
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of hydrophobic alkyl chain length on the mechanical properties of latex particle hydrogels
Yang Gaoa,
Lijie Duanb,
Shuang Guanb,
Guanghui Gao a,
Ya Chenga,
Xiuyan Ren*a and
Yuanrui Wang*a
a,
Ya Chenga,
Xiuyan Ren*a and
Yuanrui Wang*a
aSchool of Chemical Engineering, Changchun University of Technology, Changchun 130012, China. E-mail: xyren_bio@163.com; wyr@ccut.edu.cn
bSchool of Chemistry and Life Science, Changchun University of Technology, Changchun 130012, China
First published on 18th September 2017
Abstract
Herein, different long alkyl chains (C1, C6, C12, and C16) were introduced as hydrophobic segments to enhance the performance of hydrogels reinforced by latex particles (LP-Gel). Poly(butyl acrylate) (PBA) latex particles (LPs) were employed as hydrophobic association cross-linking centers. First, the PBA latex particles were prepared via emulsion polymerization, and then, LP-Gel with high mechanical strength was prepared via one-pot free radical polymerization using acrylamide as a monomer, LP as a cross-linking center, and methacrylate as a hydrophobic molecule. It was found that the length of the hydrophobic alkyl chains from methacrylate has a significant effect on the mechanical performance and swelling degree of the hydrogels. The short alkyl chains exhibited weak hydrophobic interactions, and the resulting LP-Gel had a low mechanical strength. However, the long alkyl chains can effectively entangle with LPs through strong hydrophobic interactions, which significantly enhance the mechanical strength of the hydrogels. As a result, the LP-Gel exhibits a maximum fracture stress of 1.2 MPa and elongation of 2336%. This study will have a profound impact on the understanding of hydrogels toughened by hydrophobic alkyl chains of different lengths.
Introduction
Hydrogels are typically soft and wet materials with a three-dimensional polymer network and a large amount of water. In recent decades, hydrogels have attracted significant attention in various fields such as in tissue engineering,1,2 sensors,3,4 and actuators.5–7 However, traditional hydrogels are weak and brittle; this limits their wide application. Thus, extensive efforts have been devoted towards exploring tough hydrogels, including slide-ring hydrogels,8–10 nanocomposite hydrogels,11–14 double-network hydrogels,15–18 macromolecular microsphere composite hydrogels,19–23 and hydrophobic association hydrogels.24–28For hydrophobic association hydrogels, the hydrophobic segments can be self-assembled into micelles as cross-linking centers; thus, the mechanical strength of the hydrogels is enhanced. Okay et al. developed hydrophobic association hydrogels with a large strain, high mechanical strength, and self-healing performance; their properties could be tuned by modulating the length of the hydrophobic alkyl chains. However, the maximum mechanical strength of these hydrogels was not more than 500 kPa.29,30 Liu et al. have prepared a novel hydrophobic association hydrogel using acrylamide and fatty alcohol polyoxyethylene acrylate molecules. The mechanical strength could be tuned by adjusting the ratio of monomers and the concentration of surfactants. However, the maximum tensile strength was 318 kPa.31 Wang and co-workers successfully fabricated hydrophobically associated hydrogels based on acrylamide and anionic surfactant monomers. The anionic surfactant segment formed multi-micellar aggregates and acted as cross-linking centers to endow the hydrogels with high mechanical strength.32 Furthermore, a series of double-network hydrogels and hybrid hydrogels was fabricated by introducing hydrophobic segments. Chen et al. designed a novel double-network physical cross-linking hydrogel. The hydrophobic association polyacrylamide hydrogel acted as a second network to effectively dissipate energy and improve strength.16 Moreover, Cui and co-workers constructed a hybrid hydrogel cross-linked by graphene oxide sheets and hydrophobically associated domains that exhibited anti-fatigue and self-healing properties. However, the fracture stress of the hydrogel was only 260 kPa.33
In our previous study, a novel method was proposed to improve the mechanical properties of hydrogels via the introduction of latex particles (LPs) as hydrophobic association cross-linking centers. Hexadecyl methacrylate (HMA) as a hydrophobic monomer could be adsorbed on the surface of the LPs due to hydrophobic interactions; this forms the LP-Gel.34 However, the length of the hydrophobic alkyl chains could not be considered to enhance the mechanical strength of hydrogels. We considered that the hydrophobic segments could exert a great influence on the interaction between the hydrophobic segments and latex particles and affect the mechanical behavior of the LP-Gel. Therefore, it was crucial to study the effects of different long hydrophobic chains on the mechanical properties based on the system of latex particles toughening hydrogels.
Herein, we introduced different hydrophobic monomers to enhance the mechanical strength of hydrogels reinforced by LPs as cross-linking centers for hydrophobic association. The mechanical performance of hydrogels can be measured by tuning the contents of LPs and the length of hydrophobic alkyl chains. Subsequently, the swelling behavior and rheological performance of the hydrogels were also investigated by varying the length of the hydrophobic alkyl chains. These results are meaningful for the design of a new generation of novel latex particle hydrogels.
Experimental
Materials
Acrylamide (AAm; 99.0%), potassium persulphate (KPS; 99.5%), sodium dodecyl sulfate (SDS; 97%), N,N,N′,N′-tetramethylethylenediamine (TMEDA; 99.5%), methyl methacrylate (MMA; 99.5%), hexyl methacrylate (HMA; 98%), lauryl methacrylate (LMA; 96%), isocyanuric acid triallyl ester (TAIC; 98%), and sodium carbonate (Na2CO3; 99.0%) were supplied by Aladdin. Hexadecyl methacrylate (HDMA; 95%) was purchased from Zhejiang Kangde New Materials Co. Ltd., China. MMA and HMA were distilled under reduced pressure, and LMA and HDMA were passed three times through a column packed with basic alumina. Butyl acrylate (BA) was bought from Aladdin and further purified by distillation under reduced pressure. Sodium chloride (NaCl; 99.5%) was obtained from Beijing Chemical Factory. Deionized water was used in all the experiments.Preparation of PBA LMs
SDS (0.4 g) and Na2CO3 (0.24 g) were dissolved in deionized water (35 mL) in a three-necked boiling flask. Subsequently, BA (60 g) and TAIC (0.3 g) were added to the reactor and stirred for 30 min under the protection of oxygen gas. Finally, a certain amount of KPS (0.24 g) was dissolved in 5 mL deionized water, and the solution was added to the reactor. The reaction was carried out at 65 ± 1 °C for 2 h to achieve high conversation rate. The obtained yield was 97.4%. Finally, the PBA LPs were obtained and could be utilized directly in the following process.Preparation of hydrogels
The hydrogels were synthesized by free radical polymerization of AAm and different hydrophobic monomers in the presence of the abovementioned latex particles (LPs) as cross-linking centers for hydrophobic association. For example, the P(AAm/LMA)-LP hydrogel was prepared as follows: SDS (0.21 g) and NaCl (0.168 g) were dissolved in deionized water (27 mL) under stirring at 25 °C for 15 min to obtain a transparent solution. LMA (0.07 g) and LPs (0.0175, 0.035, 0.07, and 0.105 g) were added to the abovementioned SDS-NaCl solution under stirring for 6 h to form a homogeneous solution. AAm (7 g) and 1 mL of initiator KPS (0.03 g mL−1) were added and stirred for 10 min. Subsequently, TMEDA (60 μL) was added to the abovementioned uniform solution. Finally, the resulting solution was injected into a mold consisting of a pair of parallel glass plates and a silicone spacer (thickness: 3 mm), and polymerization was conducted at 30 °C for 12 h. For comparison, the P(AAm/LMA) hydrogel and PAAm-LP hydrogel were fabricated according to the abovementioned procedure. P(AAm/MMA)-LP, P(AAm/HMA)-LP, and P(AAm/HDMA)-LP hydrogels were also obtained by the same experimental process. The mole ratio of hydrophobic monomer was fixed at 0.28 mol% of AAm.Dynamic light scattering measurements
The dynamic light scattering (American Brookhaven 90Plus Particle Size Analyzer) experiments were performed on the LPs and the mixed solution of LPs and hydrophobic monomers (MMA, HMA, LMA, and HDMA) to characterize their particle size and size distribution. The laser light-scattering angle was fixed at 90°. The samples were diluted in deionized water with a concentration of 0.0002 mg mL−1 for analysis. Each sample was tested at least five times, and the average particle size was obtained.Mechanical tests
The mechanical tests were performed on the as-prepared samples using a tensile tester (SHIMADZU, model AGS-X, 100N, Japan) at room temperature. The hydrogel specimens were cut into a dumbbell shape with a gauge length of 30 mm, width of 4 mm, and a depth of 3 mm. The crosshead speed used in all the experiments was fixed at 100 mm min−1. The value of toughness was obtained by calculating the area below the stress–strain curve of each sample, whereas the elastic modulus was determined by fitting the initial linear region (between 0% and 20% for elongation) of the stress–strain curve. To ensure accuracy, five specimens of each sample were tested, and the average values were obtained. For the cyclic tensile experiments, the samples were first loaded to the strain of 1000% with a constant velocity of 100 mm min−1 and then unloaded at the same velocity. The dissipated energies were estimated by the area between loading–unloading curves.Rheological measurements
The rheological measurements of the hydrogels were carried out using a TA AR 2000ex rheometer in the oscillatory mode at 25 °C. The measuring device was equipped with a parallel plate configuration (25 mm in diameter and 2 mm between gap distance). The storage modulus G′ and loss modulus G′′ were subjected to frequency sweeping tests in the angular frequency range from 0.1 to 100 rad s−1. The gelation reactions of the four different hydrogels were monitored by the rheometer at 30 °C up to a reaction time of about 40 min.Swelling behavior
The hydrogel samples prepared using different hydrophobic monomers (MMA, HMA, LMA, and HDMA) with a thickness of 2 mm and a diameter of 15 mm were dried in a vacuum oven at 60 °C for 24 h to a constant weight. The dried hydrogels were then immersed in an excess of deionized water (50 mL). Deionized water was changed regularly during the test. Swollen hydrogels were periodically removed from deionized water and weighed after blotting the surface water with filter paper. The swelling ratio was calculated by the following equation:| SR = ((Ws − Wd)/Wd) | (1) |
Morphological observation
The hydrogel morphology was observed by scanning electron microscopy (SEM, JSM6510, JEOL). The samples were freeze-dried under vacuum using a freeze dryer (FDU-2110, Eyela). Before the measurement, all the samples were broken by inserting into liquid nitrogen for 3 min and then sputtered with platinum. The magnification factor was 200 times.Results and discussion
In this investigation, poly(butyl acrylate) (PBA) latex particles (LPs) were designed as association cross-linking centers for hydrophobic segments to improve the mechanical strength of the hydrogels. PBA LPs were synthesized by an emulsion polymerization method using TAIC as a cross-linker and KPS as an initiator. The particle size and size distribution of the PBA LPs were characterized by DLS. As shown in Fig. 1, the mean diameter was 293.2 nm, and the size distribution was 0.005. The particle size of LPs upon absorption of hydrophobic methacrylate was also measured by DLS. As expected, the DLS results indicated that the particle size of the LP-adsorbed hydrophobic monomer presented monodisperse distribution. In other words, the methacrylates (MMA, HMA, LMA, and HDMA) could be adsorbed onto LPs. Simultaneously, the mean diameter gradually increased with the increase of the hydrophobic alkyl length. The mean diameters of the LPs with MMA, HMA, LMA, and HDMA were 294.2, 297.8, 301.4, and 309.9 nm, respectively.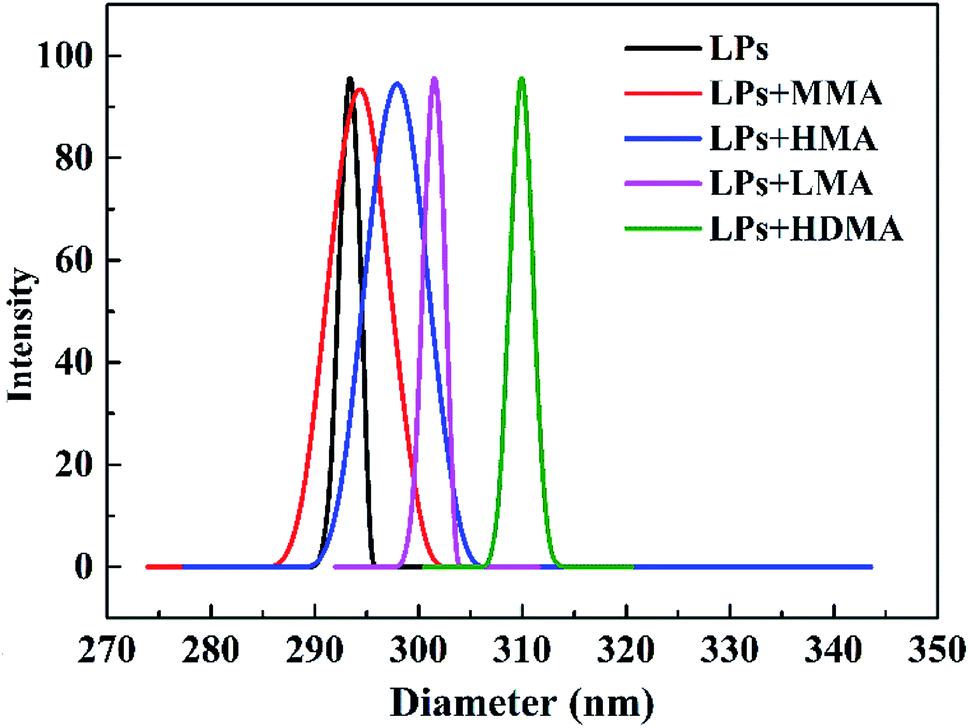 | ||
| Fig. 1 The particle size of PBA LPs and methacrylate-absorbed LPs. | ||
The hydrogels were synthesized by the radical polymerization of AAm and methacrylates with different length of hydrophobic chains in the presence of the PBA LPs. As illustrated in Fig. 2, the hydrogels were fabricated with LPs as association cross-linking centers for hydrophobic monomers. As a result, four samples, i.e. P(AAm/MMA)-LPs, P(AAm/HMA)-LPs, P(AAm/LMA)-LPs, and P(AAm/HDMA)-LPs, with different hydrophobic chains were obtained.
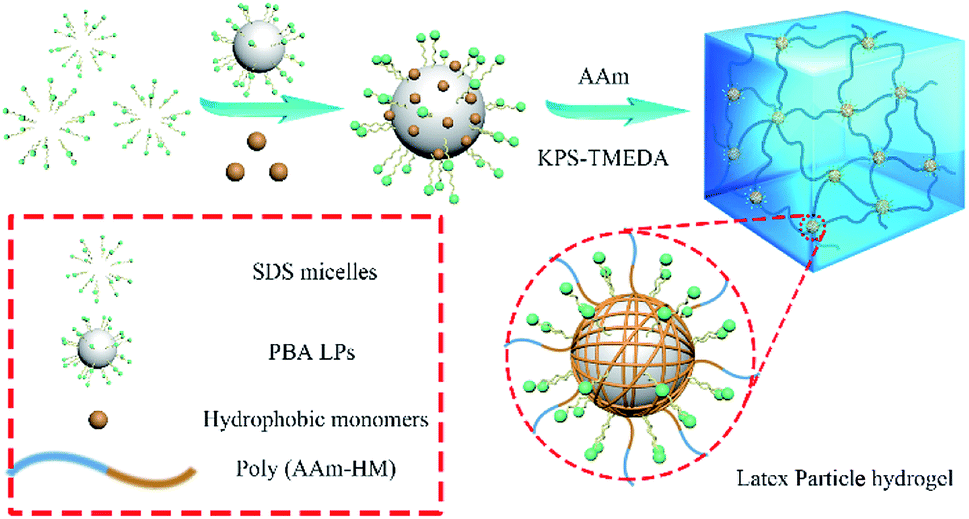 | ||
| Fig. 2 Schematic of the LP-toughened hydrogels with different hydrophobic monomers. | ||
For observing the validity of the LP-reinforced hydrogels, first, the typical tensile mechanical performance of PAAm-LP, P(AAm/LMA), and P(AAm/LMA)-LP hydrogels was determined (Fig. 3). The tensile strength, toughness, and elastic modulus of the PAAm-LPs hydrogels were only 19 kPa, 0.41 MJ m−3, and 25 kPa, respectively. Moreover, the related values of P(AAm/LMA) hydrogels were obtained, which were only 560 kPa, 3.26 MJ m−3, and 40 kPa. In contrast, when the LPs were introduced into the P(AAm/LMA) hydrogel as association cross-linking centers for hydrophobic segments, the P(AAm/LMA)-LP hydrogels exhibited excellent mechanical performance. The tensile strength, toughness, and elastic modulus of the hydrogels were 1.2 MPa, 8.26 MJ m−3, and 50 kPa, respectively, much higher than those of the PAAm-LPs and P(AAm/LMA) hydrogels; this suggested that the introduction of the LPs could significantly improve the mechanical performance of the hydrophobic association hydrogels.
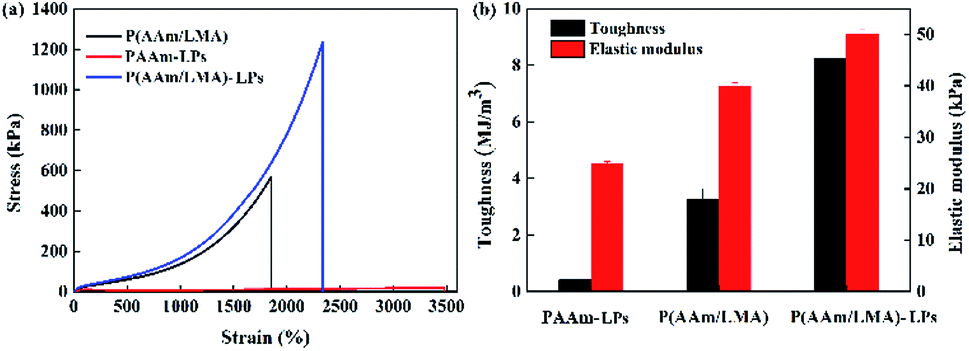 | ||
| Fig. 3 (a) Tensile stress–strain curves and (b) toughness and elastic modulus of the P(AAm/LMA), PAAm-LPs, and P(AAm/LMA)-LPs hydrogels. | ||
Subsequently, the influence of LP content on the mechanical properties of the P(AAm/LMA)-LP hydrogels was systematically investigated. As observed from Fig. 4, the tensile strength and toughness of P(AAm/LMA)-LPs obviously increased and then decreased when the LP concentration varied from 0.25 to 1.5 wt% for AAm. When the LP concentration for AAm was 0.5 wt%, the tensile strength and toughness of the P(AAm/LMA)-LP hydrogel became 1.2 MPa and 8.26 MJ m−3, respectively. With the increase of the LP concentration, more hydrophobic association cross-linking centers were formed, and the P(AAm/LMA)-LP hydrogels showed a significant increase of tensile strength and toughness. However, the excess of LPs led to extremely high cross-linking density in the polymer network. The number of cross-linking centers increased and the content of hydrophobic monomers adsorbed on each LPs was relatively reduced; this caused the hydrogels to exhibit low strength and fracture elongation. Therefore, an appropriate content of LPs was crucial to enhance the mechanical property of the LP-Gel.
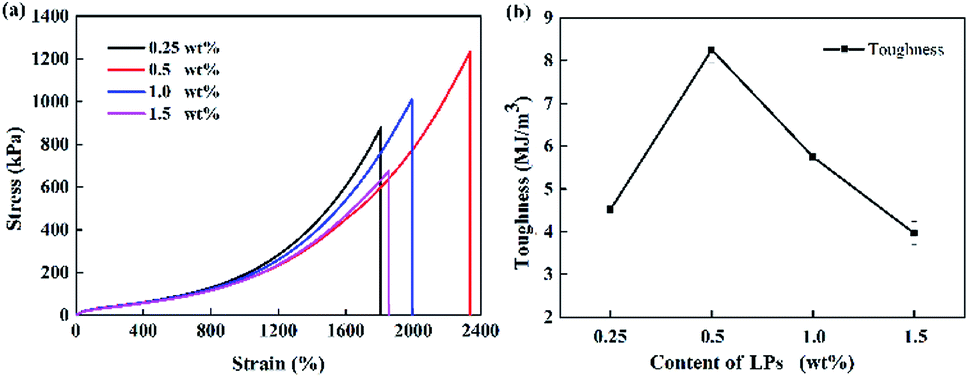 | ||
| Fig. 4 (a) Tensile stress–strain curves and (b) toughness of P(AAm/LMA)-LPs hydrogels with different concentrations of LPs. | ||
The effect of the hydrophobic alkyl monomers (C1, C6, C12, and C16) on the mechanical properties of the hydrogels was investigated, as illustrated in Fig. 5(a) and (b). It was shown that the hydrogels with MMA and HMA as the hydrophobic monomers exhibited large elongation ratio (3800% and 4664%), but weak tensile strength (23.9 kPa and 35.4 kPa), respectively. However, the hydrogels with LMA and HDMA displayed a moderate elongation ratio (2336% and 2716%) and high tensile strength (1.2 MPa and 471 kPa), respectively. Apparently, the tensile strength, toughness, and elastic modulus of the hydrogels considerably increased and then decreased with the increasing alkyl chain length. It is worth noting that the mechanical performance of the P(AAm/LMA)-LP hydrogels is much better than that of the P(AAm/HDMA)-LP hydrogels. Possibly, HDMA with very long alkyl chains was difficult to be absorbed on LPs; this led to a weak cross-linking internal network structure of the hydrogels. As a result, the P(AAm/HDMA)-LP hydrogels displayed poor mechanical performance as compared to the P(AAm/LMA)-LP hydrogels. To display the toughness of the hydrogels with various alkyl chain length, the P(AAm/MMA)-LP and P(AAm/LMA)-LP hydrogels were subjected to a 90% strain compressive test. As shown in Fig. 5(c), the P(AAm/MMA)-LP hydrogel was compressed into a thin slice, whereas the P(AAm/LMA)-LP hydrogel recovered its original length after compression without damage.
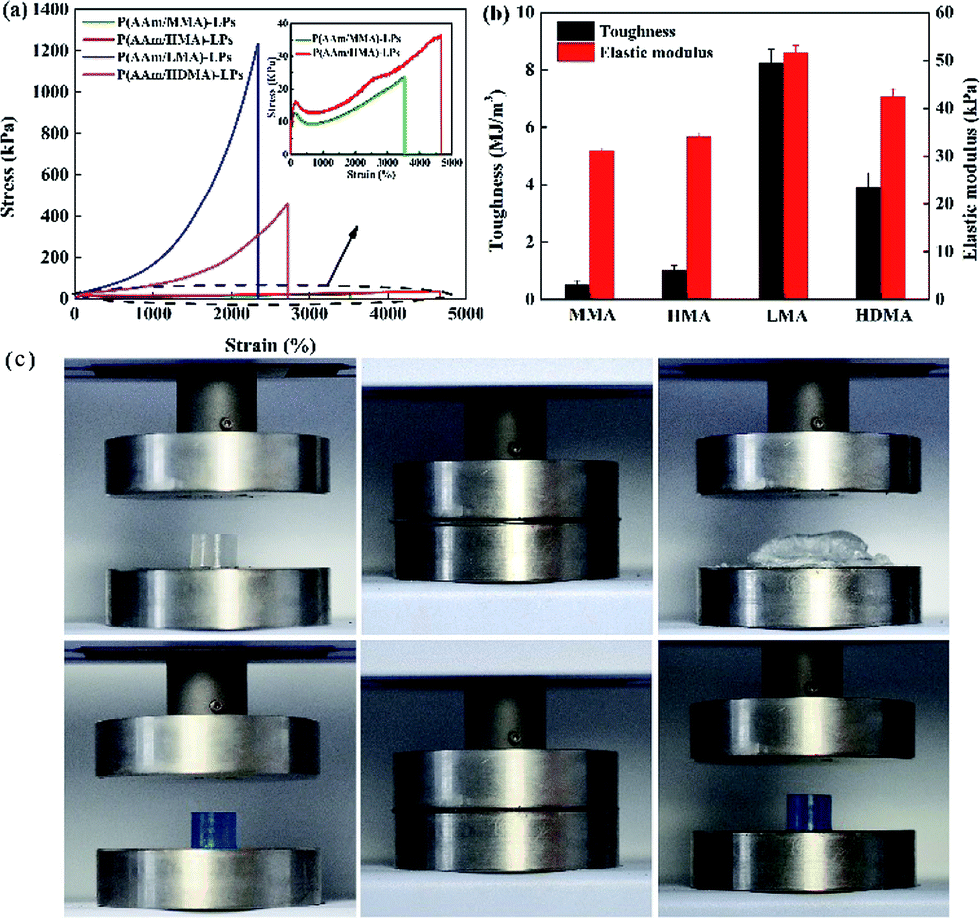 | ||
| Fig. 5 (a) Tensile stress–strain curves and (b) toughness and elastic modulus of the hydrogels with different hydrophobic segments of MMA, HMA, LMA, and HDMA. (c) The toughness exhibition of P(AAm/MMA)-LP hydrogels (white) and P(AAm/LMA)-LP hydrogels (blue). | ||
The following process was proposed as the toughening mechanism of LP-reinforced hydrogels with different hydrophobic alkyl chains (Fig. 6). For the P(AAm/MMA)-LP and P(AAm/HMA)-LP hydrogels, the short hydrophobic chains were prone to disentangle from LPs under initial deformation due to the weak hydrophobic interaction. The network structure was easy to collapse, resulting in the poor tensile strength of hydrogels. However, the LMA and HDMA hydrophobic segments of hydrogels hardly disentangled from the LPs at the initial deformation. When the external force was applied, the hydrophobic chains would first transform from curling to stretching due to the efficient aggregation of hydrophobic chains on LPs. As the external load increased, the long chains would slide on the surface of LPs to dissipate a large amount of energy.
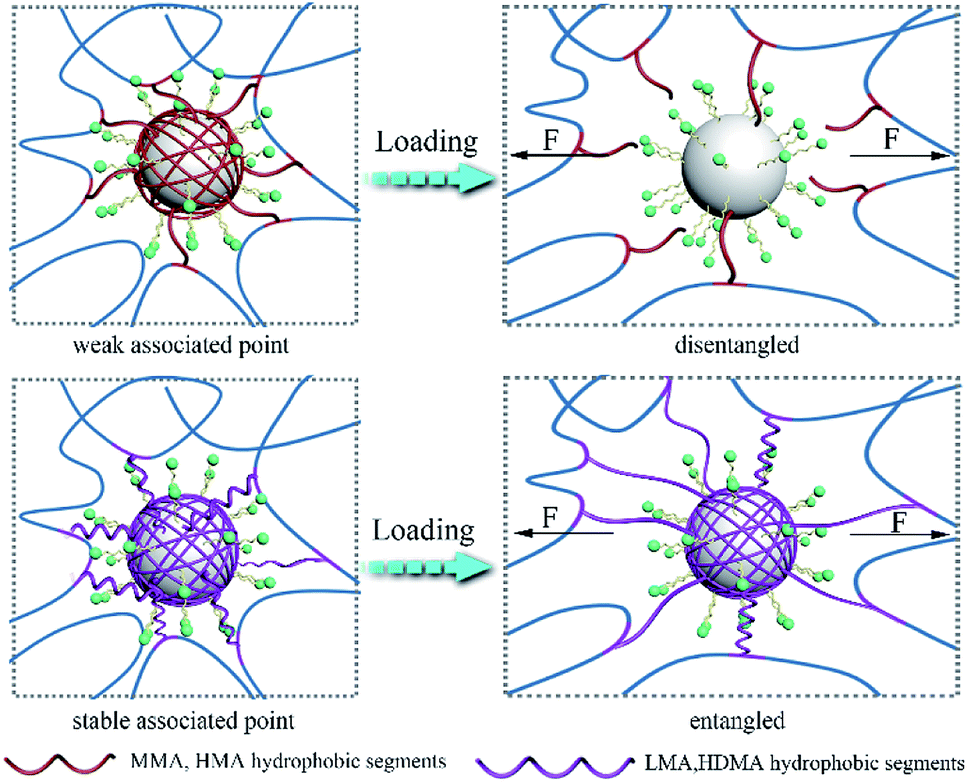 | ||
| Fig. 6 The toughening mechanism for LP-reinforced hydrogels with different hydrophobic chain lengths. | ||
Furthermore, hydrophobic monomers with different alkyl chain lengths have a significant influence on the energy dissipated by hydrogels. Dissipated energy was an indicator to analyze the toughness of the hydrogels. Fig. 7 shows the hysteresis loops and dissipated energy condition for the four hydrogels with different hydrophobic segments at a strain of 1000%. The P(AAm/LMA)-LP and P(AAm/HDMA)-LP hydrogels displayed prominent hysteresis loops, but inconspicuous hysteresis loops were exhibited for P(AAm/MMA)-LP and P(AAm/HMA)-LP hydrogels. Accordingly, the dissipated energy of the P(AAm/LMA)-LP hydrogel and P(AAm/HDMA)-LP hydrogel was up to 129.7 and 107.5 KJ m−3, which were much higher than those of the P(AAm/MMA)-LP hydrogel (53.24 kJ m−3) and P(AAm/HMA)-LP hydrogel (65.38 kJ m−3). The results indicated that the disentanglement between LPs and longer alkyl chains could effectively dissipate a great amount of energy under the external force; this endowed the hydrogels with excellent toughness.
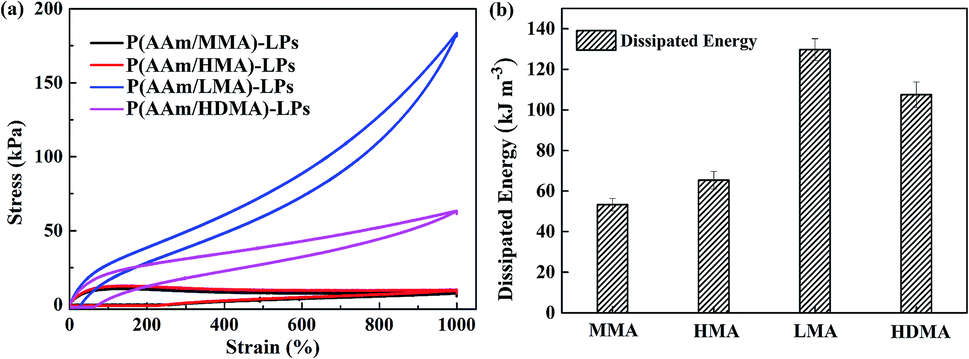 | ||
| Fig. 7 (a) Loading–unloading curves and (b) dissipated energy of hydrogels with MMA, HMA, LMA, and HDMA alkyl chains at a strain of 1000%. | ||
Furthermore, the successive loading–unloading test was carried out to evaluate the mechanical properties of hydrogels in response to the change of alkyl chain length (Fig. 8). For the four hydrogel samples, although a significant hysteresis occurred in the first loading–unloading curve, a smaller hysteresis was observed in the second to fifth cycle, and they followed the path of the first unloading curve. The results indicated that unrecovered damage occurred in the first curve that generated a permanent residual elongation.30 Especially, the stress of hydrogels with MMA and HMA slightly increased. This was ascribed to the fact that the short chains disentangled from the LPs in the first loading process and rearranged in the following stretching process; this led to the slight improvement of stress.
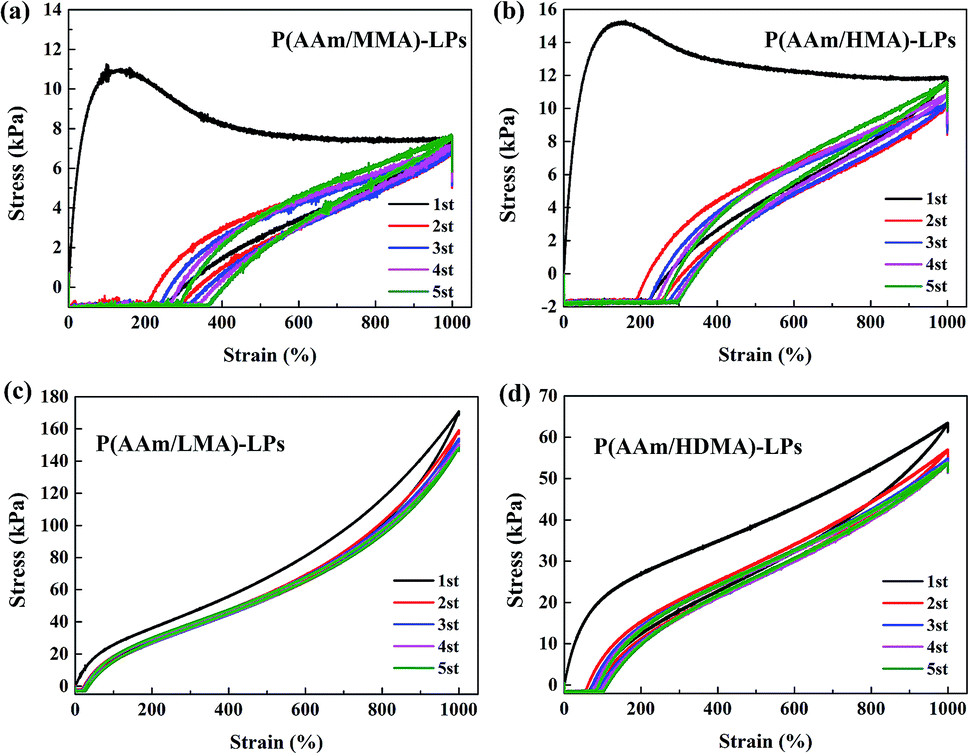 | ||
| Fig. 8 The successive loading–unloading tests of (a) P(AAm/MMA)-LPs, (b) P(AAm/HMA)-LPs, (c) P(AAm/LMA)-LPs, and (d) P(AAm/HDMA)-LPs. | ||
The swelling characteristic is one of the important factors for studying the properties of hydrogels. Fig. 9(a) shows the swelling behavior of hydrogels containing different hydrophobic segments. Obviously, the P(AAm/MMA)-LP and P(AAm/HMA)-LP hydrogels collapsed after being soaked in water for 24 h. The swelling ratio of the P(AAm/LMA)-LP and P(AAm/HDMA)-LP hydrogels at the swelling equilibrium state was about 17.2 and 38.6, respectively. As previously discussed, the entanglement between the LPs and shorter alkyl chains was loose, and the internal network structure of the hydrogels was unstable; this resulted in the destruction of the hydrogels during the process of swelling. On the contrary, the longer hydrophobic chains of LMA and HDMA were difficult to disentangle from LPs in the swelling process and could maintain the network of the hydrogels; therefore, the corresponding hydrogels could keep the original shape without fracture. The swelling ratio was related to the cross-linking density of the hydrogels. As shown in the SEM image (Fig. 9(b)), the cross-linking density of P(AAm/LMA)-LP and P(AAm/HDMA)-LP hydrogels was higher than that of P(AAm/MMA)-LP and P(AAm/HMA)-LP hydrogels; this was consistent with the swelling result. The higher cross-linking density would lead to a lower swelling degree.
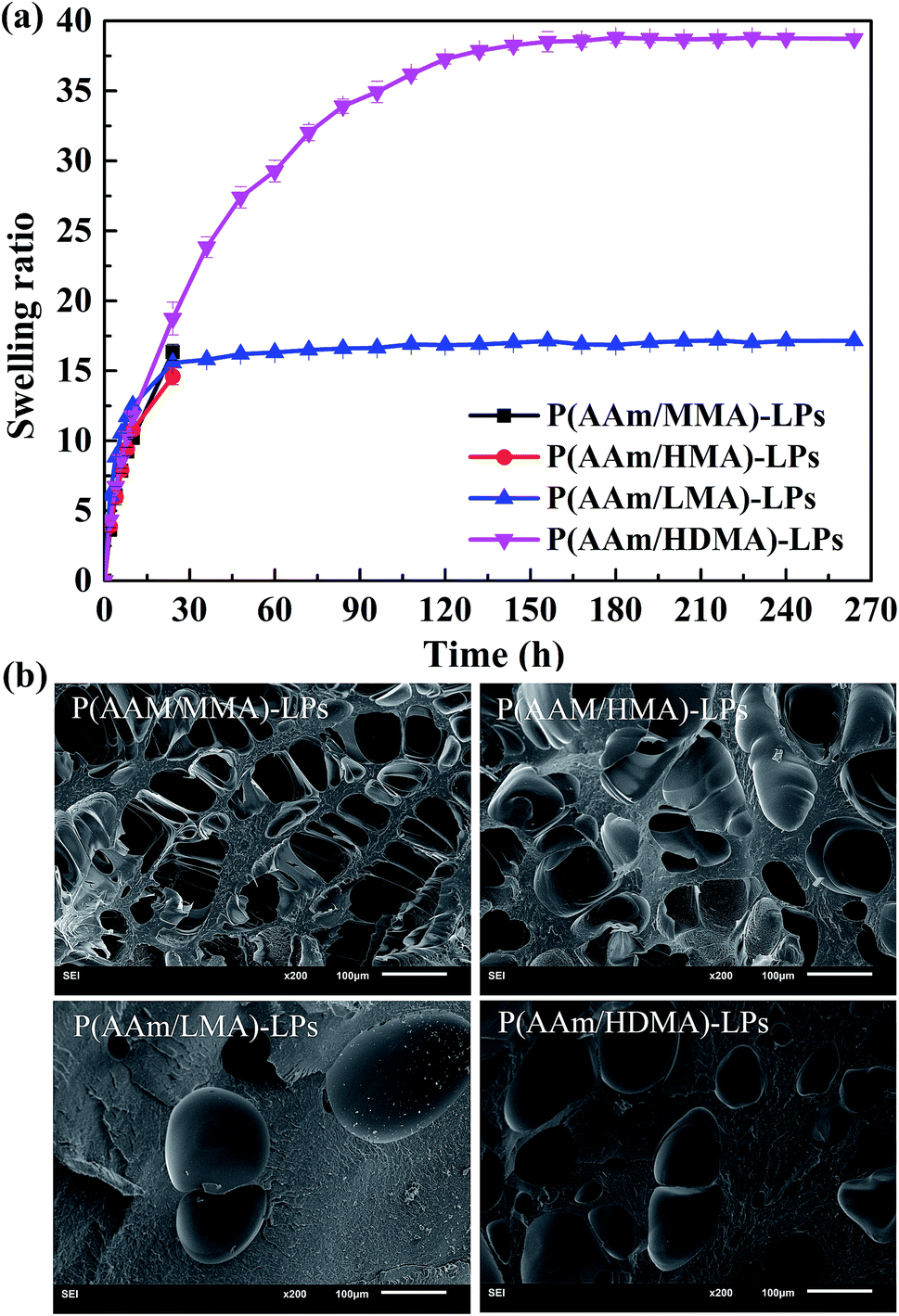 | ||
| Fig. 9 (a) Swelling behavior and (b) SEM images of the hydrogels with hydrophobic segments of MMA, HMA, LMA, and HDMA. | ||
The content of LPs has a great influence on the swelling ratio of the P(AAm/LMA)-LP hydrogels. Fig. 10 shows that the swelling ratio quickly increased at the beginning and then gradually reached a steady value. When the content of LPs varied from 0 to 0.5 wt%, the swelling ratio decreased significantly and reached the minimum value at the concentration of 0.5 wt%. This is because the introduction of LPs changed the cross-linking structure of the hydrophobic association hydrogels. The homogeneity and the effective cross-linking density of the hydrogel network will significantly increase with the increase of LP concentration; therefore, the hydrogels exhibited the lowest swelling ratio at the LP concentration of 0.5 wt%. Furthermore, the hydrogels with 0.5 wt% LPs exhibited the best tensile strength. However, the swelling ratio increased with the increase in the LP contents from 0.5 to 1.5 wt%. A possible reason was that the excessive LPs resulted in the heterogeneity of the hydrogel network structure and the effective cross-linking density reduced; hence, the swelling ratio of the hydrogels increased.
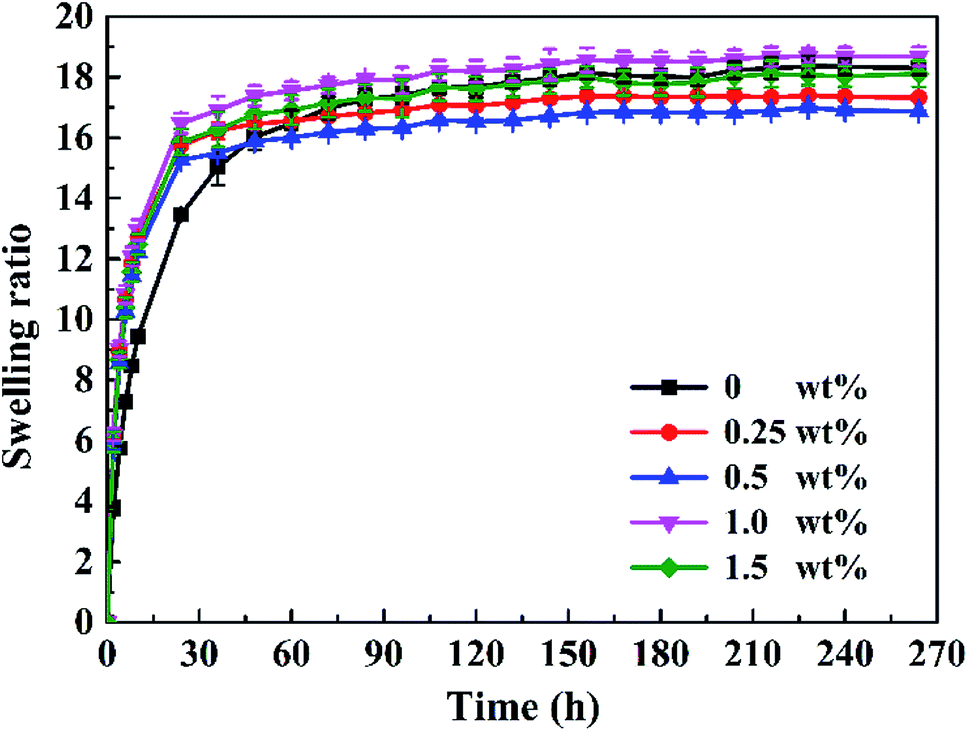 | ||
| Fig. 10 The influence of the LP content on the swelling ratio of P(AAm/LMA)-LP hydrogels. | ||
The kinetics of the copolymerization process was monitored by rheological experiments, as shown in Fig. 11(a–e). It was clearly observed from the gelation profile that a certain induction period existed in the four hydrogel samples. The gelation time of polyacrylamide with MMA, HDMA, LMA, and HDMA was 409, 347, 480, and 614 s, respectively. The gelation time first decreased and then increased with the increase of the alkyl chain length owing to the different hydrophobicities and steric hindrances of the hydrophobic monomers. The steric hindrance increased because the alkyl chain length increased, such that the gelation time increased from HMA to HDMA. However, the gelation time of AAm and MMA was longer than that for HMA. This was attributed to the fact that the hydrophobicity of MMA was weaker than that of HMA. Simultaneously, it was found that AAm and HMA formed a gel more easily possibly due to appropriate their hydrophobicity and steric hindrance. Then, both the storage modulus G′ and loss modulus G′′ of the hydrogel samples increased sharply with the increase of reaction time and reached a plateau value. The copolymerization process was considered to be completed for the system.
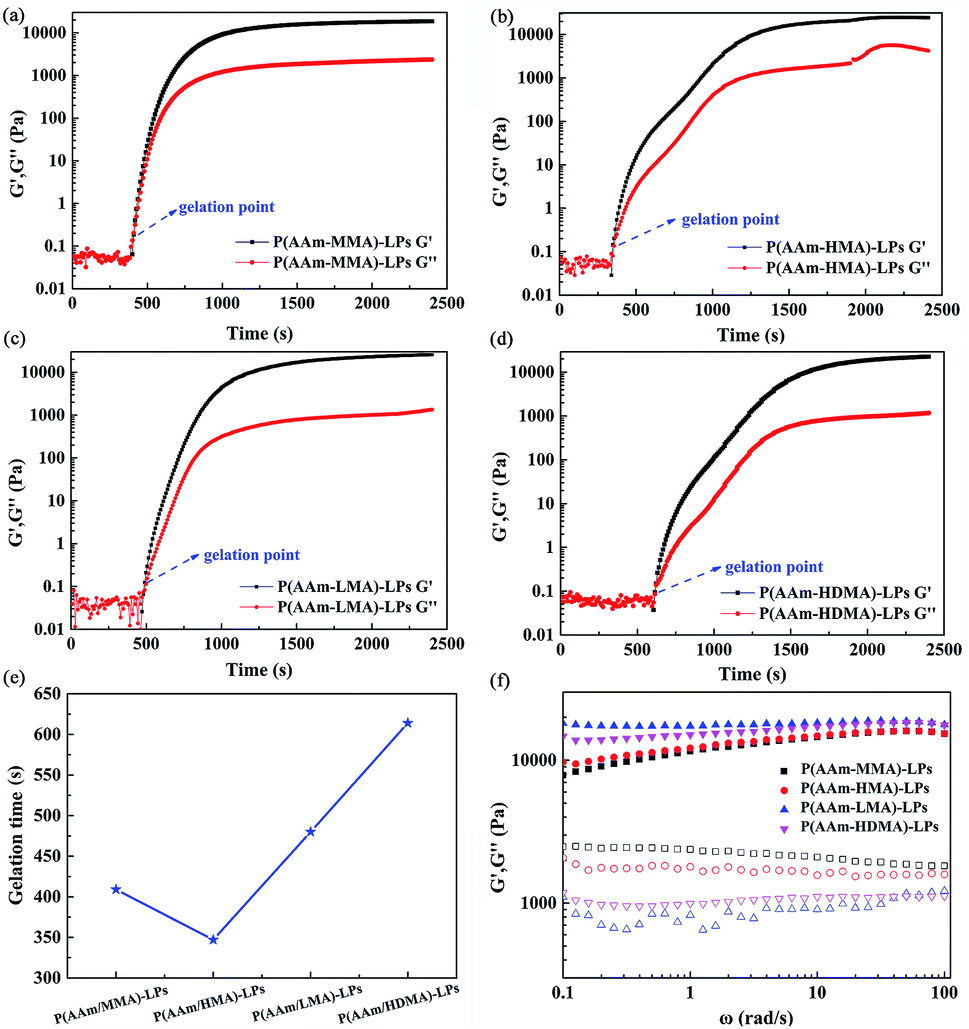 | ||
| Fig. 11 Storage modulus (G′) and loss modulus (G′′) shown as a function of reaction time during the copolymerization of AAm with (a) MMA, (b) HMA, (c) LMA, and (d) HDMA. (e) The gelation time for four different hydrogels. (f) Storage modulus (G′) (filled symbols) and loss modulus (G′′) (open symbols) of hydrogels shown as a function of the frequency. | ||
The frequency sweeping test was carried out at the frequencies varying from 0.01 to 100 rad s−1. As shown in Fig. 11(f) , the P(AAm/MMA)-LP and P(AAm/HMA)-LP hydrogels exhibited a viscoelastic behavior. As expected, both the storage modulus G′ and loss modulus G′′ are dependent on frequency. With the increase of frequency, the storage modulus G′ increased, but the loss modulus G′′ decreased. This is characteristic of viscoelastic materials. However, the P(AAm/LMA)-LP and P(AAm/HDMA)-LP hydrogels exhibited a gel-like behavior. The storage modulus G′ is higher than the loss modulus G′′ in the abovementioned frequency range, and both are nearly independent of frequency. Moreover, note that the G′ curves of all hydrogels are smooth, but the G′′ curves are relatively rough. Obviously, this feature was similar to that of the elastic materials.
Conclusions
A series of hydrogels with different long alkyl chains were successfully fabricated by introducing LPs as hydrophobic association centers. LPs played a significant role in improving the mechanical strength of the hydrogels, and methacrylate could adsorb on the surface of LPs due to hydrophobic interactions. The mechanical properties of the LP-Gel could be tuned by adjusting the concentration of LPs and the length of alkyl chains of hydrophobic monomers. The degree of hydrophobic interaction was different between methacrylate and the LPs; this caused the hydrogels to display different mechanical strengths and swelling ratios. The optimal P(AAm/LMA)-LP hydrogel possessed a high fracture of 1.2 MPa and a large elongation of 2336%. Therefore, the current study may provide a new thought and theoretical support for the design of a novel latex particle hydrogel and further expand the applications of hydrogels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a grant received from the National Natural Science Foundation of China (NSFC) (No. 51473023 and 51103014), Science and Technology Department of Jilin Province (No. 20150204064G), Education Department of Jilin Province (No. 201582), and Changchun Municipal Science and Technology Bureau (No. 14KG054).Notes and references
- S. Hong, D. Sycks, H. F. Chan, S. Lin, G. P. Lopez, F. Guilak, K. W. Leong and X. Zhao, Adv. Mater., 2015, 27, 4035 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673 CrossRef CAS PubMed.
- L. Dong, A. K. Agarwal, D. J. Beebe and H. Jiang, Nature, 2006, 442, 551 CrossRef CAS PubMed.
- J. Yan, V. A. Pedrosa, A. L. Simonian and A. Revzin, ACS Appl. Mater. Interfaces, 2010, 2, 748 CAS.
- E. Wang, M. S. Desai and S. W. Lee, Nano Lett., 2013, 13, 2826 CrossRef CAS PubMed.
- J. Duan, X. Liang, K. Zhu, J. Guo and L. Zhang, Soft Matter, 2016, 13, 345 RSC.
- B. Xue, M. Qin, T. Wang, J. Wu, D. Luo, Q. Jiang, Y. Li, Y. Cao and W. Wang, Adv. Funct. Mater., 2016, 26, 9053 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485 CrossRef CAS.
- C. Katsuno, A. Konda, K. Urayama, T. Takigawa, M. Kidowaki and K. Ito, Adv. Mater., 2013, 25, 4636 CrossRef CAS PubMed.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef PubMed.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- G. Gao, G. Du, Y. Sun and J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 5029 CAS.
- D. Wu, M. Yi, H. Duan, J. Xu and Q. Wang, Carbon, 2016, 108, 394 CrossRef CAS.
- S. Pacelli, P. Paolicelli, G. Moretti, S. Petralito, S. Di Giacomo, A. Vitalone and M. A. Casadei, Eur. Polym. J., 2016, 77, 114 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS.
- Q. Chen, L. Zhu, H. Chen, H. Yan, L. Huang, J. Yang and J. Zheng, Adv. Funct. Mater., 2015, 25, 1598 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171 CrossRef CAS PubMed.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, Adv. Mater., 2007, 19, 1622 CrossRef CAS.
- C. He, K. Jiao, X. Zhang, M. Xiang, Z. Li and H. Wang, Soft Matter, 2011, 7, 2943 RSC.
- T. Cai, G. Wang, S. Thompson, M. Marquez and Z. Hu, Macromolecules, 2008, 41, 9508 CrossRef CAS.
- Y. Tan, K. Xu, P. Wang, W. Li, S. Sun and L. Dong, Soft Matter, 2010, 6, 1467 RSC.
- J. Zhao, K. Jiao, J. Yang, C. He and H. Wang, Polymer, 2013, 54, 1596 CrossRef CAS.
- G. Jiang, C. Liu, X. Liu, G. Zhang, M. Yang and F. Liu, Macromol. Mater. Eng., 2009, 294, 815 CrossRef CAS.
- G. Jiang, C. Liu, X. Liu, Q. Chen, G. Zhang, M. Yang and F. Liu, Polymer, 2010, 51, 1507 CrossRef CAS.
- S. Abdurrahmanoglu, M. Cilingir and O. Okay, Polymer, 2011, 52, 694 CrossRef CAS.
- C. Bilici, V. Can, U. Nöchel, M. Behl, A. Lendlein and O. Okay, Macromolecules, 2016, 49, 7442 CrossRef CAS.
- T. Zhao, M. Tan, Y. Cui, C. Deng, H. Huang and M. Guo, Polym. Chem., 2014, 5, 4965 RSC.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449 CrossRef CAS.
- D. C. Tuncaboylu, A. Argun, M. Sahin, M. Sari and O. Okay, Polymer, 2012, 53, 5513 CrossRef CAS.
- T. T. Gao, N. Niu, Y. D. Liu, X. L. Liu, G. Gao and F. Q. Liu, RSC Adv., 2016, 6, 43463 RSC.
- W. Li, H. An, Y. Tan, C. Lu, C. Liu, P. Li, K. Xu and P. Wang, Soft Matter, 2012, 8, 5078 RSC.
- W. Cui, J. Ji, Y.-F. Cai, H. Li and R. Ran, J. Mater. Chem. A, 2015, 3, 17445 CAS.
- X. Y. Ren, Z. Yu, B. Liu, X. J. Liu, Y. J. Wang, Q. Su and G. H. Gao, RSC Adv., 2016, 6, 8956 RSC.
| This journal is © The Royal Society of Chemistry 2017 |