
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation of aquatic sulfadiazine by Fe0/persulfate: kinetics, mechanisms, and degradation pathway†
Shidong Yang * and
Di Che
* and
Di Che
School of Civil Engineering and Architecture, Northeast Electric Power University, Jilin 132012, PR China. E-mail: ysd_nedu@163.com; Fax: +86-432-64806481; Tel: +86-432-64806481
First published on 1st September 2017
Abstract
Effects of treatment factors on the kinetics of sulfadiazine (SDZ) removal by Fe0/persulfate (Fe0/PS) were studied at an initial pH of 7.0. The kinetics of SDZ degradation by Fe0/PS were divided into a lag phase and a rapid reaction. The presence of the lag phase was ascribed to the slow release of Fe(II) in the heterogeneous Fe0/PS system. The rapid phase was simulated by pseudo first-order kinetics model. With increasing Fe0 or PS ranging from 0.25 to 2 mM, the kobs (min−1) of SDZ degradation increased and remained stable at a high level of 5 mM Fe0 or PS. But increasing SDZ inhibited the SDZ removal rate for the scavenging of reactive oxygen species (ROS). SDZ degradation by Fe0/PS in neutral or weak alkaline solutions exhibited higher removal rates than in weak acid solutions. Common aquatic materials including sulfate, nitrate, chloride, perchlorate, and HA all showed negative effects on SDZ degradation by Fe0/PS following a trend of Cl− < ClO4− < SO42− < NO3− < HCO3− < HA. The dominating ROS in the Fe0/PS system was identified as ˙SO4− by chemical quenching experiments in the presence of methanol or tert-butyl alcohol. And the chemical detection of dimethyl pyridine N-oxide (DMPO)-˙SO4− and DMPO–˙OH by electron paramagnetic resonance (EPR) spectrum confirmed the presence of ˙SO4−. Besides, strongly negative effects of 1,10-phenanthroline, ethylenediaminetetraacetic acid (EDTA), and dissolving oxygen (DO) on SDZ degradation in the Fe0/PS process proved that ˙SO4− was not generated by an one-step reaction between Fe0 and PS but via the indirect oxidation of Fe(II) by PS. Finally, degradation pathways of SDZ by Fe0/PS were proposed based on theoretical reactive sites attacked by radicals and intermediate products.
Introduction
Sulfonamide drugs have been synthesized and commercially used since 1930s.1 This important category of antibiotics has been extensively used to treat and prevent various infectious diseases of humans and animals due to its broad antimicrobial spectrum.2 According to the reports of Zhang et al.,1 7136 tons of 9 typical sulphonamides were consumed in China in 2013, about 94.3% of which were used as veterinary antibiotics for pigs, chicken, and other animals. The over-use of sulfonamides, especially in the industry of livestock feeding,3 has increased the potential contamination of sulfonamides in water and soil environments. Most sulfonamides are excreted from the human body and animal organisms partially in unmetabolised form.4 Many studies have revealed that the expired and unused sulfonamides exposed to humans have shown various adverse effects towards human health.5 Although sulfonamides in ground and surface water are detected at low levels,6 residual sulfonamides can be accumulated in various organisms of a food chain, increasing antibiotic resistance of pathogenic bacteria in aquatic environments.7Traditional wastewater treatment plants (WWTPs) using biological technologies as the main processes may be ineffective to sulphonamides for the aquatic sulphonamides exhibit high resistance to biological degradation as antibiotics.8 Many researchers2,9,10 have reported that the removal rates of various sulfonamides by traditional WWTPs are limited, thus adopting effective treatment alternatives is essential to eliminate the contamination of sulfonamides.
Of various chemical techniques, such as advance oxidation process (AOPs),11 ozonation,12 and permanganate,13 AOPs have been frequently employed to remove many antibiotics in water and wastewater treatment processes since hydroxyl radical (˙OH) produced by AOPs possesses stronger redox potential (E0 = 1.9–2.7 V),14 higher performance, and superior mineralization rate than traditional chemical oxidants.15 In recent years, new AOPs based on sulfate radical (˙SO4−) have been developed to destroy organic pollutants include antibiotics16 and dyes17 in surface water,18,19 hospital effluents,20 and waste water.21 Sulfate radical has been known as a strong oxidant for its higher redox potential (E0 = 2.5–3.1 V)14 than hydroxyl radical. Moreover, Neta et al.22 has reported that sulfate radical is a more effective oxidant than hydroxyl radical to eliminate many organic compounds by hydrogen abstraction and addition in a wide pH range. Thus, the AOP based on sulfate radical was an effective strategy to degradative aquatic sulfonamides.
The activation of persulfate (PS) and peroxymonosulfate (PMS) have been considered as the most portable methods to generate sulfate radical in aquatic environment. PS occupying higher redox potential (E0 = 2.01 V)23 have been widely used to generate ˙SO4− when it is activated by transition metals and heterogeneous catalysts. Among various transition metal ions, Fe(II) is advantageous since it is cheap, non-toxic, naturally abundant and environmental friendly.19 Classical Fe(II) activating PS process has present potential efficiencies on rapidly reducing and even mineralizing organics for the generation of sulfate radical via eqn (1), but a higher removal rate of the target compound requires the continuous addition of dissolved Fe(II) in a homogeneous system.19 Meanwhile, excessive Fe(II) can quickly consume PS or sulfate radical in solution, which can seriously inhibit removal efficiency of target organics (Eq. (2)). Recently, zero valent iron (ZVI, Fe0), as a green reductive reagent,24–28 in lieu of Fe(II) can also induce heterogeneous activated PS, and some studies of sulfamethoxazole degradation by Fe0/PS have been reported by Ghauch's group.29,30 The application of Fe0 not only overcome the disadvantages of sulfate radical and PS consumption by excessive Fe(II) but also avoid the addition of other anions (Cl− or SO42−) to the solution (Eq. (1) and (3)). Meanwhile, the recycle of Fe(II) by the reaction between Fe(II) and Fe0 at the Fe0 surface has been provided (Eq. (4)). According to Oh et al.,31 Fe0 not only is the source of dissolved Fe(II), but also directly activates PS to produce sulfate radical which did not transform to Fe(II) (Eq. (5)). Besides, Guo et al.12,32 firstly reported that common oxidants could enhance the reactivity of Fe0 by cleaning the precipitation of ferric hydroxides at the Fe0 surface. Lai et al.33,34 also reported that the PS could accelerate the reductive rate of 4-nitrophenol by Fe0. Thus Fe0 exhibiting more reactivity in the presence of PS also can generate ˙OH rather than ˙SO4− with dissolved oxygen in aquatic chemistry. Thus, the dominating ROS in the Fe0/PS process should be identified, and the reactive mechanisms between Fe0 and PS need to be reinvestigated.
| S2O82− + Fe2+ → ˙SO4− + Fe3+ + SO42− | (1) |
| ˙SO4− + Fe2+ → Fe3+ + SO42− | (2) |
| Fe0 − 2e− → Fe2+ | (3) |
| Fe0 + 2Fe3+ → 3Fe2+ | (4) |
| 2S2O82− + Fe0 → 2˙SO4− + Fe2+ + 2SO42− | (5) |
Hence, sulfadiazine (SDZ) as a typical sulfonamide was selected as the target containment, and kinetics, mechanisms, and degradation pathway of SDZ degradation in the Fe0/PS process were investigated. The aims of this study are to (1) assess the effects of initial Fe0, PS, and SDZ concentration on SDZ degradation by the Fe0/PS process, (2) determine the effects of several background materials in water on SDZ degradation by the Fe0/PS process, (3) identify the dominating ROS in the Fe0/PS process, and (4) clarify SDZ degradation pathways in the Fe0/PS process.
Experimental
Materials
Sulfadiazine of 99% purity was supplied by TCI Co. LLC. (Tokyo, Japan). Fe0 powder of 97% purity, dimethyl pyridine N-oxide (DMPO) of 97% purity, and humic acid (HA) were purchased from Sigma-Aldrich Co. LLC. (St. Louis, MO, USA). Potassium persulfate were supplied by Sinopharm Chemicals Reagent Co., Ltd. (Shanghai, China). Other chemicals of analytical grade were provided by Sinopharm Chemicals Reagent Co., Ltd. All chemicals were not further purified and solutions were prepared with deionized (DI) water.Batch experiments
Kinetic experiments were carried out in an organic glass reactor open to the air at 20 ± 1.0 °C, and 0.5 L solution containing SDZ was completely mixed by digital display electric blender at 600 rotation rate (rpm). For experiments carried out under anoxic and oxygen conditions, solutions were purged for 30 min including 20 min of preparation time and 10 min of reaction time with pure nitrogen and oxygen at a flow rate of 1 L min−1 controlled by VAT-315 rotary flowmeter (Dwyer Instruments Inc., US), respectively. The initial pH of solution was adjusted by H2SO4 and NaOH, and then experiments were initiated after addition of Fe0 and PS into the reactor. For the kinetic study, at fixed time intervals, 2 mL sample was rapidly transferred into the sample beaker that was immediately quenched with 20 μL of sodium hyposulfite, filtered with 0.22 μm membrane and collected into sample vials quickly.Chemical analysis
A Mettler-Toledo high-performance FE20-FiveEasy pH meter with a saturated KCl solution as electrolyte produced (Switzerland) was employed to measure solution pH and daily calibration with standard buffers (pH 4.00, 6.86 and 9.18) was done to ensure its accuracy. Electron paramagnetic resonance (EPR) spectrometry (Bruker, Germany) with the magnetic field of 3400–3500 G was employed.A Merlin Compact scanning electron microscopy (SEM) (Carl Zeiss, Germany) coupled with an X-Max energy dispersive X-ray spectrum (Oxford Instrument, UK) was employed to characterize iron particles and analysis the elemental composition which were depicted in Fig. S1 (see ESI†). The concentrations of Fe(II) and total ions (after reduction to Fe(II) with hydroxylamine hydrochloride) were determined at 510 nm after complexing with 1,10-phenanthroline by an UV-2600 UV-Vis spectrophotometer (Shimadzu, Japan).
SDZ was determined by a Waters ACQUITY ultra-performance liquid chromatography (UPLC) system including a binary solvent manager and a sample manager with a TUV detector (Milford MA, United States). The water samples were extracted by an off line solid-phase extraction (SPE) with Oasis HLB (200 mg) cartridges which was used to enrich and to clean up the aqueous sample. Separation was accomplished with an Agilent SB-C18 column (2.1 × 50 mm, 1.8 μm; Agilent, United States) at 30 ± 1.0 °C with a mobile phase of two effluents (effluent A: 30% acetonitrile; effluent B: 70% H2O with 0.1% formic acid) at a flow rate of 0.1 mL min−1. Concentrations of SDZ were determined by comparing the peak area at 265 nm with that of standards. The intermediate products of SDZ degradation were separated by the Agilent 1290 Infinity but interfaced with a triple quadrupole mass detector (6400) (UHPLC-MS) (Santa Clara CA, United States). The separated sample analysis by mass spectra was conducted in positive and negative mode electrospray ionization ((+)ESI and (−)ESI) over a mass range of 50–500 m/z. Separation was accomplished with an Agilent Proshell-C18 column (2.1 × 100 mm, 1.8 μm; Agilent, United States). The fragment used was 90 V conducted in auto full scan mode (MS). The fragment and collision energy used in product ion mode were 90 V and 15 eV, respectively.
Results and discussion
Effects of treatment factors on kinetics of SDZ removal in the Fe0/PS process
The effect of initial Fe0 loading ranged from 0.25 to 5 mM on kinetics of SDZ degradation in the Fe0/PS process is shown in Fig. 1(a). SDZ degradative curve by Fe0/PS exhibited an autocatalytic shape, which was divided into a lag phase and a rapid degradation phase in the Fe0/PS process similar to the Fe0/H2O2 process.35 The lag phase was explained by heterogeneous reactions between Fe0 and PS, and was shortened with increasing Fe0 loading. Then rapid reactions between Fe(II) and PS was initialled by the releasing of Fe(II) after the lag phase. The degradative rate constant of SDZ by Fe0/PS in the rapid phase is calculated by a pseudo-first-order kinetic model (Eq. (6)),8 and fitting results are shown as solid lines in Fig. 1.
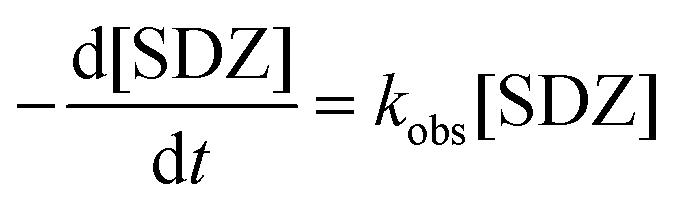 | (6) |
 | ||
| Fig. 1 Effect of initial Fe0 loading (a), PS concentration (b), and SDZ concentration (c) on SDZ degradative kinetics by Fe0/PS. Experimental conditions: initial pH = 7.0, rpm = 600, and T = 20 ± 1 °C. | ||
The rate constant of SDZ removal linearly increases with Fe0 loading ranged from 0.25 mM to 2 mM (see Fig. S2(a)†) for more Fe0 could release more Fe(II). The final removal rate of SDZ at 10 min also increased with initial Fe0 loading. When the concentration of Fe0 reached 5 mM, the rate constant of SDZ removal did not increase but even slightly decreased from 0.63 to 0.58 min−1 that was ascribed to the consumption of ROS by excess Fe0. Similarly, the final removal rate of SDZ at 10 min merely rose about 2% from with increasing Fe0 loading from 2 to 5 mM. As shown in Fig. 1(b), the influence of initial PS concentration ranged from 0.25 to 5 mM in SDZ removal kinetics by Fe0/PS was investigated. The lag phase was seriously shortened to 1 min with PS of high concentration above 1 mM. Increasing PS could rapidly eliminate the passivating film on the surface of Fe0 and improved the reactivity of Fe0. According to eqn (1), more PS could accelerate reactions between Fe(II) and PS by generating more ROS. Thus, the rate constant of SDZ removal linearly increase with increasing PS concentration from 0.25 mM to 2 mM (see Fig. S2(a)†). In addition, the degradative rate constant of SDZ in rapid phase and the removal rate of SDZ was not increased with increasing PS from 2 to 5 mM for the limitation of Fe0 loading.
Influence of SDZ concentration on SDZ removal by Fe0/PS also are studied in Fig. 1(c). At low SDZ concentration, the rate constant of SDZ removal was not changed. With increasing SDZ concentration above 10 μM, the rate constant and removal rate of SDZ degradation were seriously inhibited for the amount of ROS was limited to concentrations of Fe0 and PS. Thereby, removal of aquatic SDZ of high concentrations might require more Fe0 and PS.
Meanwhile, a comparison of SDZ removal kinetics in the Fe0/PS system and the Fe(II)/PS system has been conducted in Fig. S3† at the optimal experimental conditions. Fe(II) showed the high activity towards PS in the first 1 min, which was different from the lag phase in the Fe0/PS system. But the final removal rate of SDZ by Fe(II)/PS was lower than Fe0/PS for the consumption of ROS by excess dissolving Fe(II), which was in agreement with other reports.29
Effect of initial pH
Generally, pH value plays a key role in the application of traditional AOPs.19 Hence, the effect of initial pH on SDZ degradation in the Fe0/PS process should be clarified. As illustrated in Fig. 2, SDZ degradative kinetics by Fe0/PS in a wide range of initial pH value from 5.0 to 9.0 are investigated. Meanwhile, the variation of pH values in the reaction has been monitored in Fig. S4†(a). The acid solution could shorten the lag phase and accelerate the rate constant of SDZ degradation since Fe0 showed more activity in weak acid conditions. Nevertheless, the final removal rate of SDZ were only 70.4% at pH 5.0 and 69.4% at pH 6.0, which were much lower than that of 83.5% at neutral pH. Quick release of Fe(II) in acid solutions might enhance reactions between Fe(II) and PS but excess Fe(II) seriously compete for ROS or PS with SDZ. On the contrary, alkaline solutions at pH 8.0 and 9.0 can restrain the passivation of Fe0 surface and extend the lag phase of SDZ. The degradative kinetics and the rate constant of SDZ removal in the rapid phase at pH 8.0–9.0 still reach 0.28 min−1 which is lower than 0.40 min−1 at pH 7.0 (see Fig. S4†(b)). Nevertheless, the presence of PS could accelerate the passivation of Fe0 as reported by Lai,33,34 and the Fe0/PS system in alkaline solutions still developed reactive ability to degrade SDZ. Therefore, the removal rate at 10 min did not decrease with increasing pH. Removal rates of SDZ by Fe0/PS in alkaline and neutral solutions were even higher than that at acid pH, which indicated that alkaline solutions did not decrease the total concentration of ROS employed to destroy SDZ. So slow release of Fe(II) rather than rapid dissolving Fe(II) from Fe0 enhanced the degradative reactions between ROS and SDZ in the Fe0/PS process. Neutral and weak alkaline pH are more suitable to SDZ degradation by Fe0/PS, which increase the potential for engineering applications in organics removal in the Fe0/PS process. Furthermore, sludge generation by the end of the reaction was well controlled in neutral and weak alkaline pH by comparing with the concentration of dissolved iron species in solution at each pH (see Fig. S4(b)†).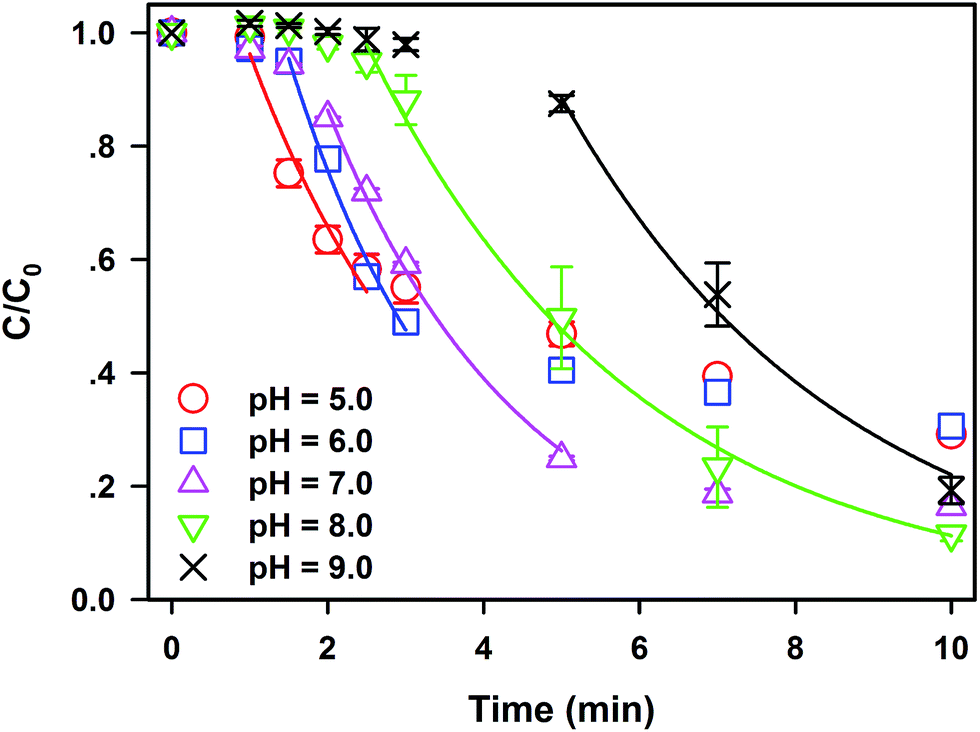 | ||
| Fig. 2 Effect of initial pH on SDZ degradative kinetics by Fe0/PS. Experimental conditions: [Fe0]0 = 1 mM, [PS]0 = 1 mM, [SDZ] = 20 μM, rpm = 600, and T = 20 ± 1 °C. | ||
Effects of background materials in water on SDZ removal by Fe0/PS
Batch experiments were carried out to investigate SDZ degradative kinetics by Fe0/PS in the presence of aquatic background materials including Cl−, SO42−, NO3−, ClO4−, HCO3−, and HA. The raw time courses of SDZ removal by Fe0/PS are shown and modelled by eqn (1) in Fig. S5.† The summary of rate constants are exhibited in Fig. 3. With increasing sulfate, the inhibition on SDZ degradation increased for the reactivity of ZVI for the precipitation of acicular α-FeOOH and precipitation of basic ferric sulfate on iron surface.36 Meanwhile, the presence of SO42− might also decrease the redox potential of ˙SO4−/SO42−, and weaken the oxidative activity of PS.37 Nitrate anion always exhibited serious inhibiting effects on SDZ removal by Fe0/PS with increasing nitrate.24 Nitrate was easily reduced by Fe0 and the reactivity between Fe0 and PS was decreased for the competition of nitrate.38 Besides, more nitrate radical (NO3˙, E0 = 2–2.2 V) showing less reactivity towards SDZ than ˙SO4− could be generated by NO3− and ˙SO4− in the presence of nitrate.39 The effect of chloride on SDZ degradation in the Fe0/PS process was more complicated than other water matrixes. Generally, chlorate exhibited the reaction with ˙SO4− at the rate constant of (1.3–3.1) × 108 M−1 s−1 which could compete for ˙SO4− with SDZ,8 and SDZ degradative rate was decreased in the presence of chloride. However, chloride could be oxidized to halogenated reactive species such as Cl˙ and Cl2˙− which still exhibited strong degradative ability to SDZ.40,41 Thus, the presence of chloride did not seriously inhibit SDZ removal by Fe0/PS, and a higher concentration of chloride decreased the inhibiting effect of chloride on SDZ degradation. The perchlorate anions inhibited SDZ removal by Fe0/PS since the perchlorate could occupy some reactivity sites of Fe0 reacting with PS, which decreased SDZ removal rate.24 However, the degradative rate of SDZ was not affected as the perchlorate increased to 5 mM. The presence of bicarbonate ranged from 0 to 5 mM significantly inhibited the degradation of SDZ by Fe0/PS. With increasing HCO3−, the final removal rate and kobs both sharply decreased. The negative effect of HCO3− on SDZ degradation was most serious among other tested anions. This achieved result was accompany with other reports using activated persulfate process to degrade organic pollutants.22,30 As Ghauch et al.30 reported, HCO3− delayed iron corrosion and limit Fe(II) release into the solution, which could inhibit the generation of ROS in the Fe0/PS system.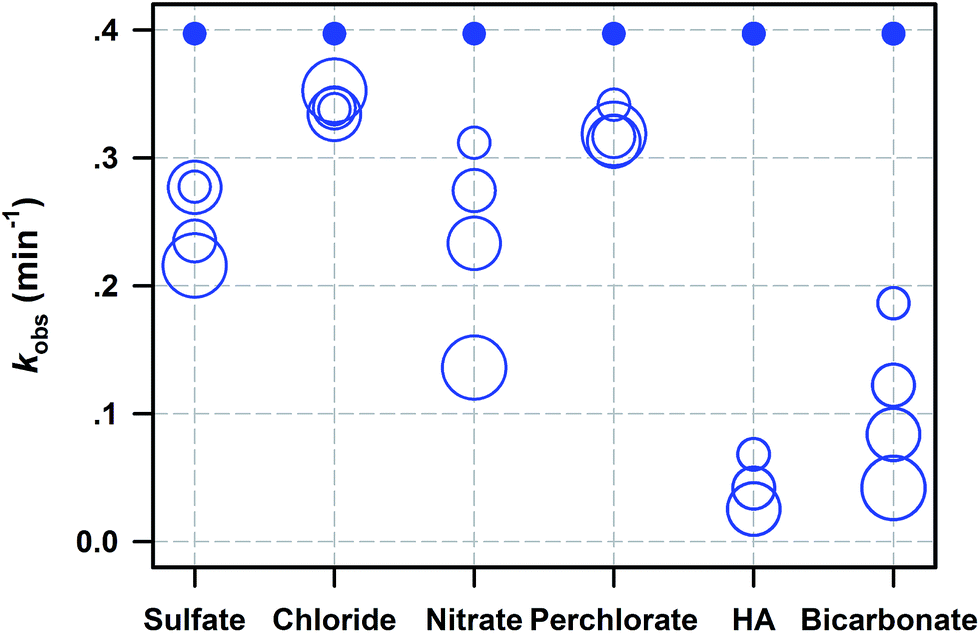 | ||
| Fig. 3 Summary of kobs for SDZ degradation by Fe0/PS with background materials (dissolved anions and HA). The circle size represents the concentration of each background material. Values of kobs for all these data and different levels of concentrations are given in Table S1.† Reaction conditions: [Fe0]0 = 1 mM, [PS]0 = 1 mM, [SDZ] = 20 μM, initial pH = 7.0, rpm = 600, and T = 20 ± 1 °C. | ||
On the other side, HCO3− was considered as an active quencher for ˙SO4− and ˙OH which could compete ROS with SDZ. HA as the most important natural organic matters in surface water and ground water played a critical role on SDZ degradative kinetics by Fe0/PS. HA was consider as a competitive organic to SDZ in the Fe0/PS process, and extensively decreased the removal rate of SDZ. Meanwhile, HA also was a strong ligand13 which could quickly complex dissolving Fe(II) in the Fe0/PS process and inhibited the reaction between Fe(II) and PS. Overall, all the chosen background materials including Cl−, SO42−, NO3−, ClO4−, HCO3−, and HA inhibited SDZ removal by Fe0/PS following a trend of Cl− < ClO4− < SO42− < NO3− < HCO3− < HA.
Identification of the ROS in the Fe0/PS process
Quenching experiments were carried out to identify ROS generated in the Fe0/PS process. Generally, ˙SO4− is considered as the main ROS in the reactions between PS and Fe(II). Meanwhile, ˙OH may also be generated by ˙SO4− described in eqn (7) and (8) with the second order rate constant of 103–104 M−1 s−1 and 4.6–9.1 M−1 s−1, respectively. Besides, the second order constants for methanol (METH) towards ˙OH and ˙SO4− are 9.7 × 108 M−1 s−1 and 2.5 × 107 M−1 s−1, and for tert-butyl alcohol (TBA) are (3.8–7.6) × 108 M−1 s−1 and (4.0–9.1) × 105 M−1 s−1, respectively.42 So METH and TBA were applied to identify the contribution of ˙OH or ˙SO4− to SDZ degradation by Fe0/PS.Degradative kinetics of SDZ by Fe0/PS in the presence of 500 mM METH and TBA were shown in Fig. 4(a). In the presence of 500 mM METH, the degradation of SDZ was entirely inhibited, and the final removal rate only reached 10.9%. Contrarily, the degradative rate of SDZ by Fe0/PS decreased to 67.4% with 500 mM TBA. Although TBA exhibited much lower rate constant to ˙SO4−, high concentration of TBA could also scavenge partial ˙SO4− in solution. In addition, the difference of inhibiting effects between TBA and METH was considered as the contribution of ˙SO4−. Thus, ˙SO4− was considered as the dominated ROS in the Fe0/PS process.
 | ||
| Fig. 4 Inhibition effect of radical quenchers on SDZ degradation in the Fe0/PS process (a) and ESR spectra obtained from Fe0, PS, and the Fe0/PS process with the existence of DMPO (▲ represents the DMPO–˙OH adduct, ● represents the DMPO–˙SO4− adducts, and ◆ represents the HDMPO–OH adduct) (b). Reaction conditions: [Fe0]0 = 1 mM, [PS]0 = 1 mM, [SDZ] = 20 μM, [TBA]0 or [METH]0 = 500 mM, initial pH = 7.0, rpm = 600, and T = 20 ± 1 °C. EPR experimental conditions: [DMPO]0 ≈ 0.1 M, [Fe0]0 = 10 mM, [PS]0 = 10 mM, initial pH = 7.0, T = 20 ± 1 °C. | ||
To further confirm the presence of ˙SO4−, EPR spectroscopy was used to verify the specific adduct between DMPO and ROS generated in the Fe0/PS process. As shown in Fig. 4(b), three apparent signals of DMPO–˙OH, DMPO–˙SO4−, and HDMPO–OH (the oxidative products of HDMPO by ˙OH) adducts at different time interval in the Fe0/PS process were detected by EPR. However, no signals of DMPO–˙OH, DMPO–˙SO4−, HDMPO–OH, and DMPOX (the oxidative products of DMPO by ˙OH) were determined by EPR in Fe0 or PS solution, which indicated that no ROS could be generated by Fe0 or PS in experimental conditions. The special hyperfine coupling constants (a(N) 1.49 mT, a(H) 1.49 mT, all ±0.05 mT, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 quartet) were consistent with that of the DMPO–˙OH adduct, while the special hyperfine coupling constants (a(N) 1.38 mT, a(H) 1.02 mT, a(H) 0.14 mT, a(H) 0.08 mT, all ±0.05 mT) were in accordance with that of the DMPO–˙SO4− adduct.14 HDMPO was generated by the reaction of Fe(III) and DMPO which was always observed in Fenton reactions.43 According to literatures,23,42 the signal of DMPO–˙SO4− adducts usually accompanied with the signal of DMPO–˙OH but hardly to be detected alone in aquatic solution. Besides, the intensity of DMPO–˙SO4− was much lower than that of DMPO–˙OH. This behaviour was ascribed to the fast transformation from DMPO–˙SO4− adducts to DMPO–˙OH adducts via nucleophilic substitution.35
:2:2:1 quartet) were consistent with that of the DMPO–˙OH adduct, while the special hyperfine coupling constants (a(N) 1.38 mT, a(H) 1.02 mT, a(H) 0.14 mT, a(H) 0.08 mT, all ±0.05 mT) were in accordance with that of the DMPO–˙SO4− adduct.14 HDMPO was generated by the reaction of Fe(III) and DMPO which was always observed in Fenton reactions.43 According to literatures,23,42 the signal of DMPO–˙SO4− adducts usually accompanied with the signal of DMPO–˙OH but hardly to be detected alone in aquatic solution. Besides, the intensity of DMPO–˙SO4− was much lower than that of DMPO–˙OH. This behaviour was ascribed to the fast transformation from DMPO–˙SO4− adducts to DMPO–˙OH adducts via nucleophilic substitution.35
| ˙SO4− + H2O → ˙OH + SO42− + H+ | (7) |
| ˙SO4− + OH− → ˙OH + SO42− | (8) |
Discussion on the role of Fe0 in the Fe0/PS process
As mentioned above, ˙SO4− might be produced via two different reactions of eqn (1) and eqn (5). Hereon, the key role of Fe0 in the Fe0/PS process should be clarified to understand mechanisms of SDZ removal by Fe0/PS. As shown in Fig. 5(a), 1,10-phenanthroline as an excellent complex with Fe(II) has been added to study SDZ degradative kinetics by Fe0/PS.44 A very critical inhibition of SDZ removal was observed in the presence of 1 mM 1,10-phenanthroline, and as 1,10-phenanthroline increased to 5 mM, the degradation of SDZ almost was completely stopped by blocking the reaction of eqn (1) since the complex of Fe(II) with 1,10-phenanthroline extensively decreased the concentration of dissolved Fe(II). Meanwhile, the presence of 1,10-phenanthroline did not inhibit the reaction between Fe0 and PS (Eq. (5)) which indicated that ˙SO4− might not be produced via the direct oxidation of Fe0 by PS. Although EDTA was a good ligand for Fe(II) and Fe(III), EDTA was considered as a common promoter in reactions based on ˙OH especially in Fe0/O2 reactions.27,45 As illustrated in Fig. 5(b), various level of EDTA concentrations ranged from 0.05 to 5 mM all inhibited SDZ degradation by Fe0/PS. EDTA was the complex of Fe(II) which exhibited the similar effects on SDZ degradation to 1,10-phenanthroline. EDTA was an organics which could compete ˙SO4− with SDZ. Therefore, the negative effect of EDTA in the Fe0/PS process is different from that in Fe0/O2 reactions. | ||
| Fig. 5 Effects of 1,10-phenanthroline (a), EDTA (b), and DO (c) on SDZ degradation in the Fe0/PS process. Reaction conditions: [Fe0]0 = 1 mM, [PS]0 = 1 mM, [SDZ] = 20 μM, initial pH = 7.0, rpm = 600, and T = 20 ± 1 °C. | ||
Dissolving oxygen (DO) in solution also played a significant role in the Fe0/PS system29 by changing the corrosion of Fe0. Kinetics experiments of SDZ removal by Fe0/PS conducted in the presence of N2, air, and O2 are shown in Fig. 5(c). Anoxic conditions could enhanced SDZ removal by Fe0/PS; nevertheless, the presence of excess oxygen could depress the SDZ degradation. According to the mechanisms of Fe0/PS via the indirect reaction between Fe(II)/PS, the release of Fe(II) was the rate-limiting step. By comparing to the experiments with air or oxygen, dissolving Fe(II) was more stable without the generation of iron oxides on Fe0 surface in the presence of N2, which could promote generation of ROS via eqn (1). Meanwhile, the lower SDZ removal rate in the presence of air and oxygen indicated that the generation of ˙OH by Fe0 and O2 might be ignored in the Fe0/PS process. Besides, the lag phase with purging oxygen increased to 3 min. Although the inhibition of DO on SDZ removal might be assumed to the lack of Fe0 which decreased the generation of ROS via eqn (5), the consumption of Fe0 could not extend the lag phase of reactions. Thus, the extending lag phase increasing with the concentration of DO was ascribed to the generation of iron oxides by the oxidation of Fe(II) by oxygen on the iron surface, which blocked the reactions between Fe(II) and PS.
Possible degradation pathways of SDZ by Fe0/PS
Many studies have identified the degradation intermediates of SDZ by various processes including ozonation,5 Fenton,46 and US/Fe0/PS systems.2 The species of reaction intermediates and degradation pathways of SDZ in different AOPs identified by GC-MS, LC-MS, and HPLC were different. In this paper, off line SPE-UHPLC-MS was utilized to detect the reaction intermediates of SDZ by Fe0/PS at pH 7.0, based on which the SDZ degradation pathways were proposed.As literatures reported, N7, N11, N13 and N17 in SDZ might be reactive sites for oxidation. To confirm the theoretical reactive sites in SDZ molecule, Fukui function calculations of fi0 was employed to assign the most vulnerable sites of SDZ by the attack of ˙SO4− radical. fi0 was calculated by eqn (9)47 which obtained by Multiwfn software based on Hirshfeld charges.48 Thus, Hirshfeld charges and calculated values of condensed Fukui function (fi0) of the optimized SDZ molecule (N + 1 and N − 1) are listed in Table 1. According to the value of condensed Fukui function (fi0), atoms of S8, O8, O9, C13, C11, N17, C1, N6, and N7 were the most reactive sites for ˙SO4− radical attack.
 | (9) |
| Atom, i | qN−1 | qN+1 | fi0 |
|---|---|---|---|
| C1 | 0.069 | −0.007 | 0.038 |
| C2 | −0.087 | 0.036 | −0.062 |
| C3 | 0.170 | 0.201 | −0.016 |
| N4 | −0.278 | −0.212 | −0.033 |
| C5 | 0.342 | 0.390 | −0.024 |
| N6 | −0.272 | −0.320 | 0.024 |
| N7 | −0.210 | −0.251 | 0.020 |
| S8 | 1.304 | 0.527 | 0.388 |
| O9 | −0.752 | −0.867 | 0.058 |
| O10 | −0.740 | −0.851 | 0.055 |
| C11 | 0.350 | 0.007 | 0.171 |
| C12 | −0.010 | 0.031 | −0.021 |
| C13 | −0.040 | −0.389 | 0.174 |
| C14 | 0.169 | 0.400 | −0.115 |
| C15 | −0.030 | −0.047 | 0.008 |
| C16 | 0.034 | 0.004 | 0.015 |
| N17 | −0.076 | −0.297 | 0.111 |
By combination of full scan and product ion mode, 20 reaction intermediates and SDZ were detected in the process of SDZ removal by Fe0/PS, which were all illustrated from Fig. S6 to Fig. S26.† Based on the detected intermediates specified in this study, four proposed transformation pathways (A, B, C, and D) of SDZ degradation in the Fe0/PS process were presented in Fig. 6. Pathway A was formed via the break of S8–N7 bond by ˙SO4− radical, and 4-aminobenzenesulfonic acid (Int 1) and pyrimidin-2-amine (Int 2) were generated. By the continuous attacking of ˙SO4− to Int 1, 4-amino-2,3,5-trihydroxybenzenesulfonic acid (P4, m/z+ 222) was produced. Meanwhile, 2-nitropyrimidine (P6, m/z+ 126) was formed by the oxidation of Int 2 by ˙SO4−, and hydroxy(nitro(nitroso)methyl)carbamic acid (P7, m/z+ 166) and N-((E)-2-hydroxyprop-1-en-1-yl)formimidic acid (P8, m/z+ 102) also were detected as the oxidative products of P6. Besides, the formation of 4-(2-iminopyrimidin-1(2H)-yl) aniline or N-(pyrimidin-2-yl) benzene-1,4-diamine (P1, m/z+ 187) between Int 1 and Int 2 was also a common reaction in SDZ degradation by ˙SO4−.49 In addition, the oxidative pathway of P1 by ˙SO4− was also established based on the detection of P2 (m/z+ 226) and P3 (m/z+ 242).
 | ||
| Fig. 6 Possible degradation pathways of SDZ by Fe0/PS. | ||
In pathway B and C, direct attacking of ˙SO4− on N6 and N17 produced 4-amino-N-carbamimidoyl benzenesulfonamide (P9, m/z+ 215) and 4-nitro-N-(pyrimidin-2-yl)benzenesulfonamide (P11, m/z+ 281),2 respectively. And P9 was oxidized to 4-amino-N-(hydroxy(hydroxyamino)methyl)benzenesulfonamide (P10, m/z+ 234) by ˙SO4−. P11 was the most frequently oxidative product of SDZ by ˙SO4−.2 Besides, abundant oxidative products of P11 by ˙SO4− are also detected and illustrated in Fig. 6.
Among three pathways of SDZ degradation via ˙SO4−, the products generated by pathway A was more abundant than pathway B and C. As shown in Fig. S27,† four initial products of P1, P4, P5, and P6 in pathway A were detected in the first 2 min. All four products increased with reaction time, and then decreased, which revealed that pathway A could continuously consume ˙SO4−. Besides, the area of P11 in pathway B increased with reaction time, which proved that pathway B, was also important to the removal of SDZ. However, P9 of pathway C was firstly detected at 5 min, which indicated the contribution of attacking on N6 by ˙SO4− was ignored in the first 5 min.
Pathway D was a classical oxidative pathway of SDZ by ˙OH but was hardly to be formed in oxidative systems based on ˙SO4−. The generation of OH-SDZ (P19, m/z+ 267)2 by substituting H at C13 with hydroxyl indicated that ˙OH also joined the degradation of SDZ in the Fe0/PS process. In addition, the contribution of pathway D to SDZ removal was consistent with the experiments of identifying ROS, which was about 16.1%.
Conclusions
Batch experiments were carried out to investigate effects of some key factors on SDZ removal by Fe0/PS. Initial concentrations of Fe0 and PS increased from 0.25 to 5 mM both increased the removal rate of SDZ. But increasing SDZ inhibited the SDZ removal rate for the limitation of ROS. Neutral or weak alkaline solutions exhibited higher removal rate of SDZ than weak acid pH, which indicated that the Fe0/PS process was a potential technology for engineering applications in organics removal. Common aquatic matrixes including sulfate, nitrate, chloride, perchlorate, bicarbonate, and HA all showed negative effects on SDZ degradation in the Fe0/PS process following a trend of Cl− < ClO4− < SO42− < NO3− < HCO3− < HA. To verify the dominating ROS in the Fe0/PS system, chemical quenching experiments in the presence of METH and TBA were conducted. Results of quenching experiments implied that ˙SO4− was the dominating ROS in the Fe0/PS system. The chemical detection of DMPO–˙SO4− and DMPO–˙OH by EPR spectra also confirmed the presence of ˙SO4−. Besides, strongly negative effects of 1,10-phenanthroline and EDTA on SDZ degradation in the Fe0/PS process proved that ˙SO4− was not generated by an one-step reaction between Fe0 and PS but via the indirect oxidation of Fe(II) by PS. Meanwhile, the negative effect of DO on SDZ removal also proved that the generation of ˙SO4− was dominated by reactions between Fe(II) and PS. Finally, three pathways via ˙SO4− attack and one pathway via ˙OH substitute of SDZ degradation by Fe0/PS were proposed based on the reactive sites attacking by radicals and intermediate products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Mr Juanshan Du (State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin, PR China) for the kind supporting of chemical calculations and EPR experiments.References
- Q. Q. Zhang, G. G. Ying, C. G. Pan, Y. S. Liu and J. L. Zhao, Environ. Sci. Technol., 2015, 49, 6772–6782 CrossRef CAS PubMed.
- X. Zou, T. Zhou, J. Mao and X. Wu, Chem. Eng. J., 2014, 257, 36–44 CrossRef CAS.
- L. Y. He, Y. S. Liu, H. C. Su, J. L. Zhao, S. S. Liu, J. Chen, W. R. Liu and G. G. Ying, Environ. Sci. Technol., 2014, 48, 13120–13129 CrossRef CAS PubMed.
- W. Q. Guo, R. L. Yin, X. J. Zhou, J. S. Du, H. O. Cao, S. S. Yang and N. Q. Ren, Ultrason. Sonochem., 2015, 22, 182–187 CrossRef CAS PubMed.
- W. Guo, Z. Yang, J. Du, R. Yin, X. Zhou, S. Jin and N. Ren, RSC Adv., 2016, 6, 57138–57143 RSC.
- W. Baran, E. Adamek, A. Sobczak and A. Makowski, Appl. Catal., B, 2009, 90, 516–525 CrossRef CAS.
- Y. Luo, D. Mao, M. Rysz, Q. Zhou, H. Zhang, L. Xu and P. J. J. Alvarez, Environ. Sci. Technol., 2010, 44, 7220–7225 CrossRef PubMed.
- T. Zhou, X. Zou, J. Mao and X. Wu, Appl. Catal., B, 2016, 185, 31–41 CrossRef CAS.
- O. Gonzalez, C. Sans and S. Esplugas, J. Hazard. Mater., 2007, 146, 459–464 CrossRef CAS PubMed.
- M. Clara, B. Strenn, O. Gans, E. Martinez, N. Kreuzinger and H. Kroiss, Water Res., 2005, 39, 4797–4807 CrossRef CAS PubMed.
- X. M. Xiong, Y. K. Sun, B. Sun, W. H. Song, J. Y. Sun, N. Y. Gao, J. L. Qiao and X. H. Guan, RSC Adv., 2015, 5, 13357–13365 RSC.
- X. Guo, Z. Yang, H. Dong, X. Guan, Q. Ren, X. Lv and X. Jin, Water Res., 2016, 88, 671–680 CrossRef CAS PubMed.
- J. S. Du, B. Sun, J. Zhang and X. H. Guan, Environ. Sci. Technol., 2012, 46, 8860–8867 CrossRef CAS PubMed.
- J. Zou, J. Ma, L. Chen, X. Li, Y. Guan, P. Xie and C. Pan, Environ. Sci. Technol., 2013, 47, 11685–11691 CrossRef CAS PubMed.
- A. Ghauch, J. Adv. Oxid. Technol., 2017, 20, 20160197–20160198 Search PubMed.
- S. Naim and A. Ghauch, Chem. Eng. J., 2016, 288, 276–288 CrossRef CAS.
- A. Ghauch, A. M. Tuqan, N. Kibbi and S. Geryes, Chem. Eng. J., 2012, 213, 259–271 CrossRef CAS.
- Y. Wang, J. B. Liang, X. D. Liao, L.-s. Wang, T. C. Loh, J. Dai and Y. W. Ho, Ind. Eng. Chem. Res., 2010, 49, 3527–3532 CrossRef CAS.
- X. Xiong, B. Sun, J. Zhang, N. Gao, J. Shen, J. Li and X. Guan, Water Res., 2014, 62, 53–62 CrossRef CAS PubMed.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2015, 279, 861–873 CrossRef CAS.
- A. Ghauch, A. Baalbaki, M. Amasha, R. El Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- C. Qi, X. Liu, Y. Li, C. Lin, J. Ma, X. Li and H. Zhang, J. Hazard. Mater., 2017, 328, 98–107 CrossRef CAS PubMed.
- J. Du, D. Che, X. Li, W. Guo and N. Ren, RSC Adv., 2017, 7, 18231–18237 RSC.
- L. P. Liang, X. H. Guan, Z. Shi, J. L. Li, Y. N. Wu and P. G. Tratnyek, Environ. Sci. Technol., 2014, 48, 6326–6334 CrossRef CAS PubMed.
- L. P. Liang, W. Sun, X. H. Guan, Y. Y. Huang, W. Y. Choi, H. L. Bao, L. N. Li and Z. Jiang, Water Res., 2014, 49, 371–380 CrossRef CAS PubMed.
- X. H. Guan, Y. K. Sun, H. J. Qin, J. X. Li, I. M. Lo, D. He and H. R. Dong, Water Res., 2015, 75, 224–248 CrossRef CAS PubMed.
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045–2053 CrossRef CAS PubMed.
- G. Ayoub and A. Ghauch, Chem. Eng. J., 2014, 256, 280–292 CrossRef CAS.
- A. Ghauch, G. Ayoub and S. Naim, Chem. Eng. J., 2013, 228, 1168–1181 CrossRef CAS.
- S. Y. Oh, S. G. Kang and P. C. Chiu, Sci. Total Environ., 2010, 408, 3464–3468 CrossRef CAS PubMed.
- X. Guo, Z. Yang, H. Liu, X. Lv, Q. Tu, Q. Ren, X. Xia and C. Jing, Sep. Purif. Technol., 2015, 146, 227–234 CrossRef CAS.
- Z. Xiong, B. Lai, P. Yang, Y. Zhou, J. Wang and S. Fang, J. Hazard. Mater., 2015, 297, 261–268 CrossRef CAS PubMed.
- Y. Yuan, B. Lai and Y. Y. Tang, Chem. Eng. J., 2016, 283, 1514–1521 CrossRef CAS.
- T. Zhou, Y. Li, J. Ji, F. S. Wong and X. Lu, Sep. Purif. Technol., 2008, 62, 551–558 CrossRef CAS.
- T. Sugimoto and Y. S. Wang, J. Colloid Interface Sci., 1998, 207, 137–149 CrossRef CAS PubMed.
- X. Wu, X. Gu, S. Lu, Z. Qiu, Q. Sui, X. Zang, Z. Miao and M. Xu, Sep. Purif. Technol., 2015, 147, 186–193 CrossRef CAS.
- W. Z. Yin, J. H. Wu, P. Li, X. D. Wang, N. W. Zhu, P. X. Wu and B. Yang, Chem. Eng. J., 2012, 184, 198–204 CrossRef CAS.
- P. Neta and R. E. Huie, J. Phys. Chem., 1986, 90, 4644–4648 CrossRef CAS.
- R. Yuan, S. N. Ramjaun, Z. Wang and J. Liu, J. Hazard. Mater., 2011, 196, 173–179 CrossRef CAS PubMed.
- C. Liang, Z. S. Wang and N. Mohanty, Sci. Total Environ., 2006, 370, 271–277 CrossRef CAS PubMed.
- G. Fang, J. Gao, D. D. Dionysiou, C. Liu and D. Zhou, Environ. Sci. Technol., 2013, 47, 4605–4611 CrossRef CAS PubMed.
- J. M. Fontmorin, R. C. Burgos Castillo, W. Z. Tang and M. Sillanpaa, Water Res., 2016, 99, 24–32 CrossRef CAS PubMed.
- L. P. Liang, W. J. Yang, X. H. Guan, J. L. Li, Z. J. Xu, J. Wu, Y. Y. Huang and X. Z. Zhang, Water Res., 2013, 47, 5846–5855 CrossRef CAS PubMed.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 6936–6941 CrossRef CAS PubMed.
- J. F. Yang, S. B. Zhou, A. G. Xiao, W. J. Li and G. G. Ying, J. Environ. Sci. Health, Part B, 2014, 49, 909–916 CrossRef CAS PubMed.
- J. Oláh, C. Van Alsenoy and A. B. Sannigrahi, J. Phys. Chem. A, 2002, 106, 3885–3890 CrossRef.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- Y. Feng, D. Wu, Y. Deng, T. Zhang and K. Shih, Environ. Sci. Technol., 2016, 50, 3119–3127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07920f |
| This journal is © The Royal Society of Chemistry 2017 |