DOI:
10.1039/C7RA08836A
(Paper)
RSC Adv., 2017,
7, 45401-45407
Sterically bulky amido magnesium methyl complexes: syntheses, structures and catalysis†
Received
10th August 2017
, Accepted 19th September 2017
First published on 25th September 2017
Abstract
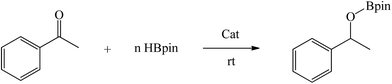
A series of bulky secondary amines were treated with two equiv. Grignard reagent MeMgI at low temperature to yield the corresponding magnesium methyl complexes (1–4) in high yields. The prepared complexes have been comprehensively characterized. All four complexes exhibited very high reactivity as efficient pre-catalysts in the catalytic hydroboration of ketones with pinacolborane under mild conditions and at low catalyst loadings.
Introduction
The catalytic hydroboration of unsaturated substrates is an important transformation in organic chemistry. Recently a variety of unsaturated organic compounds such as alkenes, alkynes, imines, aldehydes and ketones have been hydroborated to get the corresponding organoboranes compounds using transition metals and main group compounds as catalysts.1–9 Even carbon dioxide could be reduced to a C1-building block such as methanol, formaldehyde and formic acid via the catalytic hydroboration.10,11 For example, Nozaki et al. reported the reduction of CO2 with HBpin catalyzed by N-heterocyclic carbene supported Cu(I) complex [(IPr)Cu(OtBu)] recently.10
The reduction of carbonyl compounds is a facile and efficient synthetic route to functionalized alcohols which are industrially important compounds that are produced in bulk. Compared to hydrogenation and hydrosilylation protocols, the hydroboration of carbonyl compounds is more efficient and can normally proceed at room temperature and with very low catalyst loading.12–15 For instance, quite recently Gade et al. reported that a manganese alkyl complex containing a chiral bis(oxazolinyl-methylidene)isoindoline pincer ligand has been used as a pre-catalyst for the enantioselective hydroboration of ketones under mild reaction conditions.14 When compared to the transition metal catalyzed hydroboration of aldehydes and ketones which has been extensively investigated, the research on the catalytic hydroboration of carbonyl compounds using main group catalysts is less developed but has recently received considerable attention by many research groups.12–27
Compared to the precious and relatively toxic transition metals, amongst the main group metals, magnesium is earth-abundant, non-toxic and inexpensive. The Mg-catalyzed hydroboration has been relatively well explored and has shown high reactivity in recent studies.28–35 For example, the Hill group has reported the hydroboration of pyridines, aldehydes, ketones, imines, isonitriles, nitriles, carbodiimides and isocyanates with pinacolborane (HBpin) using the β-diketiminato magnesium n-butyl complex [(DippNacnac)MgnBu] as an efficient pre-catalyst.29 Harder et al. found that the β-diketiminato magnesium hydride tetramer also could be used as a catalyst in the catalytic hydroboration of pyridines.30 Hydroboration of esters to two alkoxyborates was successfully achieved using a tris(pyrazolyl)borate magnesium methyl complex as a catalyst by Sadow and co-workers. The same catalyst can also catalyze the hydroboration of amides for their deoxygenation to amines.31 However, literature reports about the Mg-catalyzed hydroboration of ketones are relatively few and the catalysts are mainly the corresponding bidentate β-diketiminato, metal–organic framework and ion pair magnesium complexes respectively.29c,32–34 Herein we reported the syntheses and characterizations of a series of bulky secondary amidomagnesium methyl complexes which can act as new monodentate magnesium pre-catalysts for the hydroboration of a range of ketones.
Results and discussion
Syntheses and structures of magnesium methyl complexes
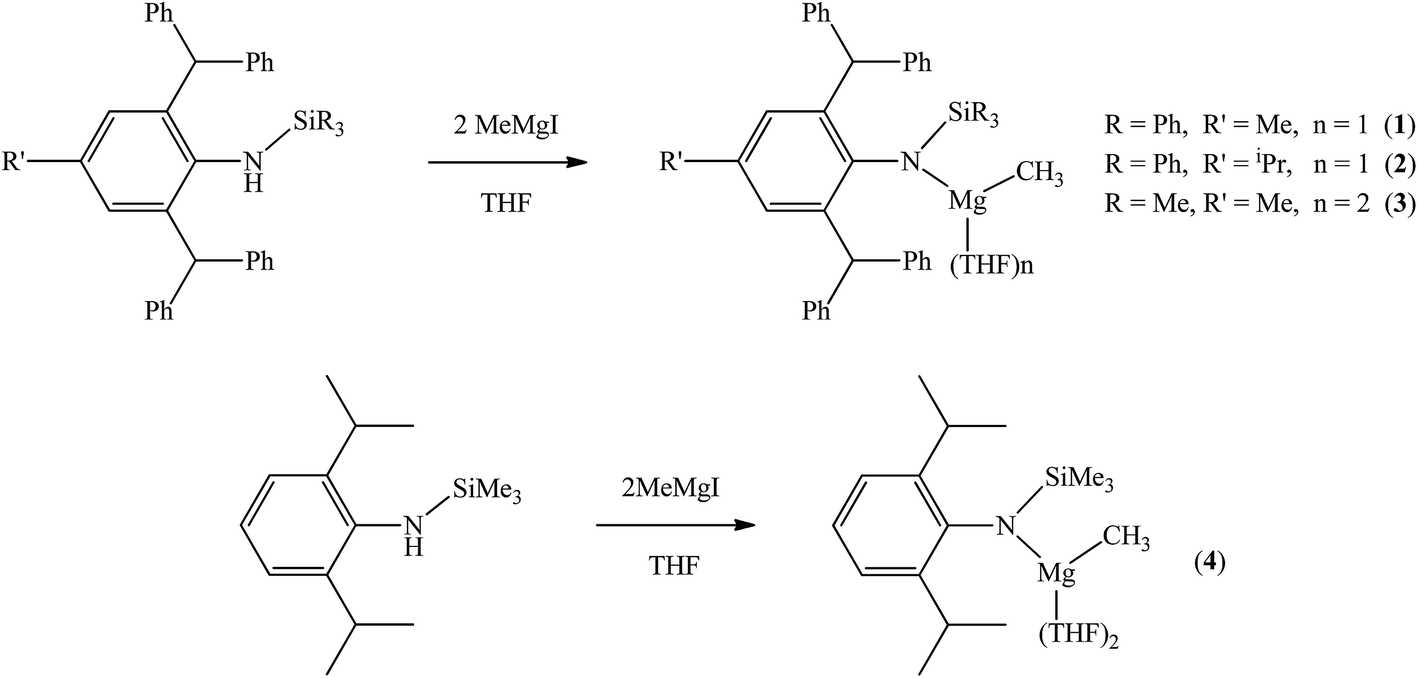
The bulky secondary amine ArNH(SiPh3) (Ar = C6H2Me(CHPh2)2-4,2,6) was treated with one equiv. MeMgI in Et2O or THF, we initially attempted to prepare the corresponding magnesium iodide complex, namely the precursor of the magnesium(I) complex. During the process of this work Jones et al. published the corresponding magnesium(I) complexes.36 However, an intractable mixture was observed. According to the 1H NMR spectroscopic analysis of the obtained white solid, we ascertained that the corresponding magnesium iodide and magnesium methyl complexes could be in the mixture because there are two sets of new peaks in the 1H NMR spectrum of the solid mixture besides signals that can be attributed to the ligand. Furthermore, a new singlet signal at δ −1.60 ppm was also observed which could be the characteristic methyl group resonance because the previously reported magnesium methyl complexes have also shown the similar high field signal characteristic of the magnesium bound methyl from −0.65 to −2.00 ppm in 1H NMR spectra.37–39 We reasoned that when the amido ligand was treated with MeMgI, firstly the corresponding magnesium iodide was produced. However due to the high reactivity of the iodide moiety it further reacted with MeMgI to yield the magnesium methyl complex. In order to prove our speculation, the same ligand ArNH(SiPh3) was treated with two equiv. Grignard reagent MeMgI and the corresponding magnesium methyl complex 1 was indeed obtained as colorless crystals in high yield (88%) (Scheme 1). 1H NMR spectroscopic analysis further revealed that the NH signal (δ = 2.68 ppm)40 of the starting material disappeared, with the subsequent appearance of a new singlet signal at δ −1.60 ppm which is assigned to the Mg–CH3 characteristic resonance in 1. The 13C{1H} NMR spectrum of complex 1 further confirmed our speculation. It also displayed a characteristic high field Mg–CH3 resonance at δ −14.5 ppm as observed in similar magnesium methyl complexes.37–39 In the 29Si{1H} NMR spectrum of complex 1 the SiPh3 signal (δ = −18.8 ppm) of the starting secondary amine ArNH(SiPh3) disappeared and a new singlet signal at δ −26.4 ppm was found which is similar to analogous magnesium n-butyl complex (δ = −24.7 ppm).36 Finally, single crystal X-ray diffraction, NMR and CHN analysis confirmed the successful synthesis of 1.
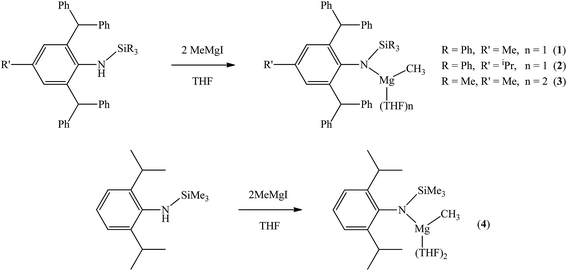 |
| | Scheme 1 Syntheses of complexes 1–4. | |
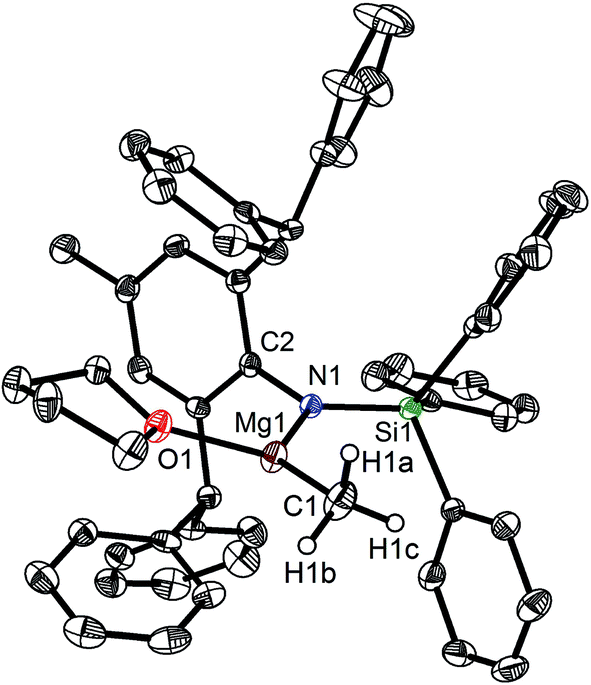
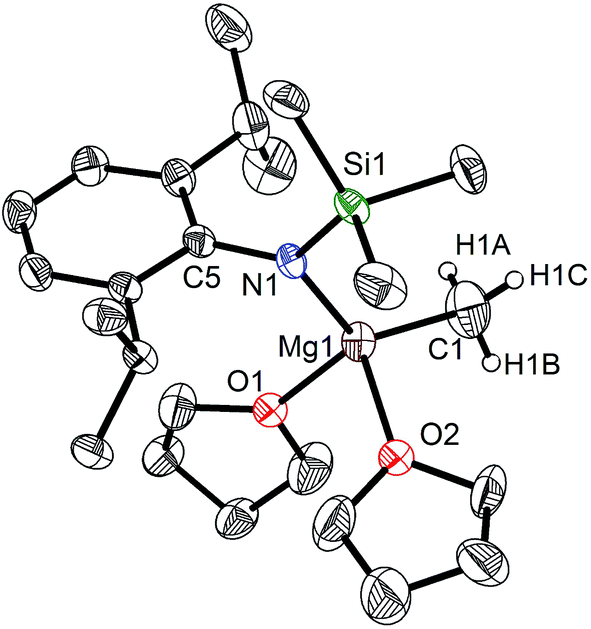
When the similar secondary amines Ar*NH(SiPh3) (Ar* = C6H2iPr(CHPh2)2 -4,2,6), ArNH(SiMe3) or DippNH(SiMe3) (Dipp = 2,6-diisopropylphenyl) were treated with one equiv. MeMgI respectively, unfortunately the intractable mixtures were once again obtained which consisted of the corresponding magnesium iodide, methyl complexes and ligands etc. As expected, the reaction between the secondary amines and two equiv. MeMgI also produced the corresponding magnesium methyl complexes 2–4 in high yield as seen earlier in the case of complex 1. The high field Mg–CH3 characteristic resonances of complexes 2–4 were observed as singlets at δ −1.47, −0.79, −0.94 ppm in the 1H NMR spectra and at δ −14.4, −12.6, −15.0 ppm in the 13C{1H} NMR spectra respectively. These chemical shift values are similar to those of the previously reported magnesium methyl and analogous magnesium butyl complexes.36–39 The solid state structure of complexes 2–4 were also determined by single crystal X-ray diffraction. The molecular structures of complexes 1–4 are shown in Fig. 1–4. Selected bond lengths and angles for complexes 1–4 are presented in Tables 1–4. X-ray crystal structure analyses further revealed that complexes 1–4 were monomeric. In complexes 1 or 2, the geometry of three-coordinated magnesium atom was slightly elongated trigonal planar which the nitrogen atom of amido ligand, oxygen atom of THF solvent and carbon atom of new methyl group occupying three vertices of the planar triangle. However, the geometry of four-coordinated magnesium metal in complexes 3 or 4 was the slightly distorted tetrahedron because there were two coordinated THF solvents. The magnesium-methyl group [Mg–C(1), 2.114(4)–2.1844(16) Å] bond lengths in complexes 1–4 are similar to those of the reported magnesium methyl complexes which have been structurally characterized.37–39 Furthermore when a series of similar amino ligands such as ArNH(SiMe3) and Ar*NH(SiiPr3) were reacted with less reactive Grignard reagent MeMgBr, the corresponding magnesium bromide or methyl complexes could not be isolated.
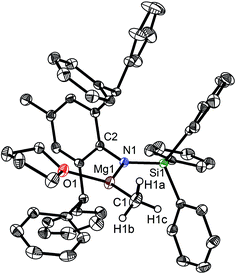 |
| | Fig. 1 Molecular structure of 1. | |
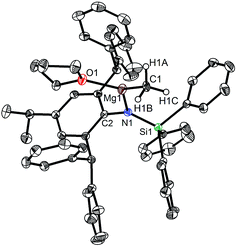 |
| | Fig. 2 Molecular structure of 2. | |
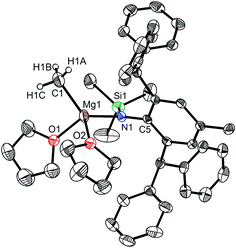 |
| | Fig. 3 Molecular structure of 3. | |
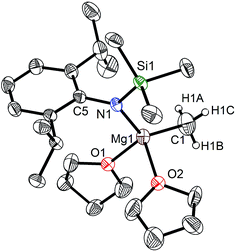 |
| | Fig. 4 Molecular structure of 4. | |
Table 1 Selected bond lengths [Å] and angles [°] for 1
| Mg(1)–C(1) |
2.114(4) |
Mg(1)–N(1) |
1.997(3) |
| Mg(1)–O(1) |
2.010(2) |
N(1)–C(2) |
1.421(4) |
| N(1)–Si(1) |
1.704(2) |
|
|
| N(1)–Mg(1)–C(1) |
132.34(13) |
O(1)–Mg(1)–C(1) |
106.09(12) |
| N(1)–Mg(1)–O(1) |
116.45(10) |
Si(1)–N(1)–Mg(1) |
121.77(14) |
| C(2)–N(1)–Si(1) |
123.0(2) |
C(2)–N(1)–Mg(1) |
115.07(18) |
Table 2 Selected bond lengths [Å] and angles [°] for 2
| Mg(1)–C(1) |
2.1844(16) |
Mg(1)–N(1) |
1.9973(12) |
| Mg(1)–O(1) |
2.0123(12) |
N(1)–C(2) |
1.4141(18) |
| N(1)–Si(1) |
1.7056(12) |
|
|
| N(1)–Mg(1)–C(1) |
130.89(6) |
O(1)–Mg(1)–C(1) |
107.07(5) |
| N(1)–Mg(1)–O(1) |
115.94(5) |
C(2)–N(1)–Mg(1) |
112.43(9) |
| Si(1)–N(1)–Mg(1) |
122.55(7) |
C(2)–N(1)–Si(1) |
125.00(9) |
Table 3 Selected bond lengths [Å] and angles [°] for 3
| Mg(1)–C(1) |
2.173(4) |
Mg(1)–N(1) |
2.046(2) |
| Mg(1)–O(1) |
2.065(2) |
Mg(1)–O(2) |
2.038(2) |
| N(1)–C(5) |
1.410(3) |
N(1)–Si(1) |
1.709(2) |
| N(1)–Mg(1)–C(1) |
130.21(11) |
O(1)–Mg(1)–C(1) |
102.14(11) |
| N(1)–Mg(1)–O(1) |
111.21(10) |
O(2)–Mg(1)–C(1) |
107.08(12) |
| O(2)–Mg(1)–N(1) |
105.02(8) |
O(2)–Mg(1)–O(1) |
96.02(9) |
| C(5)–N(1)–Si(1) |
124.55(15) |
C(5)–N(1)–Mg(1) |
119.36(15) |
| Si(1)–N(1)–Mg(1) |
115.01(10) |
|
|
Table 4 Selected bond lengths [Å] and angles [°] for 4
| Mg(1)–C(1) |
2.129(4) |
Mg(1)–N(1) |
2.019(2) |
| Mg(1)–O(1) |
2.052(2) |
Mg(1)–O(2) |
2.072(2) |
| N(1)–Si(1) |
1.699(2) |
N(1)–C(5) |
1.415(3) |
| N(1)–Mg(1)–O(1) |
108.12(10) |
O(1)–Mg(1)–O(2) |
91.95(10) |
| N(1)–Mg(1)–O(2) |
107.76(10) |
O(1)–Mg(1)–C(1) |
105.15(13) |
| N(1)–Mg(1)–C(1) |
127.14(14) |
O(2)–Mg(1)–C(1) |
100.91(14) |
| C(5)–N(1)–Si(1) |
123.07(18) |
C(5)–N(1)–Mg(1) |
116.91(17) |
| Si(1)–N(1)–Mg(1) |
119.08(13) |
|
|
Catalytic hydroboration of ketones
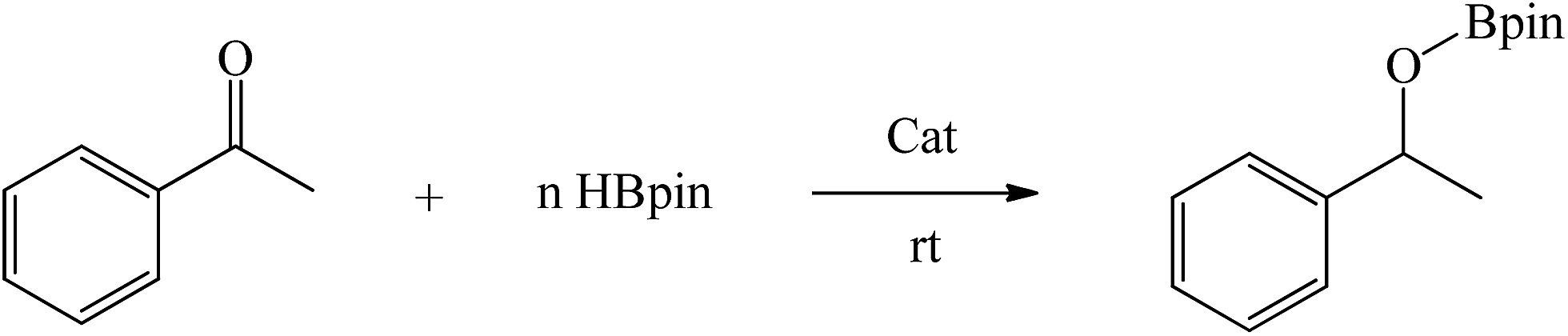
Encouraged by the recent success of employing alkaline earth metal complexes as catalysts for the hydroboration of unsaturated molecules1–6,28 and with the newly prepared and characterised magnesium complexes (1–4) in hand, we initially examined their catalytic activity towards the hydroboration of ketone. In a simple screening reaction, we tested the room temperature catalytic performance of 1–4 when using acetophenone as a test substrate in combination with pinacolborane (HBpin). The results are summarized in Table 5. We found that acetophenone was clearly hydroborated with almost quantitative conversion by 1.5 equiv. of HBpin in the presence of 0.1 mol% of complexes 1–4 in few hours (entries 1–4, Table 5). It should be noted that the reactivity of 1–4 is higher than that of the reported β-diketiminato magnesium n-butyl complex [(DippNacnac)MgnBu] which needed 1 mol% catalyst loading and 4 hours to get 94% conversion in the acetophenone hydroboration.29 For complex 4, only 30 min was needed at room temperature, other magnesium methyl complexes 1–3 needed a little longer time to achieve near full conversion. It appears that the catalytic activity of 1–4 is in contrast with the steric hindrance associated with their structures. Since complex 4 was noted to be the most active, the HBpin loading was reduced from 1.5 to 1 equiv. However, the hydroboration reaction time was prolonged from 0.5 to 2.5 h (entry 5, Table 5). When the catalyst loading of 4 was increased from 0.1 to 0.5 mol%, the reaction time did not decrease appreciably, and still required half an hour for conversion (entry 6, Table 5).
Table 5 Preliminary catalytic screening results
|
| Entry |
Cat |
Cat (mol%) |
n |
Time (h) |
Yielda (%) |
| The reaction was monitored by 1H NMR spectroscopy. |
| 1 |
1 |
0.1 |
1.5 |
2.5 |
99 |
| 2 |
2 |
0.1 |
1.5 |
3 |
99 |
| 3 |
3 |
0.1 |
1.5 |
1.5 |
99 |
| 4 |
4 |
0.1 |
1.5 |
0.5 |
>99 |
| 5 |
4 |
0.1 |
1 |
2.5 |
98 |
| 6 |
4 |
0.5 |
1 |
0.5 |
>99 |
To further extend the utility of the prepared magnesium complexes, we explored the catalytic hydroboration of ketones bearing different functionalities. With 0.1 mol% of 4 and HBpin as the reductant, the hydroboration of acetophenones with F–, O2N–, NC–, MeO– and H2N– substituents (entries 1–8, Table 6) were completed in very high yield. Since both electron-donating and electron-withdrawing groups appeared to make the hydroboration reaction slightly sluggish, we used a higher catalyst loading (1 mol%) in those instances to shorten the reaction time (entries 4, 6, 8, Table 6). Compared to 4′-nitroacetophenone (entry 2, Table 6), 4′-aminoacetophenone (entry 8, Table 6) required a slightly longer time possibly because it will react with HBpin leading to the uncatalysed amine-borane blocking catalytic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydroboration pathway.41 The reaction showed tolerance towards fluoride group which ortho-substituted F group lead to an increase in reactivity compared to the parent acetophenone (entry 1, Table 6). The incorporation of a cyano group however necessitated the need for a higher catalyst loading (1 mol%) to ensure a high conversion in a short time (entries 3 and 4, Table 6). Complex 4 was also active towards more sterically hindered aryl-substituted ketones, although in these cases, a slightly longer reaction time was required to achieve a high yield at room temperature (entries 9 and 10, Table 6). For the benzophenone hydroboration, it showed equal efficiency with the above-mentioned [(DippNacnac)MgnBu]29 and was slightly faster than the same amide ligand complexes LMH (M = Ge or Sn).16 However, the hydroboration of the bulky 2,4,6-trimethylacetophenone was slow (24 h) even at 60 °C and required a higher catalyst loading (1 mol%) to reach a high conversion (entries 11 and 12, Table 6). This is slightly slower than that of [(DippNacnac)MgnBu].29 Dialkyl ketones were hydroborated in quantitative yield at room temperature in a short time (entries 13 and 14, Table 6).
O hydroboration pathway.41 The reaction showed tolerance towards fluoride group which ortho-substituted F group lead to an increase in reactivity compared to the parent acetophenone (entry 1, Table 6). The incorporation of a cyano group however necessitated the need for a higher catalyst loading (1 mol%) to ensure a high conversion in a short time (entries 3 and 4, Table 6). Complex 4 was also active towards more sterically hindered aryl-substituted ketones, although in these cases, a slightly longer reaction time was required to achieve a high yield at room temperature (entries 9 and 10, Table 6). For the benzophenone hydroboration, it showed equal efficiency with the above-mentioned [(DippNacnac)MgnBu]29 and was slightly faster than the same amide ligand complexes LMH (M = Ge or Sn).16 However, the hydroboration of the bulky 2,4,6-trimethylacetophenone was slow (24 h) even at 60 °C and required a higher catalyst loading (1 mol%) to reach a high conversion (entries 11 and 12, Table 6). This is slightly slower than that of [(DippNacnac)MgnBu].29 Dialkyl ketones were hydroborated in quantitative yield at room temperature in a short time (entries 13 and 14, Table 6).
Table 6 Hydroboration of ketones under the catalysis of the complex 4
Conclusions
In conclusion, we have successfully prepared a series of new monodentate magnesium methyl complexes via the use of sterically bulky secondary amines and two equiv. MeMgI. The fully characterized complexes were found to be efficient pre-catalysts for the hydroboration of a variety of aromatic and aliphatic ketones under mild conditions and at low catalyst loadings and showed moderate efficiency on the hydrosilylation of acetophenone. Investigation of the detailed mechanism of the catalytic cycle is in progress in our laboratory.
Experimental section
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of high purity dinitrogen or argon. Toluene, THF, diethyl ether and hexane were distilled from molten sodium. 1H, 13C{1H} and 29Si{1H} NMR spectra were recorded at 25 °C on Bruker Avance III 600 MHz spectrometer in deuterated solvents and were referenced to the resonances of the solvent used or external SiMe4. Microanalyses were performed by the Elemental Analysis Laboratory of the Advanced Analysis and Testing Center at Nanjing Forestry University. Melting points were determined in sealed capillaries under dinitrogen and are uncorrected. The starting materials ArNH(SiPh3), ArNH(SiMe3), Ar*NH(SiPh3) and DippNH(SiMe3) were prepared according to literature procedures.36,40,42 All other reagents were used as received.
Synthesis of complex 1
MeMgI (0.96 mL, 3 M in Et2O, 2.88 mmol) was added dropwise to a solution of ArNH(SiPh3) (1.00 g, 1.44 mmol) in THF (25 mL) at −60 °C. The reaction mixture was warmed to room temperature and stirred overnight, filtered. Removal of the solvents gave 1 as solid which was then recrystallized from toluene and hexane to give the complex as colorless crystals at room temperature (1.02 g, yield: 88%). Mp: 181–183 °C. 1H NMR (600 MHz, 298 K, C6D6): δ = −1.60 (s, 3H, Mg–CH3), 0.86 (br., 4H, CH2 (THF)), 1.98 (s, 3H, CH3), 2.60 (br., 4H, OCH2 (THF)), 6.69 (s, 2H, CHPh2), 6.77–7.88 (m, 37H, Ar–H). 13C{1H} NMR (151 MHz, 298 K, C6D6): δ = −14.5 (Mg–CH3), 21.0 (CH3), 24.3 (CH2 (THF)), 51.6 (CHPh2), 68.4 (OCH2 (THF)), 125.5, 128.0, 128.8, 129.3, 129.8, 129.9, 136.1, 136.6, 139.8, 140.6, 145.5, 145.9, 150.4 (Ar–C). 29Si{1H} NMR (119 MHz, 298 K, C6D6) δ = −26.4. Anal. calc. for C56H53MgNOSi: C, 83.20; H, 6.61; N, 1.73. Found: C, 83.51; H, 6.83; N, 1.41.
Synthesis of complex 2
Complex 2 was prepared using a similar procedure that employed for 1, but using Ar*NH(SiPh3) as a starting material. Colorless crystals (yield: 90%). Mp: 167–169 °C. 1H NMR (600 MHz, 298 K, C6D6): δ = −1.47 (s, 3H, Mg–CH3), 0.83 (br., 4H, CH2 (THF)), 1.00 (d, 3JHH = 6.6 Hz, 6H, CH(CH3)2), 2.48 (br., 4H, OCH2 (THF)), 2.58 (sept, 3JHH = 6.6 Hz, 1H, CH(CH3)2), 6.71 (s, 2H, CHPh2), 6.80–7.88 (m, 37H, Ar–H). 13C{1H} NMR (151 MHz, 298 K, C6D6): δ = −14.4 (Mg–CH3), 24.1 (CH(CH3)2), 24.2 (CH2 (THF)), 33.4 (CH(CH3)2), 51.6 (CHPh2), 68.4 (OCH2 (THF)), 125.5, 126.6, 127.0, 127.6, 128.0, 128.8, 129.2, 129.3, 129.6, 136.7, 139.4, 139.7, 140.3, 145.4, 146.2, 151.1 (Ar–C). 29Si{1H} NMR (119 MHz, 298 K, C6D6) δ = −26.5. Anal. calc. for C58H57MgNOSi: C, 83.28; H, 6.87; N, 1.67. Found: C, 83.61; H, 7.14; N, 1.35.
Synthesis of complex 3
Complex 3 was prepared using a similar procedure that employed for 1, but using ArNH(SiMe3) as a starting material. Colorless crystals (yield: 92%). Mp: 131–133 °C. 1H NMR (600 MHz, 298 K, C6D6): δ = −0.79 (s, 3H, Mg–CH3), 0.25 (s, 9H, Si(CH3)3), 1.10 (br., 8H, CH2 (THF)), 2.07 (s, 3H, CH3), 3.24 (br., 8H, OCH2 (THF)), 6.88 (s, 2H, CHPh2), 6.97–7.50 (m, 22H, Ar–H). 13C{1H} NMR (151 MHz, 298 K, C6D6): δ = −12.6 (Mg–CH3), 4.6 (Si(CH3)2), 21.3 (CH3), 24.9 (CH2 (THF)), 51.5 (CHPh2), 69.2 (OCH2 (THF)), 125.9, 126.0, 126.5, 128.2, 128.3, 128.4, 128.6, 130.0, 130.2, 130.4, 130.5, 140.1, 144.7, 146.3, 147.3 (Ar–C). 29Si{1H} NMR (119 MHz, 298 K, C6D6): δ = −8.09. Anal. calc. for C45H55MgNO2Si: C, 77.84; H, 7.98; N, 2.02. Found: C, 78.16; H, 8.32; N, 1.73.
Synthesis of complex 4
Complex 4 was prepared using a similar procedure that employed for 1, but using DippNH(SiMe3) as a starting material. Colorless crystals (yield: 91%). Mp: 90–92 °C. 1H NMR (600 MHz, 298 K, C6D6): δ = −0.94 (s, 3H, Mg–CH3), 0.38 (s, 9H, Si(CH3)3), 1.18 (br., 8H, CH2 (THF)), 1.25 (d, 3JHH = 6.6 Hz, 6H, CH(CH3)2), 1.42 (d, 3JHH = 6.6 Hz, 6H, CH(CH3)2), 3.32 (br., 8H, CH2 (THF)), 4.22 (sept, 2H, CH(CH3)2), 6.96 (t, 3JHH = 7.2 Hz, 1H, Ph-H), 7.14 (d, 3JHH = 7.2 Hz, 2H, Ph-H). 13C{1H} NMR (151 MHz, 298 K, C6D6): δ = −15.0 (Mg–CH3), 3.4 (Si(CH3)3), 24.7 (CH2 (THF)), 24.9 (CH(CH3)2), 25.2 (CH(CH3)2), 26.8 (CH(CH3)2), 68.4 (OCH2 (THF)), 118.9, 122.9, 144.8, 152.6 (Ar–C). 29Si{1H} NMR (119 MHz, 298 K, C6D6): δ = −8.14. Anal. calc. for C24H45MgNO2Si: C, 66.72; H, 10.50; N, 3.24. Found: C, 66.95; H, 10.76; N, 2.94.
X-ray crystal structure determination
Crystallographic data for complexes 1–4 are given in Table 7. Diffraction data were collected on a Bruker D8 VENTURE PHOTON 100 diffractometer using a graphite-monochromated MoKα radiation (0.71073 Å) at 135 K in the ω-2θ scan mode. In all cases, an empirical absorption correction by SADABS was applied to the intensity data. The structures were solved by direct methods and refined on F2 by full-matrix least-squares methods using the SHELXTL crystallographic software package. All non-hydrogen atoms were refined anisotropically with hydrogen atoms included in calculated positions (riding model). CCDC 1554843–1554846 contain the supplementary crystallographic data for complexes 1–4.†
Table 7 Crystallographic data for complexes 1–4
| |
1 |
2 |
3 |
4 |
| Formula |
C56H53MgNOSi |
C58H57MgNOSi |
C45H55MgNO2Si |
C24H45MgNO2Si |
| Fw |
808.39 |
836.45 |
694.30 |
432.01 |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
| a (Å) |
10.8664(4) |
11.0079(6) |
14.9209(9) |
9.8194(7) |
| b (Å) |
38.1625(15) |
11.3921(5) |
18.7056(12) |
16.3480(11) |
| c (Å) |
11.1840(4) |
20.2197(10) |
14.3767(9) |
16.4988(11) |
| α (°) |
90 |
75.7510(10) |
90 |
92.119(2) |
| β (°) |
106.5270(10) |
87.559(2) |
93.052(2) |
94.112(2) |
| γ (°) |
90 |
71.6810(10) |
90 |
90.371(2) |
| V (A3) |
4446.3(3) |
2331.4(2) |
4006.9(4) |
2639.8(3) |
| Z |
4 |
2 |
4 |
4 |
| T (K) |
135(2) |
135(2) |
135(2) |
135(2) |
| λ (Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Dcalcd (g cm−3) |
1.208 |
1.192 |
1.151 |
1.087 |
| μ (mm−1) |
0.108 |
0.105 |
0.111 |
0.131 |
| F (000) |
1720 |
892 |
1496 |
952 |
| R1 (obsd data) |
0.0453 |
0.0503 |
0.0736 |
0.0762 |
| wR2 (obsd data) |
0.0986 |
0.1525 |
0.1669 |
0.1950 |
| GOF on F2 |
1.017 |
1.197 |
1.006 |
1.119 |
General procedure for catalytic hydroboration of ketones
In a glove box, catalyst 4 (0.1 mol%) was added to a solution of ketone (1 mmol) and pinacolborane (1.5 mmol) in a J. Young NMR tube equipped with a Teflon screw cap, which was charged with C6D6 (0.5 mL). The progress of the reaction was monitored by 1H NMR and 11B NMR, which indicated the completion of the reaction by the appearance of a new CH resonance.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21372117, 21772093) and the Natural Science Foundation of Jiangsu Province, China (Grant No. BK20141468) for support of this research.
References
- K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840 RSC.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238 CrossRef CAS.
- S. J. Geier, C. M. Vogels and S. A. Westcott, ACS Symp. Ser., 2016, 1236, 209 CrossRef CAS.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC.
- M. Arrowsmith, Encycl. Inorg. Bioinorg. Chem., 2016, p. 1 Search PubMed.
- W. Li, X. Ma, M. G. Walawalkar, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2017 DOI:10.1016/j.ccr.2017.03.017.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595 CrossRef CAS PubMed.
- S. Chakrabarty and J. M. Takacs, J. Am. Chem. Soc., 2017, 139, 6066 CrossRef CAS PubMed.
- S. Yu, C. Wu and S. Ge, J. Am. Chem. Soc., 2017, 139, 6526 CrossRef CAS PubMed.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459 CrossRef CAS.
- T. J. Hadlington, C. E. Kefalidis, L. Maron and C. Jones, ACS Catal., 2017, 7, 1853 CrossRef CAS.
- V. L. Weidner, C. J. Barger, M. Delferro, T. L. Lohr and T. J. Marks, ACS Catal., 2017, 7, 1244 CrossRef CAS.
- S. Chen, D. Yan, M. Xue, Y. Hong, Y. Yao and Q. Shen, Org. Lett., 2017, 19, 3382 CrossRef CAS PubMed.
- V. Vasilenko, C. K. Blasius, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2017, 56, 8393 CrossRef CAS PubMed.
- R. Arévalo, C. M. Vogels, G. A. MacNeil, L. Riera, J. Péreza and S. A. Westcott, Dalton Trans., 2017, 46, 7750 RSC.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028 CrossRef CAS PubMed.
- C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190 CrossRef CAS PubMed.
- D. Mukherjee, H. Osseili, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2016, 138, 10790 CrossRef CAS PubMed.
- H. Osseili, D. Mukherjee, K. Beckerle, T. P. Spaniol and J. Okuda, Organometallics, 2017 DOI:10.1021/acs.organomet.7b00308.
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225 CrossRef CAS PubMed.
- V. K. Jakhar, M. K. Barman and S. Nembenna, Org. Lett., 2016, 18, 4710 CrossRef CAS PubMed.
- Y. Wu, C. Shan, Y. Sun, P. Chen, J. Ying, J. Zhu, L. Liu and Y. Zhao, Chem. Commun., 2016, 52, 13799 RSC.
- M. K. Bisai, S. Pahar, T. Das, K. Vanka and S. S. Sen, Dalton Trans., 2017, 46, 2420 RSC.
- S. Yadav, S. Pahar and S. S. Sen, Chem. Commun., 2017, 53, 4562 RSC.
- J. Schneider, C. P. Sindlinger, S. M. Freitag, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2017, 56, 333 CrossRef CAS PubMed.
- D. Wu, R. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Chem, 2017, 3, 134 CAS.
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017 10.1039/c7gc01632h.
- R. Rochat, M. J. Lopez, H. Tsurugi and K. Mashima, ChemCatChem, 2016, 8, 10 CrossRef CAS.
-
(a) M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 555 Search PubMed;
(b) C. Weetman, M. S. Hill and M. F. Mahon, Polyhedron, 2016, 103, 115 CrossRef CAS;
(c) M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. Kociok-Köhn, Chem. Commun., 2012, 48, 4567 RSC;
(d) M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Chem.–Eur. J., 2013, 19, 2776 CrossRef CAS PubMed;
(e) C. Weetman, M. S. Hill and M. F. Mahon, Chem. Commun., 2015, 51, 14477 RSC;
(f) C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628 RSC;
(g) C. Weetman, M. S. Hill and M. F. Mahon, Chem.–Eur. J., 2016, 22, 7158 CrossRef CAS PubMed;
(h) Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, B. Weetman and M. S. Hill, Chem. Sci., 2017, 8, 3529 RSC;
(i) M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009 CrossRef CAS PubMed;
(j) M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036 CrossRef CAS PubMed.
- J. Intemann, M. Lutz and S. Harder, Organometallics, 2014, 33, 5722 CrossRef CAS.
-
(a) D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959 RSC;
(b) N. L. Lampland, M. Hovey, D. Mukherjee and A. D. Sadow, ACS Catal., 2015, 5, 4219 CrossRef CAS.
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashimab and J. Okuda, Chem. Commun., 2016, 52, 13155 RSC.
- L. Fohlmeister and A. Stasch, Chem.–Eur. J., 2016, 22, 10235 CrossRef CAS PubMed.
- K. Manna, P. Ji, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2016, 138, 7488 CrossRef CAS PubMed.
- M. K. Barman, A. Baishya and S. Nembenna, Dalton Trans., 2017, 46, 4152 RSC.
- A. J. Boutland, D. Dange, A. Stasch, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2016, 55, 9239 CrossRef CAS PubMed.
- P. J. Bailey, C. M. E. Dick, S. Fabre and S. Parsons, J. Chem. Soc., Dalton Trans., 2000, 1655 RSC.
- M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed.
- J. F. Dunne, D. B. Fulton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 17680 CrossRef CAS PubMed.
- J. Li, A. Stasch, C. Schenk and C. Jones, Dalton Trans., 2011, 40, 10448 RSC.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362 CrossRef CAS PubMed.
- Y. W. Chao, P. A. Wexler and D. E. Wigley, Inorg. Chem., 1989, 28, 3860 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Jia Lia,
Xingchao Shena,
Zhijuan Yua,
Weiwei Yaob and
Sumod A. Pullarkat
*a,
Jia Lia,
Xingchao Shena,
Zhijuan Yua,
Weiwei Yaob and
Sumod A. Pullarkat