
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical investigations on the structure–property relationships of Au13 and AuxM13−x nanoclusters†
Ying Lv,
Xi Kang ,
Sha Yang,
Tao Chen,
Ao Liu,
Haizhu Yu* and
Manzhou Zhu*
,
Sha Yang,
Tao Chen,
Ao Liu,
Haizhu Yu* and
Manzhou Zhu*
Department of Chemistry, Center for Atomic Engineering of Advanced Materials, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, China. E-mail: yuhaizhu@ahu.edu.cn; zmz@ahu.edu.cn
First published on 6th November 2017
Abstract
As a fundamental building block in ultrasmall, noble metal nanoclusters, icosahedral AuxM13−x structures have recently attracted extensive research interest. In this study, density functional theory (DFT) and time-dependant DFT calculations have been carried out to investigate the structure–property (optical and electronic) relationships of a series of Au13 and AuxM13−x (M = Au, Ag, Cu, and Pd) nanoclusters co-protected by phosphine and chloride ligands. It was found that the size of the peripheral ligands significantly affects the geometric structure: the larger exterior ligands (with a larger cone angle) result in relatively longer Au–M bond distances and weaker metallic interactions within the AuxM13−x core. Therefore, the optical peak (in the UV-vis spectrum) corresponding to the HOMO → LUMO transition red-shifts accordingly. When different foreign atom(s) are incorporated, the preferential doping site is different, and the electronic and optical structures alter accordingly.
Introduction
In recent years, atomically precise ultrasmall gold nanoclusters (NCs) with diameters of <2 nm have attracted intensive research interest due to the promising applications in catalysis,1 biomimetic materials2,3 and biotherapy.4 Thanks to the atomic precision, the icosahedral AuxM13−x building block has been frequently observed in different sized NCs. Depending on the chemical environment around the AuxM13−x structure, these NCs could be roughly categorized into three groups. First, in different AuxM13−x nanoclusters, the icosahedral AuxM13−x framework is protected directly by organic ligands (such as thiolate and phosphine). Second, the vertice- or face-sharing assembly of several AuxM13−x structures might be responsible for the metal framework of some relatively larger NCs, such as Au25 (rod-like),5,6 Au38,7 and Au60.8 In the third group of NCs, the icosahedral AuxM13−x core is protected by the exterior metal–organic shell. Such a structure has been frequently observed in NCs such as Au25 (spherical),9 Au19 (ref. 10) and Au20 NCs.11 According to the recent studies, the structure and property of the metal core significantly affect the electronic, catalytic, and optical properties.12 Therefore, the systematic investigation on the icosahedral AuxM13−x core will benefit the future exploration on the property of larger NCs.Realizing the importance of the icosahedral core, chemists have extensively studied the structure–property relationships of the AuxM13−x NCs in recent years. For example, Kronik et al. successfully synthesized Au13(PPh3)4(SC12H25)4, and found that the co-protection of thiolates and phosphine ligands leads to the slight distortion of the icosahedral structure.13 Similarly, Wei and co-workers recently found that the framework of Au13(PPh3)4(SC12H25)4 is significantly disturbed by the solvent environment. They found that changing the solvent from ethanol to hexane induces the dissociation of the anionic SR ligands, and the rearrangement of the icosahedral core structure to a face-centred cubic (FCC) structure.14 Resultantly, the electronic structure remarkably changes from semiconducting to metallic state. Of note, the influence of the shell ligands on the geometric and electronic structures have also been reported elsewhere. For example, Konishi and co-workers successfully synthesized a series of Au13 NCs co-protected by bidentate phosphine (Ph2P–(CH2)m–PPh2, m = 2–5) and chloride ligands. They found that the P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cl ratio varies from 10:2 to 8:4 when m changes from 2 to 3–5, and the optical properties also change critically.15,16 After that, they further prepared another AuxM13−x NC, i.e. [Au13(dppe)5(C
:Cl ratio varies from 10:2 to 8:4 when m changes from 2 to 3–5, and the optical properties also change critically.15,16 After that, they further prepared another AuxM13−x NC, i.e. [Au13(dppe)5(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2]3+ via the ligand exchange reaction of [Au13(dppe)5Cl2]3+ (dppe is short for PPh2CH2CH2PPh2), and bore out the electronic coupling between the Au13 core and the π-orbitals of acetylide.17 Aside from the mono-metallic Au13 NCs, the alloy AuxM13−x NCs have also been successfully prepared in these years. For instance, Copley and Mingos synthesized three NCs, i.e. [Au9M4(PPh2Me)8Cl4]+ (M = Au, Ag, Cu), and found that the optical properties of these three NCs are significantly different. The characteristic peaks in the UV-vis spectrum blueshift with the incorporation of the Ag dopants, while were only slightly weakened when Cu atoms are doped.18
CPh)2]3+ via the ligand exchange reaction of [Au13(dppe)5Cl2]3+ (dppe is short for PPh2CH2CH2PPh2), and bore out the electronic coupling between the Au13 core and the π-orbitals of acetylide.17 Aside from the mono-metallic Au13 NCs, the alloy AuxM13−x NCs have also been successfully prepared in these years. For instance, Copley and Mingos synthesized three NCs, i.e. [Au9M4(PPh2Me)8Cl4]+ (M = Au, Ag, Cu), and found that the optical properties of these three NCs are significantly different. The characteristic peaks in the UV-vis spectrum blueshift with the incorporation of the Ag dopants, while were only slightly weakened when Cu atoms are doped.18
In addition to the experimental studies, theoretical methods have also been used to study the structure and properties of several Au13 NCs. For example, Remacle and co-workers recently used density functional theory (DFT) calculations to study the modeling Au13(SCH3)5(PH3)7 cluster, and found that the positions of the phosphines and thiolates significantly affect the geometric structure and electronic properties.19 In addition, comparing the UV-vis spectra of a series of Au13, Au25 and Au28 NCs, they found that the increased charge density of the metal core (Au13 for Au13, Au13 for Au25 and Au20 for Au28) is responsible for the red-shift of the optical absorption bands.
Despite the aforementioned studies, the systematic understanding on the effect of different ligands (including both the number and the structure of the protecting ligands), and especially the doping effect in the alloy AuxM13−x NCs, are rarely reported. In this study, we carried out theoretical investigation on a series of icosahedral Au13 and alloy AuxM13−x NCs with DFT and TD-DFT calculations to elucidate the structure–property relationships. The monometallic Au13 NCs including [Au13(PR3)8Cl4]+ and [Au13(PR3)10Cl2]3+ (R = Me, Ph) were chosen to examine ligand effect of the phosphine and chloride ligands.20 Meanwhile, the doping effect was explored by comparing the structure and property of the monometallic [Au13(PR3)8Cl4]+ NCs with the alloy counterparts including [Au9Ag4(PPh2Me)8Cl4]+, [Au9Cu4(PPh2Me)8Cl4]+, and [Au12Pd(PPh3)8Cl4], whose structures have been precisely elucidated by single X-ray crystallography.18,20,21 This study can provide deep understanding on the structure–property relationships in the icosahedral metal NCs, and will also benefit the future explorations on larger NCs assembled with AuxM13−x.
Computational details
The geometry optimization of all NCs was carried out with the ADF software,22 using GGA: PBE/TZP method.23 The time-dependent LB94/TZP method and the scalar relativistic were used for UV-vis spectra calculation, and the lowest 150 singlet excited states were calculated.24 The Kohn–Sham (KS) orbital analyses were performed with Gaussian 09 suite of program,25 using the long-range corrected functional CAM-B3LYP functional.26 The LanL2DZ basis set and the effective core potential were used for the metal atoms (i.e. Au, Ag, Cu, Pd), and the total electron basis set 6-31G(d) was used for all other atoms.We first compared the calculated Au–M bond distances with the experimental ones obtained from the single crystal X-ray crystallography to examine the reliability of theoretical calculations.18,20,21 As shown in Fig. 1, despite the Au–M bond distances are slightly overestimated by theoretical methods, the overall correlation between the calculated and experimental bond distances are acceptable: the linear coefficient (R) is 0.9045 for the 84 data, and the square deviation (SD) is 0.0467 Å (Fig. 1). The good correlation of the experimental and calculated Au–M (M = Au, Ag, Cu) bond distances verifies the theoretical methods.
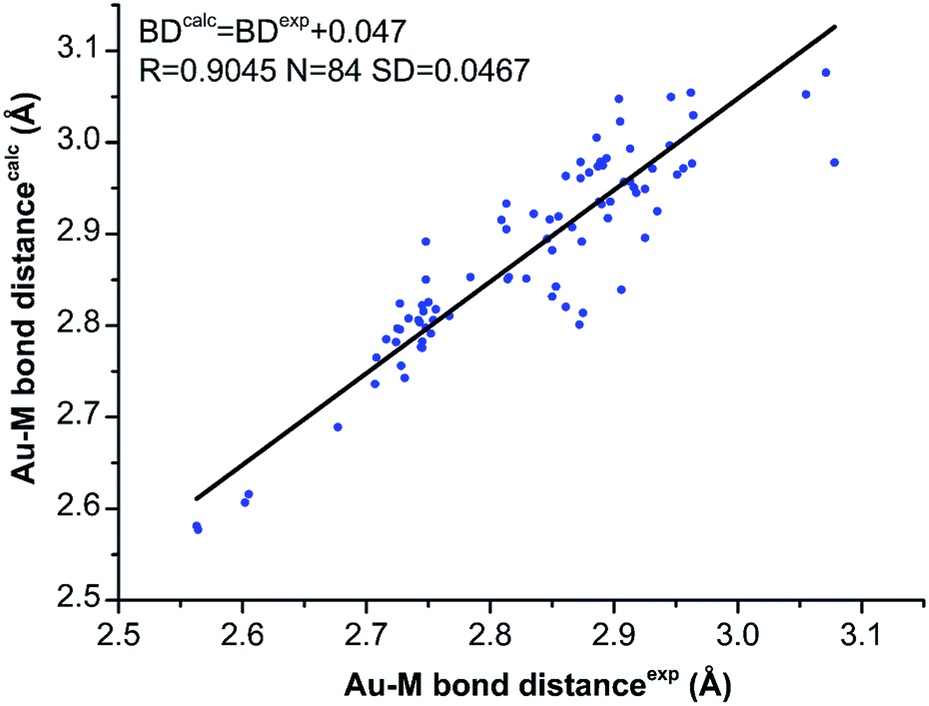 | ||
| Fig. 1 The correlation of the experimental (from single-crystal X-ray crystallography) and the calculated Au–M (M = Au, Ag, Cu) bond distances for [Au9Ag4(PPh2Me)8Cl4]+ and [Au9Cu4(PPh2Me)8Cl4]+. | ||
Results and discussions
The ligand effect in Au13 NCs co-protected by phosphine and chloride ligands
In the past decades, a series of Au13 NCs co-protected by phosphine and chloride ligands have been synthesized. For example, Mingos and co-workers used the in situ reduction of Au(PR3)Cl (with Ti(η-C6H5Me)) to prepare the Au NC, and determined the atomic structure to be [Au13(PMe2Ph)10Cl2]3+ with the single crystal X-ray analysis.20 After that, they also prepared [Au13(PMePh2)8Cl4]+ via the reaction of [Au11(PMePh2)10]3+ with AuCl(PMePh2) in CH2Cl2.18 Recently, Konishi and co-workers prepared the [Au13(POct3)8Cl4]+ (Oct = octyl) by reducing Au(POct3)Cl with NaBH4 in aqueous THF.15In accordance with the aforementioned studies, the structures, UV-vis spectra and KS-orbitals of the four Au13 NCs co-protected by chloride and phosphine ligands, i.e. [Au13(PMe3)8Cl4]+, [Au13(PPhMe2)10Cl2]3+, [Au13(PPh2Me)8Cl4]+, and [Au13(PPh3)8Cl4]+ were calculated. Note that [Au13(PMe3)8Cl4]+ was used to model the experimentally characterized [Au13(POct3)8Cl4]+, so as to reduce the computational cost. For clarity, these four NCs are designated as NC0, NC1, NC2, and NC3, respectively. The 0–3 here denote the number of phenyl groups on the PR3 ligand in each NC.
Comparing the selected structural parameters of the optimized geometries (Table 1), we found that the average Au–P and Au–Cl distances are comparable in different NCs. By contrast, the Au–Au bond distances change remarkably. The average Aucenter–Aushell bond distance in NC1 is significantly longer than those in NC0/NC2/NC3. The phenomenon is mainly caused by the stronger steric repulsion between the 10 phosphine ligands in NC1 (compared to that between the 8 phosphine ligands in all other NCs). For the same reason, the average Aucenter–AuP bond distance in NC1 is also relatively longer than those in NC0/NC2/NC3 (Table 1).
| Bond distance | NC0 | NC1 | NC2 | NC3 |
|---|---|---|---|---|
| a Note: Aushell include both the AuCl and AuP atoms, while AuCl and AuP denote the Au atoms connecting with Cl and P atoms, respectively. | ||||
| Aucenter–Aushella | 2.813 | 3.012 (2.769) | 2.822 | 2.845 |
| Aucenter–AuCl | 2.815 | 2.993 (2.715) | 2.807 | 2.776 |
| Aucenter–AuP | 2.811 | 3.016 (2.780) | 2.830 | 2.880 |
| AuCl–Cl | 2.372 | 2.362 (2.297) | 2.394 | 2.406 |
| AuP–P | 2.310 | 2.342 (2.286) | 2.335 | 2.372 |
| Cone angle (PR3) | 46.7° | 60.9° | 74.7° | 76.4° |
|
||||
| Hirshfeld charge | ||||
| Aucenter | −0.005 | −0.021 | 0.000 | 0.000 |
| Aushell | 0.014 | 0.047 | 0.037 | 0.046 |
| AuP | 0.015 | 0.044 | 0.038 | 0.046 |
| AuCl | 0.012 | 0.061 | 0.035 | 0.053 |
| Cl | −0.337 | −0.326 | −0.281 | −0.230 |
To comprehend why [Au13(PMe2Ph)10Cl2]3+ (NC1) is preferentially formed over [Au13(PMe2Ph)8Cl4]+ (analogue of NC0/NC2/NC3), we analysed the cone angle of the phosphine ligands in all these Au13 NCs. According to the previous studies, the cone angle of the surface ligands directly correlates with the composition and size of the nanoclusters.27,28 As shown in Table 1, the cone angle of PMe2Ph in NC1 is significantly smaller than the those of PPh2Me in NC2 and PPh3 in NC3. The smaller cone angle of PMe2Ph enables the incorporation of more phosphine ligands in NC1. Herein, it is noteworthy that the theoretical modelling of POct3 with PMe3 in NC0 significantly underestimate the cone angle. The small cone angle of PMe3 might strongly disfavours the small cluster sizes, and this is also the reason why only large sized NCs (with metal atoms >32)29,30 have been reported when PMe3 are dominant ligands. We suppose that the relatively larger cone angle of the PPh3 ligand also explains why PPh3 is observed as a successful ligand for [Au11(PPh3)xCly]q synthesis,31 but not yet for [Au13(PPh3)xCly]q.
According to the aforementioned discussions, the cone angle of the phosphine ligands determines the ratio of the phosphine and chloride ligands, as well as the Au–Au bond distances in the Au13 core. This conclusion is also supported by the Hirshfeld charge analysis (Table 1). The charge on the Aushell atoms of NC1 is slightly more positive than those of the other NCs, predominantly due to the weakened σ-donating ability of the phosphine ligand therein. Meanwhile, the center Au atom in these NCs is all nearly charge neutral (Table 1).
Regarding the optical properties, the absorption peaks are systematically overestimated by 100 nm due to the deficiency of DFT methods in treating the excited states. The similar observation was also reported in previous studies.32 Nevertheless, comparing the curves of different NCs (Fig. 2), we found that the UV-vis absorption spectra of NC1, NC2, NC3 are similar, while the peak splitting was observed in the spectrum of NC0. Similar separation of the optical bands induced by methyl groups has also been reported in Sun's recent study.33 As shown in Fig. 2, the first band of NC1, NC2 and NC3 appear at ∼720 nm, 650 nm and 685 nm, respectively. All these bands correspond to the HOMO → LUMO transition, and the weakness of the peaks implies that the transition is optically forbidden. In addition to the HOMO → LUMO transition, the prominent absorption peak of NC2 at 540 nm mainly arises from the transition from the 3-fold quasi-degenerate HOMO manifold to the LUMO+9, LUMO+10 and LUMO+11 set. For NC3, the prominent peak at 520 nm is mainly caused by the transition from the doubly degenerate HOMO and HOMO−1 set to the LUMO+4, LUMO+5 and LUMO+6 set. For NC0, the HOMO → LUMO transition appears at ∼640 nm, and the prominent peak (∼580 nm) corresponds to HOMO−1, HOMO−2 → LUMO+3, +4, +5 (Fig. 2).
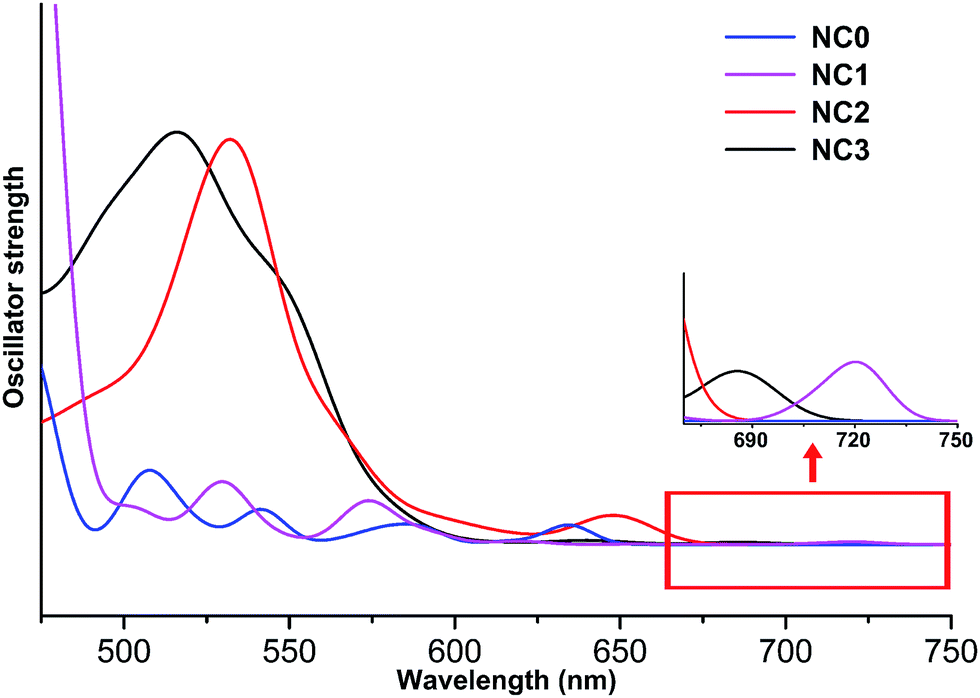 | ||
| Fig. 2 Theoretical UV-vis spectra of NC0, NC1, NC2 and NC3. | ||
Fig. 3 shows the KS-orbital analysis of all these NCs. The discrete energy levels of NC0 correlates with its separate and weak optical bands (Fig. 2 & 3a). By contrast, the introduction of phenyl substituent in the phosphine ligands results in the degeneracy of the energy levels (Fig. 3b–d). The degenerate orbitals explain the peak broadening in the UV-vis spectra of NC1, NC2 and NC3.
 | ||
| Fig. 3 The HOMO, LUMO plots and KS orbitals of NC0 (a), NC1 (b), NC2 (c) and NC3 (d). | ||
Aside from the energy levels, the atomic contributions of the frontier orbitals of all these Au13 NCs are similar. As shown in Fig. 3, Au(6sp) and Au(5d) are mainly responsible for all the HOMOs (highest occupied molecular orbitals) and LUMOs (lowest unoccupied molecular orbitals). Therefore, the active electrons mainly locate in the Au13 core. In addition, the Au(6sp) orbitals make major contribution to the higher HOMOs and LUMOs, while the Au(5d) orbitals contribute predominantly to the lower HOMOs (Fig. 3a, c and d). To this end, the first band of all the Au13 NCs correspond to interband transition of d → sp. This conclusion is consistent with the recent theoretical studies on the electronic and optical analysis of the small sized NCs (such as Au18 NC).34
Combining the key structural parameters and the optical & electronic properties of different Au13 NCs, we found that the cone angle of the PR3 ligands dominates the composition and structure of the Au13 NCs. The larger PR3 ligand encourages the structure of [Au13(PR3)8Cl4]+, while the PR3 with small cone angle facilitates the formation of [Au13(PR3)10Cl2]3+. The average Aucenter–Aushell bond distance of [Au13(PR3)10Cl2]3+ is relatively longer than that of [Au13(PR3)8Cl4]+, and the weakened bonding strength in the former case further results in the lower HOMO → LUMO transition.
For in-depth understanding on the orbital distributions of Au13 NCs, we referred to the superatom models.35 According to this model, all these Au13 NCs are 8-electron clusters with 1S2, 1P6 jellium orbitals (S, P and D orbitals in superatom correspond to the atomic s, p and d orbitals, respectively). The HOMOs of all these Au13 NCs feature the characteristic of P orbital,36,37 while the LUMOs of all these Au13 NCs feature the D orbital characteristics (Fig. 3).37
The doping effect of the foreign (Ag, Cu, and Pd) atom(s)
According to the recent experimental studies, different foreign metal (Ag, Cu, and Pd) atom(s) with the electronic structures similar to Au could be doped into the Au13 NCs.18,21 Interestingly, these metals were preferentially doped into different sites. For example, Mingos and co-workers synthesized the shell doped [Au9M4(PMePh2)8Cl4]+ (M = Cu, Ag) NCs via the reaction of [Au11(PMePh2)10]3+ with MCl(PMePh2).18 Recently, the core doped [PdAu12(PPh3)8Cl4] has been synthesized by photolysis of (Ph3P)2Pd(N3)2, Ph3PAuN3 and Ph3PAuCl in THF solvent.21 To understand the doping preference for different metal atoms, we first examined the relative energy of the AuxM13−x structures with the foreign atom doping at different site. The 13 possible doping sites on Au13 were roughly categorized into six groups: the metal center atom, the shell atom coordinating with the phosphine ligand (adjacent to three Au–Cl bonds for shell-a and adjacent to two Au–Cl bonds for shell-b/c), and the shell atom connecting with chloride (adjacent to one Au–Cl bond for shell-d, and adjacent to none Au–Cl bond for shell-e). The details are given in Fig. 4b, and the typical positions of 7, 9, 6, 11, 3, and 13 are used for the following discussion. For clarity reasons, the core-doped alloy NCs were taken as the reference, and the energy difference between the other structures and the core-doped one is used to evaluate their relatively stability. From Table 2, it can be seen that the relative energies of the shell-a/b/c doped products are comparable for each type of alloy system (note: these positions correspond to the different shell AuP sites, see Fig. 4b). Similarly, the relative energies of the shell-d and shell-e doped products are also alike (note: both positions correspond to the AuCl sites). The results imply that the differences in the adjacent atomic environment (such as the shell-a/b/c positions of AuP) hardly affect the doping facility. However, for each doping system (with specific foreign atom), the relative energy of the Aucenter doped one is distinct from that of the AuP or AuCl doped structure. Therefore, the doping easiness is mainly determined by the chemical environment (Aucenter or AuP or ACl). | ||
| Fig. 4 The UV-vis spectra of Au9Cu4, Au13 and Au9Ag4 (a); the component of KS orbitals of Au13 (b), Au9Cu4 (c) and Au9Ag4 (d). | ||
| ΔE(Au12Mshell–Au12Mcenter) | |||
|---|---|---|---|
| M = Ag | M = Cu | M = Pd | |
| Shell-a | −0.56 | −0.18 | 0.97 |
| Shell-b | −0.51 | −0.12 | 0.87 |
| Shell-c | −0.45 | −0.09 | 0.90 |
| Shell-d | −0.83 | −0.51 | 1.02 |
| Shell-e | −0.86 | −0.58 | 1.06 |
According to Table 2, Ag and Cu atoms preferentially locate on the shell sites, and especially the AuCl positions (shell-d/e). By contrast, the doping of Pd occurs preferentially on the core site, while all the shell-doping modes are thermodynamically disfavoured. The theoretical results are in good agreement with the experimental characterization.21 The distinct doping preference of Ag, Cu and Pd is mainly determined by the Au–M interactions. The intermetallic interaction of Au with different metal atoms follows the order of Cu < Ag < Au < Pd,38,39 and thus the Ag and Cu atom(s) tend to be doped in the shell while Pd tends to be doped in the center.
The geometric and electronic structure of the surface doped [(AuxM13−x)P8Cl4]+
In our study, the aforementioned four AuxM13−x NCs were selected for discussions on the doping effect of the foreign atoms, and they are labelled as Au9M4 (M = Cu, Ag), Au13 (i.e. NC2) and Au12Pd, respectively. Comparing the key structural parameters in the Au9M4 NCs (Table 3), we found that the average Aucenter–Mshell distances in Au13 and Au9Ag4 NCs are very close, while the related bond distance in Au9Cu4 is relatively shorter. This phenomenon is mainly attributed to the smaller atomic radii of Cu compared with those of Au and Ag atoms (Cu: 1.28 Å, Au/Ag: 1.44 Å). For this reason, the average Aucenter–CuCl bond distances in Au9Cu4 is significantly shorter than that of the Aucenter–AgCl in Au9Ag4 and Aucenter–AuCl in Au13 (Table 3).| Bond distance | Au9Ag4 | Au13 | Au9Cu4 |
|---|---|---|---|
| Aucenter–Mshell | 2.821(2.773) | 2.822 | 2.728(2.686) |
| MCl–Cl | 2.414(2.378) | 2.395 | 2.164(2.134) |
| Aucenter–MCl | 2.849(2.828) | 2.807 | 2.595(2.584) |
The theoretical UV-vis spectra of Au9M4 (M = Cu, Ag) and Au13 were given in Fig. 4a. Compared with the lowest-energy optical band of Au13 (∼650 nm), the HOMO → LUMO transition of Au9Ag4 red shifts to 675 nm. In contrast, the HOMO → LUMO transition of Au9Cu4 is retained at 650 nm. Therefore, the Cu dopants make little perturbation to the energy of the lowest-energy optical band.
According to the KS-orbital analysis (Fig. 4b–d), the highest occupied orbitals are predominantly constituted by Au(6sp) orbitals, which also make the major contributions to the LUMOs. Therefore, the HOMO → LUMO transitions in all these NCs correspond to the sp → sp transitions. In addition, in the UV-vis spectra of Au9Cu4, the prominent shoulder peak at ∼550 nm is mainly induced by the mixed transition of HOMO, HOMO−1 to the LUMO+6, +7, +8, +9 set (component mainly composed by ligands). The atomic contribution for such transitions is in accordance with the metal to ligand charge transfer (MLCT). With respect to the superatom model, the HOMOs of all these NCs show the P feature, while the HOMO of Au9Ag4 is slightly different (Fig. 4b) due to the electron density overlap between the two adjacent Ag atoms. Meanwhile, the LUMO of each NC figures the D orbital characteristic.
The geometric and electronic structure of the core doped Au12Pd NC
Comparing the optimized geometries of Au12Pd and its monometallic counterpart NC3, the average Aucenter–Aushell bond length in NC3 is shorter than the average Pdcenter–Aushell bond distance in Au12Pd. In addition, both the average Mcenter–AuP and Mcenter–AuCl bond length in Au12Pd are relatively longer than the related ones in NC3 (Table 4). Similar to the aforementioned discussions, the relatively longer Au–Pd distances result in weakened intermetallic interactions and the significant red shift of the first band (685 nm for NC3 to 835 nm for Au12Pd in Fig. 2 and 5a). For Au12Pd, the peak at 740 nm and the shoulder peak at 640 nm mainly arise from the transition of HOMO−1, 2 → LUMO+3, 4 and HOMO−1, 2 → LUMO+7, 8, respectively. From KS-orbital in Fig. 5, the HOMO of Au12Pd is mainly composed by Pd(4p), Au(6sp) and Au(5d) orbitals, while the Pd(4p) and Au(5d) atomic orbitals make almost no contribution to the LUMOs (Fig. 5b). Therefore, the mismatched orbital character of HOMO and LUMO results in the weakness of the first band.| Bond distance | Au12Pd | NC3 |
|---|---|---|
| Mcenter–Aushell | 2.880(2.745) | 2.845 |
| Mcenter–AuCl | 2.801(2.728) | 2.776 |
| Mcenter–AuP | 2.920(2.754) | 2.880 |
| AuCl–Cl | 2.438(2.385) | 2.406 |
| AuP–P | 2.356(2.299) | 2.372 |
|
||
| Hirshfeld charge | ||
| Mcenter | 0.121 | 0.002 |
| Aushell | 0.020 | 0.050 |
| AuP | 0.023 | 0.046 |
| AuCl | 0.018 | 0.053 |
| Cl | −0.265 | −0.230 |
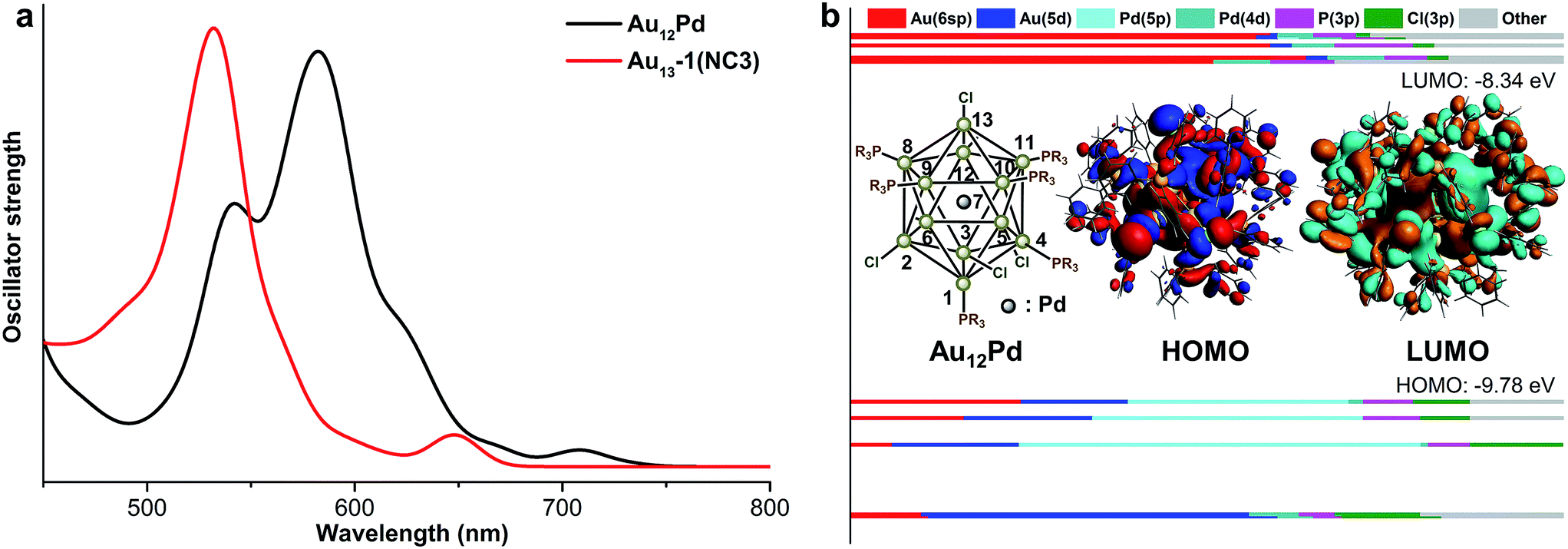 | ||
| Fig. 5 The UV-vis spectra of Au12Pd and Au13-1 (a), the component of KS orbitals of Au12Pd (b). | ||
Examining the charge distribution of Au12Pd, it can be seen that the electron density on the core Pd atom is significantly more positive than the core Au atom in NC3 (Table 4). Meanwhile, the electron density on the shell Au atoms are all increased in Au12Pd. Therefore, the results unambiguously show the charge transfer from Pd to Au. This conclusion is similar to the XPS characterization on the recently reported Au24Pd alloys.40
Conclusions
In this study, DFT and TD-DFT calculations were carried out to obtain the geometric and electronic structures of a series of Au13 and AuxM13−x (M = Au, Ag, Cu, Pd) NCs. It was found that PR3 ligands play the prominent role in the geometric structure in Au13 NCs due to the steric bulkiness. The bulkier PR3 with relatively larger cone angle favours the structure of [Au13(PR3)8Cl4]+ (and even smaller-sized NCs such an M11), while the PR3 with smaller cone angle encourages the formation of [Au13(PR3)10Cl2]3+ (or larger-sized NCs). The incorporation of more phosphine ligands results in the weaker Au–P and Au–Au interactions, associated with lowered HOMO–LUMO gap and the red shift of the related peak on UV-vis spectra. In addition, when different foreign atoms are doped into the Au13 NCs, preferential sites might be totally different. Ag and Cu atoms prefer the shell sites of the AuxM13−x core, and especially in the sites connecting with Cl atom. By contrast, Pd prefers the central site of the icosahedral core. The doping preference significantly affects the geometric structure and the frontier orbitals. The Pd dopant leads to relatively longer Au–M distances within the icosahedral core, while the Au–M bond distances are significantly shorter when Cu atoms are incorporated. The relative energy of the HOMO → LUMO transition of different AuxM13−x NCs follows the order of Au12Pd < Au9Ag4 < Au13 ∼ Au9Cu4. The present study provides deep understanding on the ligand and doping effect in the icosahedral NCs, and will hopefully benefit the future investigation on structure–property correlations of larger NCs.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge financial support from NSFC (21672001, 21372006, 21631001 & U1532141), the Ministry of Education, the Education Department of Anhui Province, 211 Project of Anhui University. We thank the National Supercomputing Centre in Shenzhen for providing the computational resources and Gaussian 09 software.Notes and references
- G. Li, H. Abroshan, Y. Chen, R. Jin and H. J. Kim, J. Am. Chem. Soc., 2015, 137, 14295 CrossRef CAS PubMed.
- J. Sun and Y. Jin, J. Mater. Chem. C, 2014, 2, 8000 RSC.
- D. An, J. Su, J. K. Weber, X. Gao, R. Zhou and J. Li, J. Am. Chem. Soc., 2015, 137, 8412 CrossRef CAS PubMed.
- B. Khlebtsov, E. Tuchina, V. Tuchin and N. Khlebtsov, RSC Adv., 2015, 5, 61639 RSC.
- Y. Song, S. Jin, X. Kang, J. Xiang, H. Deng, H. Yu and M. Zhu, Chem. Mater., 2016, 28, 2609 CrossRef CAS.
- J. Nishigaki, S. Yamazoe, S. Kohara, A. Fujiwara, W. Kurashige, Y. Negishid and T. Tsukuda, Chem. Commun., 2014, 50, 839 RSC.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210 CrossRef CAS PubMed.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430 CrossRef CAS PubMed.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500 CrossRef CAS PubMed.
- X.-K. Wan, Q. Tang, S.-F. Yuan, D.-e. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652 CrossRef CAS PubMed.
- X.-K. Wan, S.-F. Yuan, Z.-W. Lin and Q.-M. Wang, Angew. Chem., Int. Ed., 2014, 53, 2923 CrossRef CAS PubMed.
- (a) Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568 CrossRef CAS PubMed; (b) R. Ouyang and D.-e. Jiang, ACS Catal., 2015, 5, 6624 CrossRef CAS; (c) E. Pohjolainen, H. Häkkinen and A. Clayborne, J. Phys. Chem. C, 2015, 119, 9587 CrossRef CAS.
- O. Guliamov, A. I. Frenkel, L. D. Menard, R. G. Nuzzo and L. Kronik, J. Am. Chem. Soc., 2007, 129, 10978 CrossRef CAS PubMed.
- Y. Li, H. Cheng, T. Yao, Z. Sun, W. Yan, Y. Jiang, Y. Xie, Y. Sun, Y. Huang, S. Liu, J. Zhang, Y. Xie, T. Hu, L. Yang, Z. Wu and S. Wei, J. Am. Chem. Soc., 2012, 134, 17997 CrossRef CAS PubMed.
- Y. Shichibu, K. Suzuki and K. Konishi, Nanoscale, 2012, 4, 4125 RSC.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216 CrossRef CAS PubMed.
- M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konish, Chem. Commun., 2015, 51, 13519 RSC.
- R. C. B. Copley and D. M. Mingos, J. Chem. Soc., Dalton Trans., 1996, 491 RSC.
- G. Lugo, V. Schwanen, B. Fresch and F. Remacle, J. Phys. Chem. C, 2015, 119, 10969 CAS.
- C. Briant, B. C. Theobald, J. White, L. Bell and D. M. Mingos, J. Chem. Soc., Chem. Commun., 1981, 201 RSC.
- M. Laupp and J. Strähle, Z. Naturforsch., A: Phys. Sci., 1995, 50, 1369 CAS.
- G. T. Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. V. Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS.
- R. van Leeuwen and E. J. Baerends, Phys. Rev. A: At., Mol., Opt. Phys., 1994, 49, 2421 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- D. M. P. Mingos, Structure and Bonding, in Gold Clusters, Colloids, and Nanoparticles II, ed. D. M. P. Mingos, Springer International Publishing, Berlin, 2014, vol. 162, p. 1 Search PubMed.
- X.-Y. Chang, K.-H. Low, J.-Y. Wang, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 10312 CrossRef CAS PubMed.
- E. G. Mednikov and L. F. Dahl, J. Cluster Sci., 2005, 16, 287 CrossRef CAS.
- E. G. Mednikov, N. T. Tran, N. L. Aschbrenner and L. F. Dahl, J. Cluster Sci., 2007, 18, 253 CrossRef CAS.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426 CrossRef CAS PubMed.
- (a) A. Das, C. Liu, H. Byun, K. Nobusada, S. Zhao, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140 CrossRef CAS PubMed; (b) Y. Song, S. Jin, X. Kang, J. Xiang, H. Deng, H. Yu and M. Zhu, Chem. Mater., 2016, 28, 2609 CrossRef CAS; (c) Y. Wang, H. Su, C. Xu, G. Li, L. Gell, S. Lin, Z. Tang, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 4324 CrossRef CAS PubMed; (d) Y. Pe, J. Tang, X. Tang, Y. Huang and X. Zeng, J. Phys. Chem. Lett., 2015, 6, 1390 CrossRef PubMed; (e) L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-e. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970 CrossRef CAS PubMed.
- Z. Wang, R. Senanayake, C. M. Aikens, W.-M. Chen, C.-H. Tunga and D. Sun, Nanoscale, 2016, 8, 18905 RSC.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145 CrossRef CAS PubMed.
- Z. Luo and A. W. Castleman, Acc. Chem. Res., 2014, 47, 2931 CrossRef CAS PubMed.
- A. Tlahuice-Flores, R. L. Whetten and M. Jose-Yacaman, J. Phys. Chem. C, 2013, 117, 20867 CAS.
- F. Sheong, J.-X. Zhang and Z. Lin, Inorg. Chem., 2016, 55, 11348 CrossRef CAS PubMed.
- D. Mingos and Z. Lin, Comments Inorg. Chem., 1989, 9, 95 CrossRef CAS.
- (a) Y. Niihori, M. Eguro, A. Kato, S. Sharma, B. Kumar, W. Kurashige, K. Nobusada and Y. Negishi, J. Phys. Chem. C, 2016, 120, 14301 CrossRef CAS; (b) Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasab and K. Nobusada, Phys. Chem. Chem. Phys., 2016, 18, 1397 RSC; (c) B. Molinaa and A. T. Flores, Phys. Chem. Chem. Phys., 2009, 11, 7123 RSC.
- K. Kwak, Q. Tang, M. Kim, D.-e. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08421h |
| This journal is © The Royal Society of Chemistry 2017 |