys. Chem. B, 2000, 104, 869–872 CrossRef CAS.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, elemental mapping images, EDX spectra, and linear scanning spectra of the nanoparticles. See DOI: 10.1039/c7ra07721a |
| This journal is © The Royal Society of Chemistry 2017 |