
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective detection of glufosinate using CuInS2 quantum dots as a fluorescence probe
Fengyuan Zhu a,
Jing Zhu*b and
Zhengzhu Zhang*a
a,
Jing Zhu*b and
Zhengzhu Zhang*a
aState Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei 230036, Anhui, People's Republic of China. E-mail: 2408337848@qq.com; zzz@ahau.edu.cn
bDepartment of Applied Chemistry, Anhui Agricultural University, Hefei 230036, Anhui, People's Republic of China. E-mail: zhujing@ahau.edu.cn
First published on 13th October 2017
Abstract
We designed a novel method for rapid detection of glufosinate, based on quantum dots (QDs) as a fluorescence probe. To date, no studies have been published on the detection of glufosinate using QDs. The innovation of this fluorescence system, which was constructed using CuInS2 QDs and Cu(IDA)·2H2O, is based on the use of intermolecular hydrogen bonds between Cu(IDA)·2H2O and carboxyl group (–COOH) to detect glufosinate. In the fluorescence “turn-on” step, the presence of glufosinate induces the release of Cu(IDA)·2H2O from the surface of the QDs, resulting in fluorescence intensity recovery. This method was performed in relatively clean aqueous solutions. Under optimal conditions, the calibration curve of the method showed good linearity for glufosinate, with a correlation coefficient (R2) of 0.99597. The method was employed to detect glufosinate on fresh tea-leaves, with satisfactory results.
Introduction
Herbicides have been widely used in agriculture; consequently, their pollutants have become a severe problem worldwide. In 1972, a German laboratory isolated an amino acid from one species of soil bacterium and named it phosphinothricin, which was later renamed glufosinate.1 Glufosinate, registered for use in more than 40 countries, is a broad-spectrum and post-emergence organophosphorus herbicide that is applied in various applications involving annual and perennial dicotyledonous weeds. Its residues can accumulate in environmental samples and destroy the natural equilibria among soil microorganisms. Since the entry of genetically engineered herbicide-resistant crops into the market, the use of glufosinate has increased drastically. Consequently, glufosinate's lethal toxicity, teratogenicity, and potential reproductive toxicity have attracted increased research attention. Recent studies have shown that glufosinate can cause oxidative damage to human DNA.2–5 In addition, glufosinate can lead to decreased concentrations of glutamine in cerebrospinal fluid.6 With the increase in glufosinate usage, especially in tea growing areas, the monitoring of residues has attracted more and more interest.To date, several methods have been developed to detect herbicides. However, the target samples must be extracted and purified before the herbicides can be detected using high performance liquid chromatography (HPLC),7–11 gas chromatography (GC),12–16 capillary electrophoresis,17–21 ion chromatography22,23 or enzyme-linked immunosorbent assays.24–26 Druart11 used HPLC on a 300 mm C18 column to detected glufosinate and the detection limits were in 0.015 to 0.13 ppm. After protein precipitation using acetonitrile and solid-phase extraction, serum samples were derivatized and analyzed by GC-MS (gas chromatography-mass spectroscopy), Tseng12 detected glufosinate in a concentration range of 3–100 ppm. To avoid and minimize potential interfering substances, most preferred analytical systems generally require expensive instrumentation, complicated sample pretreatments, and purification procedures to remove polysaccharides, protein, and peptides through precipitation, filtration, or centrifugation. Analyzing the sample and spectral data are also time-consuming. For simplicity, photometry using a diode array detector, fluorescence detection, and MS27–30 have also been adopted. Generally, glufosinate's lack of chromophores means that it must be derivatized before fluorescence detection, mostly using fluorenylmethoxycarbonyl chloride (FMOC-Cl) to react with the amino group as a fluorogenic reagent. This process is usually completed overnight, before GC analysis. Glufosinate has many unusual physical–chemical characteristics, such as high solubility in water (1370 g L−1), insolubility in organic solvents, low volatility, and high polarity; these characteristics make it difficult to establish methods for the analysis of glufosinate at trace levels. Therefore, it is necessary to develop fast, simple, and cost-effective methods to detect glufosinate.
To this end, quantum dots (QDs)—semiconductor nanoparticles confined to 1–10 nm in three dimensions—are ideal nanomaterials to detect pesticides because of their unique optical properties. Ternary I–III–VI CuInS2 QDs have been intensively investigated for their potential to replace commonly available colloidal nanocrystals containing toxic elements in light-emitting and solar-harvesting applications. This is because of their pronounced quantum confinement effect, stable photoluminescence, and the absence of toxic elements (such as Cd, Hg or Pb).31–33 In particular, several pesticides have been detected using QD sensor systems. Some of these experiments required the use of acetylcholinesterase (AChE) enzymatic reaction for the specific selection of dichlorvos,34,35 paraoxon,36 and methyl parathion.37 Other studies used the specificity of organophosphorus hydrolase (OPH) to detect parathion,38 and parathion-methyl.39 Specific antibodies or aptamers have also been used to identify paraquat,40 and acetamiprid.41 These substances increase the costs and affect device stability and practicability. Currently, most studies have focused on the fluorescence “off” patterns; however, these sensors have low sensitivity. Therefore, new “switch-on” models, with high sensitivity and selectivity for pesticides detection, are becoming more popular. These models use surface-modified reagents to confer variable affinity and specificity of QDs towards different targets. However, no studies have yet reported a sensor system based on QDs for the highly specificity and sensitive detection of glufosinate.
In the present work, we developed an ecologically-friendly, facile, and sensitive fluorescence sensor for glufosinate detection. This fluorescence system was constructed using CuInS2 QDs and the metal–chelate, Cu(IDA)·2H2O. The schematic illustration of the measurement principle is shown in Fig. 1. L-Cysteine was modified on the surface of QDs by coordination between sulfhydryl and the metal. The amino groups (–NH2) of the surface-modified substance L-cysteine can interact with H2O molecules via hydrogen bonding, which leads to quenching of the fluorescence intensity of CuInS2 QDs.42 The fluorescence intensity of the CuInS2 QDs was quenched when Cu(IDA)·2H2O attached to the QDs through coordination with the surface-modified substance L-cysteine via hydrogen bonding between –NH2 and H2O. Glufosinate has a stronger coordinative interaction with Cu(IDA)·2H2O than L-cysteine because the hydrophilicity of –COOH is stronger than that of –NH2. In fluorescence “turn-on,” after the addition of glufosinate, Cu(IDA)·2H2O is released from the surface of QDs because intermolecular hydrogen bonds form between Cu(IDA)·2H2O and the –COOH of glufosinate. Subsequently, the fluorescence intensity recovers. The proposed method, which mostly used relatively clean aqueous solutions, was applied to detect glufosinate in fresh tea-leaves.
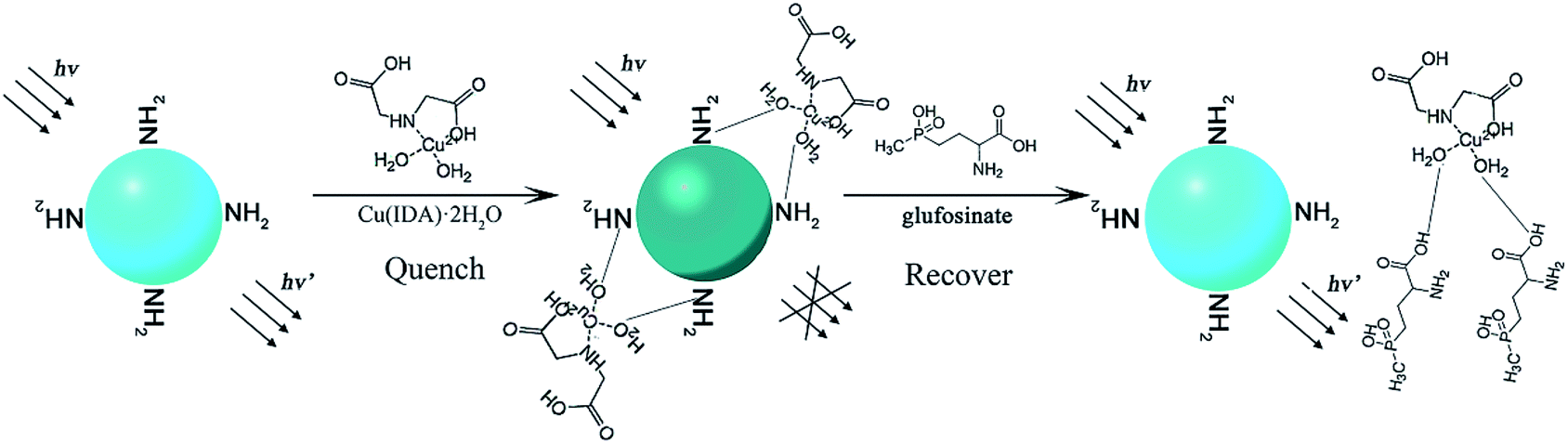 | ||
| Fig. 1 Schematic illustration of the measurement principle of CuInS2 QDs-Cu(IDA)·2H2O sensor for glufosinate detection. | ||
Experimental
Materials and devices
All chemicals and reagents used in this work were of analytical grade. Sodium hydrate (NaOH), potassium hydrate (KOH), copper(II) chloride dehydrate (CuCl2·2H2O), copper(II) hydroxide (Cu(OH)2), sulfourea (CS(NH2)2), and indium(III) chloride tetrahydrate (InCl3·4H2O) were purchased from Sinopharm Chemical Reagent Co. Ltd. Ethanol was purchased from Wuxi Zhanwang Chemical Works. The resistivity of the test water was greater than 18 MΩ cm−1. L-Cysteine (Cys) was purchased from Beijing Solarbio Science & Technology Co. Ltd. Iminodiacetic acid (IDA) was purchased from Shanghai Ryon Biological Technology Co. Ltd. L-Serine (Ser) was purchased from Sigma-Aldrich Corporation. Glufosinate and (aminomethyl)phosphonic acid were obtained from Dr Ehrenstorfer (Reference Materials for Residue Analysis). Fluorescence measurements were performed on a Carry Eclipse Fluorescence Spectrophotometer (Agilent Technologies) with a quartz cuvette path-length of 1 cm. The pH was measured using by an FE20 FiveEasy Plus pH meter (Mettler-Toledo). UV-Vis absorption spectra were recorded on a PerkinElmer UV/Vis Spectrometer Lambda 35. The centrifuge used was an Eppendorf D3024R.Syntheses of CuInS2 QDs
Hydrothermal synthesis has become a hot research topic because of its convenience and simplicity. We followed published reports42 to synthetize CuInS2 QDs modified using L-cysteine. First, L-cysteine (3.60 mmol) was added to ultrapure water (7.5 mL) and mixed to form a clear solution. Then, CuCl2·2H2O (0.15 mmol) was added to the solution with stirring for 5 min. Next, InCl3·4H2O (0.15 mmol) was added to the solution with stirring for 10 min, and the pH value was adjusted to 11.3 using an NaOH solution (4 mol L−1). Ten minutes later, CS(NH2)2 (0.30 mmol) was mixed in. Finally, the fully stirred solution was transferred into a Teflon-lined stainless steel autoclave (15 mL) and maintained at 150 °C. After 23 h, the autoclave was cooled to room temperature. The solution was purified by adding ethanol and centrifuging, and the centrifugal precipitation products were dried to obtain the CuInS2 QDs powder. The powder was dissolved in ultrapure water to obtain the CuInS2 QDs solution (182.3 μmol L−1) and stored it at 4 °C for further research.Synthesis of Cu(IDA)·2H2O
Cu(IDA)·2H2O was synthetized in an aqueous solution. First, Cu(OH)2 (0.25 mol) and IDA (0.25 mol) were successively dissolved in 200 mL of distilled water with stirring for 60 min. The Cu(OH)2 precipitates were dissolved, and the solution became light blue after the addition of IDA. The solution was evaporated, crystallized, and dried in a vacuum to obtain a blue powder. The powder was then dissolved in distilled water at a concentration of 1 mmol L−1 and stored for further experimentation.Fluorescence quenching by Cu(IDA)·2H2O
For the fluorescence quenching experiment, 100 μL of CuInS2 QDs solution and different amounts of Cu(IDA)·2H2O, without buffer solutions, were mixed in calibrated test tubes, and the mixed solution was diluted to the marker of 4 mL with ultrapure water. Ultrasonic concussion was used to thoroughly shake the test tube, and water was circulated to keep the temperature constant. Fluorescence was then measured using the Carry Eclipse Fluorescence Spectrophotometer.Detection of glufosinate
Different concentrations of glufosinate were prepared by diluting a glufosinate sample (1000 ppm) in ultrapure water and shaking thoroughly. Then, 100 μL of CuInS2 QD solution, 300 μL of Cu(IDA)·2H2O, and different concentrations of glufosinate solution (1–140 ppm) were admixed separately in calibrated test tubes, and ultrapure water was used to dilute to the marker of 4 mL. Subsequently, the mixtures were shaken thoroughly for 20 min using ultrasonic concussion before detection using the Carry Eclipse Fluorescence Spectrophotometer.Real sample detection
The proposed method was used to detect glufosinate on the surface of fresh tea leaves. The leaves were cut off from a tea plant and cleaned with ultrapure water, and then glufosinate solutions of various concentrations were placed on the leaves and allowed to dry in a natural environment. The leaves were then washed using ultrapure water and the solution collected for analysis.Results and discussion
Choosing an intermediate
The fluorescence and absorption spectra of the CuInS2 QDs are obtained and shown in Fig. 2(a). The photoluminescence quantum yield we measured is 19.6%.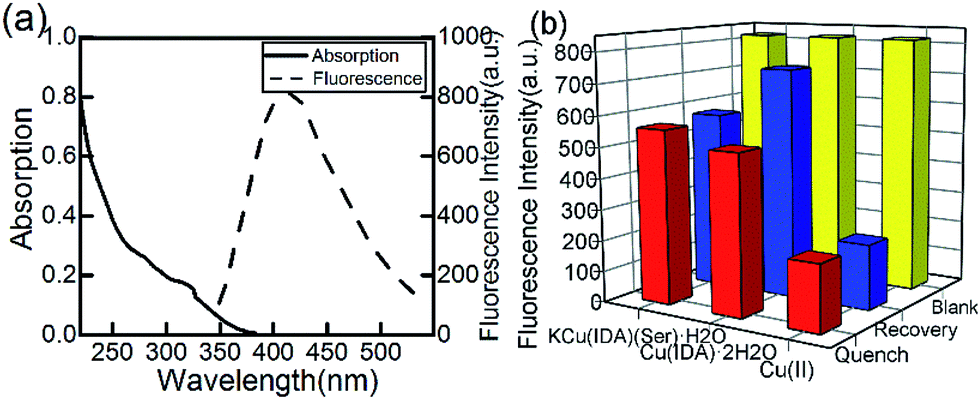 | ||
| Fig. 2 (a) Absorption and fluorescence spectra of CuInS2 QDs. (b) CuInS2 QDs quenched by intermediates and recovered by adding glufosinate. | ||
Previous studies have reported the special binding capacity of Cu(II)43 and the metal–chelate Cu-IDA.44 We synthesized another substance, KCu(IDA)(Ser)·H2O using serine, according to the main toxicity of organophosphorus pesticides resulted from covalent binding to a serine OH group. In this study, we compared the selectivity of Cu(II), Cu(IDA)·2H2O, and KCu(IDA)(Ser)·H2O to glufosinate.
Cu(IDA)·2H2O had a stronger and more sensitive coordinated interaction with glufosinate compared with the other substances (Fig. 2(b)). The fluorescence intensity of CuInS2 QDs decreased to varying degrees with the addition of 100 μmol L−1 of Cu(II), Cu(IDA)·2H2O, and KCu(IDA)(Ser)·H2O, respectively. After the addition of glufosinate, the fluorescence intensity of the CuInS2 QDs-Cu(IDA)·2H2O system recovered to a higher level than the other substances. Therefore, Cu(IDA)·2H2O had a suitable affinity for glufosinate in this study.
Quenching effect of Cu(IDA)·2H2O on CuInS2
Considering the adsorption and stability of Cu(IDA)·2H2O, ultrapure water was used in this work to formulate the solution and dilute to the marker in all experiment. We did not add any buffer solutions or other expensive biomolecular reagents. All the required substances are easily synthesized and do not harm the environment. Yan's research39 showed that in an acid environment, the structure of CuInS2 QDs would be destroyed, and in an alkaline environment, Cu(OH)2 would precipitate. Therefore, ultrapure water, whose pH value has not been adjusted, was the most suitable choice to perform the experiments conveniently and efficiently.Cu(IDA)·2H2O can compete with L-cysteine modified on QDs. It changed the photophysical properties of the QDs, resulting in the fluorescence intensity being quenched. The fluorescence intensity of CuInS2 QDs at different concentrations of Cu(IDA)·2H2O (0, 25, 50, 75, 100, 125, 150, 175 μmol L−1) are shown in Fig. 3. The decreasing trend began to ease when the concentration was greater than 75 μmol L−1, indicating that Cu(IDA)·2H2O gradually reached saturation. Therefore, we chose the working concentration of Cu(IDA)·2H2O as 75 μmol L−1.
 | ||
| Fig. 3 The fluorescence spectra upon the addition of Cu(IDA)·2H2O (0 to 175 μmol L−1). | ||
Selectivity of the CuInS2 QDs-Cu(IDA)·2H2O system
(Aminomethyl)phosphonic acid (AMPA), the major metabolite of glyphosate degradation under biological conditions, and dimethoate (DIM) a moderately toxic organophosphorus pesticide, were chosen to test the selectivity of the designed method. The fluorescence intensity of the CuInS2 QDs-Cu(IDA)·2H2O system did not show a good linear correlation with the AMPA and dimethoate concentration (Fig. 4(a and b)). The chemical structures of these three substances are shown in Fig. 4(c). These compounds share structures involving an amino group and phosphate group, but not a carboxyl group. Thus, the carboxyl group on glufosinate may be the selective group. A review of relevant literature45 revealed that the metal–chelate Cu-IDA is always applied in the biomolecular field to combine protein and amino acids because of strong affinity of the metal–chelate Cu-IDA for proteins and amino acids. The Cu(II) complex Cu(IDA)·2H2O is an independent substance that can be synthesized without involving other elements or ions. The H2O molecule on the axial copper site can form hydrogen bonds with the oxygen atoms of the carboxyl groups.46 The presence of intermolecular and intramolecular hydrogen bonds increased the stability of the material's morphology. Consequently, glufosinate, with a carboxyl group, was selectively detected by the CuInS2 QDs-Cu(IDA)·2H2O system. The H2O molecule and carboxyl group formed intermolecular hydrogen bonds between the oxygen atoms of water molecules and the oxygen atoms of the carboxyl groups. Compared with the recovery effect of the KCu(IDA)(Ser)·2H2O complex shown in Fig. 2(b), the structure of the KCu(IDA)(Ser)·2H2O affected the affinity of H2O molecules. Hence, IDA has an influential role in stimulating the affinity of H2O molecules. | ||
| Fig. 4 F1/F0 of CuInS2 QDs-Cu(IDA)·2H2O system upon the addition of (a) (aminomethyl)phosphonic acid and (b) dimethoate. (c) Chemical structure of glufosinate, dimethoate and, (aminomethyl)phosphonic acid. | ||
Although AMPA and DIM, could also cause slight changes to the system, their groups have very weak coordinative interaction with the QDs. Therefore, the addition of AMPA, and DIM caused little change in the intensity of the CuInS2 QDs-Cu(IDA)·2H2O system. Thus, the selectivity of the proposed method for glufosinate detection was confirmed using the two pesticides mentioned above.
Detection of glufosinate
The effect of reaction time on the fluorescence intensity of the CuInS2 QDs-Cu(IDA)·2H2O system after the addition of glufosinate is shown in Fig. 5(a), all the data were averaged after 5 measurements. The recovery effect of glufosinate appears stable in 20 min, and the fluorescence intensity remained stable for more than 50 min. Fig. 5(b) shows the fluorescence spectra of the CuInS2 QDs-Cu(IDA)·2H2O system upon addition of different concentrations of glufosinate. The fluorescence intensity of the CuInS2 QDs-Cu(IDA)·2H2O system recovered gradually with increasing concentrations of glufosinate. Furthermore, there was good linearity between the recovered fluorescence and glufosinate concentrations between 1 and 25 ppm (i.e., 4.65–116.18 μmol L−1). The calibration curve was F1/F0 = 1.00953 + 0.00825C[glufosinate] ppm (where F1 and F0 are the fluorescence intensity in the presence and absence of glufosinate, respectively). The corresponding regression coefficient was 0.99597 and the lower limit of detection (LOD) for glufosinate was 0.01 ppm. The detection limit is defined by the equation LOD = 3σ/s, where σ is the standard deviation of the corrected blank signals of the CuInS2 QDs-Cu(IDA)·2H2O system and s is the slope of the calibration curve. In the 30–120 ppm range (i.e., 139–464.71 μmol L−1), the calibration curve was F1/F0 = 1.16951 + 0.00188C[glufosinate] ppm and R2 = 0.98934. The reason for generating two detection ranges is that glufosinate possesses a strong affinity towards QDs, and it can create more radiative centers and stimulate blocking of nonradiative electron–hole recombination defect sites on the surface of the QDs.47 This combined action produced the first calibration curve. With the increase of glufosinate, this status reached saturation, and the second calibration curve appeared. The results showed that the CuInS2 QDs-Cu(IDA)·2H2O system can detect glufosinate in a relatively clean aqueous solution. | ||
| Fig. 5 (a) The effect of reaction time after the addition of glufosinate. (b) The effect of different concentrations of glufosinate on the fluorescence maxima. (c) The effect of different concentrations of glufosinate on total fluorescence. | ||
Most previous studies of the detection of glufosinate were based on HPLC, GC, and MS, which require derivatization. Other methods for detecting pesticides and herbicides required the corresponding antibodies, bi-enzymes, or electrochemical potential. The proposed method is more sensitive than previously reported methods, whose detection limits were in the range of 3–100 ppm used GC-MS12 or the LOD of 0.017 ppm based on antibodies.26 Our method is appreciably simpler and faster than the others, lacking steps of derivatization, antibodies, or specific conditions.
Detection of glufosinate on tea leaves
When applied to plants, glufosinate is absorbed through the leaf surface, and almost no absorption occurs through the roots or stems. Therefore, the proposed method of detecting pesticides on the surface of tea-leaves was potentially significant for practical detection. We selected several concentrations (5, 10, 40, 60, 80 ppm) to carry out the experiment. The results of the sample detection were 4.99, 10.00, 34.71, 60.80, and 78.60 ppm. These results suggested the possibility of actual detection of glufosinate on leaves.Conclusions
We designed a rapid method to detect glufosinate in pure aqueous solution, based on a CuInS2 QDs-Cu(IDA)·2H2O fluorescence sensor. No studies have yet reported on the detection of glufosinate using QDs. The H2O molecule on the axial copper site can form hydrogen bonds with –NH2 and –COOH. The hydrophilicity of –COOH is stronger than that of –NH2. Consequently, glufosinate, with –COOH, led to the recovery of the system's fluorescence intensity after it had been quenched by the Cu(IDA)·2H2O complex. Under optimum conditions, this sensor exhibited good linearity and practical range without requiring expensive antibodies, or complicated surface modification procedures. However, our method faces many challenges in the detection of complex samples, such as residues in plant-based foods with many interfering substances. Overall, this work provided a new strategy for detecting glufosinate herbicide residues in relatively clean aqueous solutions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Program for Changjiang Scholars and Innovative Research Team in University, no. IRT1101, the National Natural Science Foundation of China (NSFC, 51702004), the Scientific Research Foundation of Anhui Agricultural University (Grant No. yj2017-06), and the Youth Research Foundation of Anhui Agricultural University (2016ZR003).Notes and references
- G. Hoerlein, Rev. Environ. Contam. Toxicol., 1994, 138, 73–145 CAS.
- M. Koureas, A. Tsezou, A. Tsakalof, T. Orfanidou and C. Hadjichristodoulou, Sci. Total Environ., 2014, 496, 358–364 CrossRef CAS PubMed.
- L. Hoffman and D. Hardej, Environ. Toxicol. Pharmacol., 2012, 34, 556–573 CrossRef CAS PubMed.
- R. Paro, G. M. Tiboni, R. Buccione, G. Rossi, V. Cellini, R. Canipari and S. Cecconi, Toxicol. Appl. Pharmacol., 2012, 260, 155–161 CrossRef CAS PubMed.
- A. K. Srivastava, W. Ali, R. Singh, K. Bhui, S. Tyagi, A. A. Al-Khedhairy, P. K. Srivastava, J. Musarrat and Y. Shukla, Life Sci., 2012, 90, 815–824 CrossRef CAS PubMed.
- T. Ohtake, H. Yasuda, H. Takahashi, T. Goto, K. Suzuki, K. Yonemura and A. Hishida, Hum. Exp. Toxicol., 2001, 20, 429–434 CAS.
- G. R. Todorovic, A. Mentler, M. Popp, S. Hann, G. Kollensperger, N. Rampazzo and W. E. H. Blum, Soil Sediment Contam., 2013, 22, 332–350 CrossRef CAS.
- N. Ngaosi, K. Seebunrueng and S. Srijaranai, Anal. Methods, 2016, 8, 4254–4262 RSC.
- L. J. Liu, L. A. Xia, C. Y. Guo, C. X. Wu, G. Chen, G. L. Li, Z. W. Sun and J. M. You, Anal. Methods, 2016, 8, 3536–3544 RSC.
- M. P. Yurkova, I. P. Pozdnyakov, V. F. Plyusnin, V. P. Grivin, N. M. Bazhin, A. I. Kruppa and T. A. Maksimova, Photochem. Photobiol. Sci., 2013, 12, 684–689 CAS.
- C. Druart, O. Delhomme, A. de Vaufleury, E. Ntcho and M. Millet, Anal. Bioanal. Chem., 2011, 399, 1725–1732 CrossRef CAS PubMed.
- S. H. Tseng, Y. W. Lo, P. C. Chang, S. S. Chou and H. M. Chang, J. Agric. Food Chem., 2004, 52, 4057–4063 CrossRef CAS PubMed.
- M. Motojyuku, T. Saito, K. Akieda, H. Otsuka, I. Yamamoto and S. Inokuchi, J. Chromatogr. B, 2008, 875, 509–514 CrossRef CAS PubMed.
- E. Ueno, J. Pestic. Sci., 2011, 36, 554–558 CrossRef CAS.
- G. F. Xia, X. Y. Fang, Y. R. Wang and X. Y. Yang, Anal. Lett., 2017, 50, 787–796 CrossRef CAS.
- P. Moreira, P. G. Pinho, M. T. Baltazar, M. L. Bastos, F. Carvalho and R. J. Dinis-Oliveira, Toxicol. Lett., 2011, 205, S198 CrossRef.
- E. Orejuela and M. Silva, Electrophoresis, 2005, 26, 4478–4485 CrossRef CAS PubMed.
- S. Y. Chang and C. H. Liao, J. Chromatogr. A, 2002, 959, 309–315 CrossRef CAS PubMed.
- R. Lanaro, J. L. Costa, L. C. R. Fernandes, R. R. Resende and M. F. M. Tavares, J. Anal. Toxicol., 2011, 35, 274–279 CrossRef CAS PubMed.
- M. Chicharro, E. Bermejo, S. Ongay and A. Zapardiel, Electroanalysis, 2008, 20, 534–541 CrossRef CAS.
- K. Islam, S. K. Jha, R. Chand, D. Han and Y. S. Kim, Microelectron. Eng., 2012, 97, 391–395 CrossRef CAS.
- K. C. Wang, S. M. Chen, J. F. Hsu, S. G. Cheng and C. K. Lee, J. Chromatogr. B, 2008, 876, 211–218 CrossRef CAS PubMed.
- H. Kidwell, J. J. Jones and D. E. Games, Rapid Commun. Mass Spectrom., 2001, 15, 1181–1186 CrossRef CAS PubMed.
- Z. Q. Lv, C. H. Wang, T. T. Wang, C. C. Chen, Y. Wang, B. A. Ning, M. Liu, J. Q. Liu, J. L. Bai, Y. Peng and Z. X. Gao, Biomed. Environ. Sci., 2013, 26, 398–402 Search PubMed.
- A. Abad-Fuentes, F. A. Esteve-Turrillas, C. Agullo, A. Abad-Somovilla and J. V. Mercader, Food Chem., 2012, 135, 276–284 CrossRef CAS.
- M. G. Weller, M. Diemer, C. Wersching, R. Niessner and H. Sochor, J. Agric. Food Chem., 2003, 51, 6668–6675 CrossRef CAS PubMed.
- L. Goodwin, J. R. Startin, B. J. Keely and D. M. Goodall, J. Chromatogr. A, 2003, 1004, 107–119 CrossRef CAS PubMed.
- M. Corbera, B. M. Simonet, V. Salvado and M. Hidalgo, Rapid Commun. Mass Spectrom., 2010, 24, 2931–2937 CrossRef CAS PubMed.
- X. F. Dong, S. X. Liang, Z. H. Shi and H. W. Sun, Food Chem., 2016, 192, 432–440 CrossRef CAS PubMed.
- J. Fenoll, P. Hellin, P. Sabater, P. Flores and S. Navarro, Talanta, 2012, 101, 273–282 CrossRef CAS PubMed.
- H. Z. Zhong, Z. L. Bai and B. S. Zou, J. Phys. Chem. Lett., 2012, 3, 3167–3175 CrossRef CAS PubMed.
- T. L. Li, Y. L. Lee and H. Teng, Energy Environ. Sci., 2012, 5, 5315–5324 CAS.
- M. Michalska, A. Florczak, H. Dams-Kozlowska, J. Gapinski, S. Jurga and R. Schneider, Acta Biomater., 2016, 35, 293–304 CrossRef CAS PubMed.
- X. W. Meng, J. F. Wei, X. L. Ren, J. Ren and F. Q. Tang, Biosens. Bioelectron., 2013, 47, 402–407 CrossRef CAS PubMed.
- Z. Z. Zheng, X. Y. Li, Z. F. Dai, S. Q. Liu and Z. Y. Tang, J. Mater. Chem., 2011, 21, 16955–16962 RSC.
- Z. Z. Zheng, Y. L. Zhou, X. Y. Li, S. Q. Liu and Z. Y. Tang, Biosens. Bioelectron., 2011, 26, 3081–3085 CrossRef CAS PubMed.
- H. Liang, D. D. Song and J. M. Gong, Biosens. Bioelectron., 2014, 53, 363–369 CrossRef CAS PubMed.
- D. Du, W. J. Chen, W. Y. Zhang, D. L. Liu, H. B. Li and Y. H. Lin, Biosens. Bioelectron., 2010, 25, 1370–1375 CrossRef CAS PubMed.
- X. Yan, H. X. Li, Y. Yan and X. G. Su, Food Chem., 2015, 173, 179–184 CrossRef CAS PubMed.
- E. Valera, R. Garcia-Febrero, M. I. Pividori, F. Sanchez-Baeza and M. P. Marco, Sens. Actuators, B, 2014, 194, 353–360 CrossRef CAS.
- J. J. Guo, Y. Li, L. K. Wang, J. Y. Xu, Y. J. Huang, Y. L. Luo, F. Shen, C. Y. Sun and R. Z. Meng, Anal. Bioanal. Chem., 2016, 408, 557–566 CrossRef CAS PubMed.
- Z. P. Liu, Q. Ma, X. Y. Wang, Z. H. Lin, H. Zhang, L. L. Liu and X. G. Su, Biosens. Bioelectron., 2014, 54, 617–622 CrossRef CAS PubMed.
- Z. Q. Liu, S. P. Liu, P. F. Yin and Y. Q. He, Anal. Chim. Acta, 2012, 745, 78–84 CrossRef CAS PubMed.
- Z. el Rassi and C. Horvath, J. Chromatogr., 1986, 359, 241–253 CrossRef CAS PubMed.
- G. C. Serafica, G. Belfort and J. Pimbley, Biotechnol. Bioeng., 1994, 43, 21–36 CrossRef CAS PubMed.
- M. Tribet, B. Covelo, D. Choquesillo-Lazarte, J. M. Gonzalez-Perez, A. Castineiras and J. Niclos-Gutierrez, Inorg. Chem. Commun., 2003, 6, 343–345 CrossRef CAS.
- T. Khantaw, C. Boonmee, T. Tuntulani and W. Ngeontae, Talanta, 2013, 115, 849–856 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |