
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Template growth of Au, Ni and Ni–Au nanoclusters on hexagonal boron nitride/Rh(111): a combined STM, TPD and AES study†
Fanglue Wu *,
Dali Huang,
Yuan Yue and
Li Liu
*,
Dali Huang,
Yuan Yue and
Li Liu
Department of Materials Science and Engineering, Texas A&M University, College Station, TX 77843-3003, USA. E-mail: flwu@tamu.edu; Tel: +1-979-255-8907
First published on 12th September 2017
Abstract
The template growth of Au, Ni, and Ni–Au bimetallic nanoclusters on hexagonal boron nitride/Rh(111), i.e. h-BN/Rh(111), was investigated via scanning tunneling microscopy (STM), temperature programmed-desorption (TPD), and Auger electron spectroscopy (AES). STM study shows that template growth of Au clusters on h-BN/Rh(111) forms mainly well-dispersed monolayer clusters. In contrast, Ni forms large multilayer clusters showing a relatively high diffusivity on h-BN/Rh(111) substrate. Ni–Au bimetallic clusters are effectively formed first by Au deposition followed by Ni deposition, with the Au clusters functioning as nucleation sites for the subsequently deposited Ni. Further structural analysis was carried out via TPD and AES. The resulting TPD and AES data show the surface composition and charge transfer between Au and Ni of the bimetallic clusters. These results suggest that the h-BN/Rh(111) substrate represents a unique candidate for supporting Ni–Au bimetallic clusters in further catalytic reactions.
1. Introduction
It has been well established that both Au and Ni clusters show strong activities for a great variety of catalytic reactions. For example, Au clusters exhibit remarkable activity in low-temperature CO oxidation and hydrogenation.1,2 In particular, Au clusters of bilayer structure supported on titania have shown significantly higher activity in the catalytic oxidation of carbon monoxide,1,2 and in 1973, Bond et al.2,3 reported superior hydrogenation of olefins using these Au clusters compared to the other catalysts. In addition, the formation of butene from 1-butyne on Au films without any extra hydrogen has also been reported by Gault.4 Although the mechanism of the enhanced activity of gold nanoclusters is not fully understood, it has been discussed in detail in several review articles.5–9 On the other hand, Ni clusters demonstrate unique catalytic properties in steam and CO2 reforming,10,11 in which different Ni catalysts (on different substrates: MgO, ZnO, Al2O3 et al.) have been extensively studied. While Ni has been proven to be a common catalyst for steam reforming, graphite is also generated as a byproduct in these reactions and further poisons the Ni activity.12,13 Although it is widely known that bimetallic metals can display activities different from monometallic metals and many examples from literature have shown the promotional effects of the second metal in catalysis,14–20 there are only a few studies focused on the Ni–Au catalysts.21,22 The main reason is that Ni–Au bulk phase diagram has a big miscibility gap and therefore the formation of Ni–Au alloy becomes difficult.23,24 However, Ni–Au system remains interesting for study because Au and Ni can form surface alloys when Au is deposited on Ni surfaces.25,26 Such a surface alloy structure makes Ni–Au more resistant to carbon formation, which improves the catalytic stability in steam reforming reactions compared to that of pure Ni catalysts.10,13,26,27 Ni–Au catalysts have also demonstrated activity in low temperature CO oxidation28–30 as well as partial oxidation of methane.31In addition, other factors also influence the catalytic activities of metal clusters such as size and shape of the nanoclusters1,32–36 as well as the support effects.36 Defects in the support promote nucleation and cluster growth, and charge transfer between them has often been observed.37–41 All these results indicate that the support can never be eliminated for its effect on the morphology (size, shape and spatial distribution), electronic structure and other properties of metal clusters.42
One of the newest possibilities to control the size, shape, and spatial distribution of metal clusters is by applying template substrates. Hexagonal boron nitride (h-BN) thin films grown on transition metal surfaces have shown an exceptional potential in this regard.43–47 First, the lattice mismatch between the metal surfaces and the h-BN results in a highly corrugated superstructure. This provides regular dense arrays of defect sites for further clusters formation,44,48 for instance, the growth of h-BN on Rh(111) prepared by pyrolysis method. The lattice mismatch is around 7.6%, and the self-assembled h-BN monolayers on Rh(111) surface form the so-called “pore-wire” structure with an overall periodicity of 3.2 nm.49,50 The electronic structure of the pore parts are distinct from the wire parts, and therefore, provide a template environment for nucleation and growth of transition metals. In addition, the thin films are facile to prepare. They offer thermal stability and chemical inertness toward various environments such as ambient air or perchloric acid.51 Furthermore, h-BN films are oxygen free and can help to eliminate uncertainties when characterizing the catalytic activities in CO oxidation reactions.8,52,53
Several groups have reported the formation of Au clusters on h-BN/Rh(111).54–56 Investigation of Ni and Ni–Au clusters supported on h-BN/Rh(111), however, has not yet been investigated, according to our literature research. Herein, we demonstrated our latest results on the template growth of Ni, Au and Ni–Au clusters on h-BN/Rh(111) using STM, TPD and AES. The morphology, surface composition as well as the electronic properties of the nanoclusters were studied.
2. Experimental
STM and TPD experiments were carried out in two different ultrahigh vacuum (UHV) chambers. Each chamber is equipped with an Auger electron spectroscopy (AES) system and a low energy electron diffraction (LEED) optic to guarantee the consistency of sample preparation processes between the two chambers.2.1. Sample preparation
Rh(111) single crystal (purity 99.99%, dia. 10.00 mm × thickness 1.00 mm, orientation accuracy <0.1 deg.) was purchased from Princeton Scientific Corporation. A clean and ordered surface was obtained by repeated cycles of Ar+ sputtering and e-beam annealing to 1200 K. Surface condition was monitored by LEED and AES. Upon obtaining a clean and well-ordered Rh(111) surface, a monolayer of hexagonal boron nitride nanomesh was prepared by exposure to 80–120 Langmuir borazine vapor (liquid borazine was kept in a glass finger tube attached to a leak valve, purified before each experiment by several cycles of freeze–pump–thaw) with the sample held at 1050 K.57,58 This procedure produces a clean, well-ordered and complete h-BN film as monitored by STM, TPD, AES, and LEED. Au was deposited from a high-purity Au pellet heated up by passing current through an inner tantalum wire, and Ni was evaporated onto the sample from Ni wire wrapped around a resistively-heated tungsten wire.59 Both Au and Ni dosers were degassed thoroughly before deposition to remove any impurities and contaminants. The deposition rates for Au and Ni dosers were both calibrated using the Auger breaking point method in two chambers.60 All the depositions were carried out with the substrate at room temperature.2.2. Scanning tunneling microscopy
STM experiments were performed in an UHV chamber with a base pressure of 1.0 × 10−10 Torr. Constant-current mode was applied using homemade tungsten tips etched via an electrochemical method. The scanning parameters were reported with reference to the sample. Usually these scanning conditions resulted in dark “pores” and bright “wires”.49,57 However, a contrast reversal was often seen during our experiments with dark “wires” and bright “pores”, which was caused by adsorbed species on tips, which changed the imaging orbitals.46,61 Nevertheless, the imaging and final data analysis, including lateral size and height measurements would not be affected.2.3. Temperature programmed desorption and Auger electron spectroscopy
TPD experiments were conducted in an UHV chamber with a base pressure of 5.0 × 10−10 Torr. This chamber contains an AES (Physical Electronics Φ 15–155) system, a sputtering gun, a LEED (PRI 11-020) instrument and a quadrupole mass spectrometer (QMS, UTI-100C). The sample temperature was measured by a thermocouple (C-type) spot-welded to the sample. TPD experiments were carried out by first preparing h-BN nanomesh on Rh(111) and then vapor-depositing metals onto the sample surface with the substrate at room temperature. Next, the sample was cooled to liquid N2 temperature and then exposed to CO dosage of 30 Langmuir to ensure full saturation on metal clusters. Finally after CO adsorption, the sample was placed accurately in front of the QMS,62 and the desorption was recorded with a linear temperature ramping rate at 3.0 K s−1. The AES experiments were conducted in dN/dE mode using a single pass cylindrical mirror analyzer. The electron beam energy was set at 1.5 keV with a step voltage of 1 V. AES data were recorded every time after the sample preparation and before the TPD experiments at room temperature.3. Results and discussion
3.1. Scanning tunneling microscopy
The growth of Au, Ni, and Au–Ni bimetallic nanoclusters was investigated by STM. Fig. 1 shows a comparison of different growth modes of 0.1 monolayer (ML) pure Au (Fig. 1a) and 0.1 ML pure Ni (Fig. 1b) on h-BN/Rh(111) surface at room temperature. Fig. 1c indicates the height profile of the Au nanoclusters on h-BN/Rh(111) in Fig. 1a. The clusters with 1.5 ± 0.1 Å in height correspond to a single layer of Au (relative to the bottom of a pore), while the clusters with 3.0 ± 0.1 Å in height correspond to a bilayer of Au.46 Histograms of 0.1 ML Au on h-BN/Rh(111) in Fig. 2 show that the average diameter of Au clusters is approximately 1.75 ± 0.25 nm. Most of the Au clusters (∼94%) are single layer (0.15 nm), and the rest are bilayer clusters (∼6%). Both their nucleation and growth are confined to the nanomesh pores. Further 0.05 ML deposition coverage shows that all Au clusters form single layer clusters confined to the template pores (ESI, Fig. S1†). Unlike the growth mode of Au clusters on BN/Rh(111), the deposition of 0.1 ML Ni results in clusters with an average size of 3.92 ± 0.4 nm in diameter and 1.69 ± 0.2 nm in height (Fig. 2b and d), which are much larger compared to Au clusters. In addition, the cluster density of Au is 10.6 times greater than that of Ni (1.7 × 1012 clusters cm−2 for Au vs. 1.6 × 1011 clusters cm−2 for Ni). Both the smaller cluster size and higher cluster density of Au indicate a lower diffusion constant of Au atoms on BN/Rh(111) surface compared with Ni. This is quite different from the growth behaviors of pure Au and pure Ni previously reported on TiO2 substrate63 and 6H–SiC substrate.64 On these substrates, Au shows larger cluster size and lower cluster density, indicating a higher diffusion constant of Au atoms compared with Ni. Such a unique growth behavior of Au on BN/h-Rh(111) can be attributed to the strong Au–BN/Rh(111) interaction.54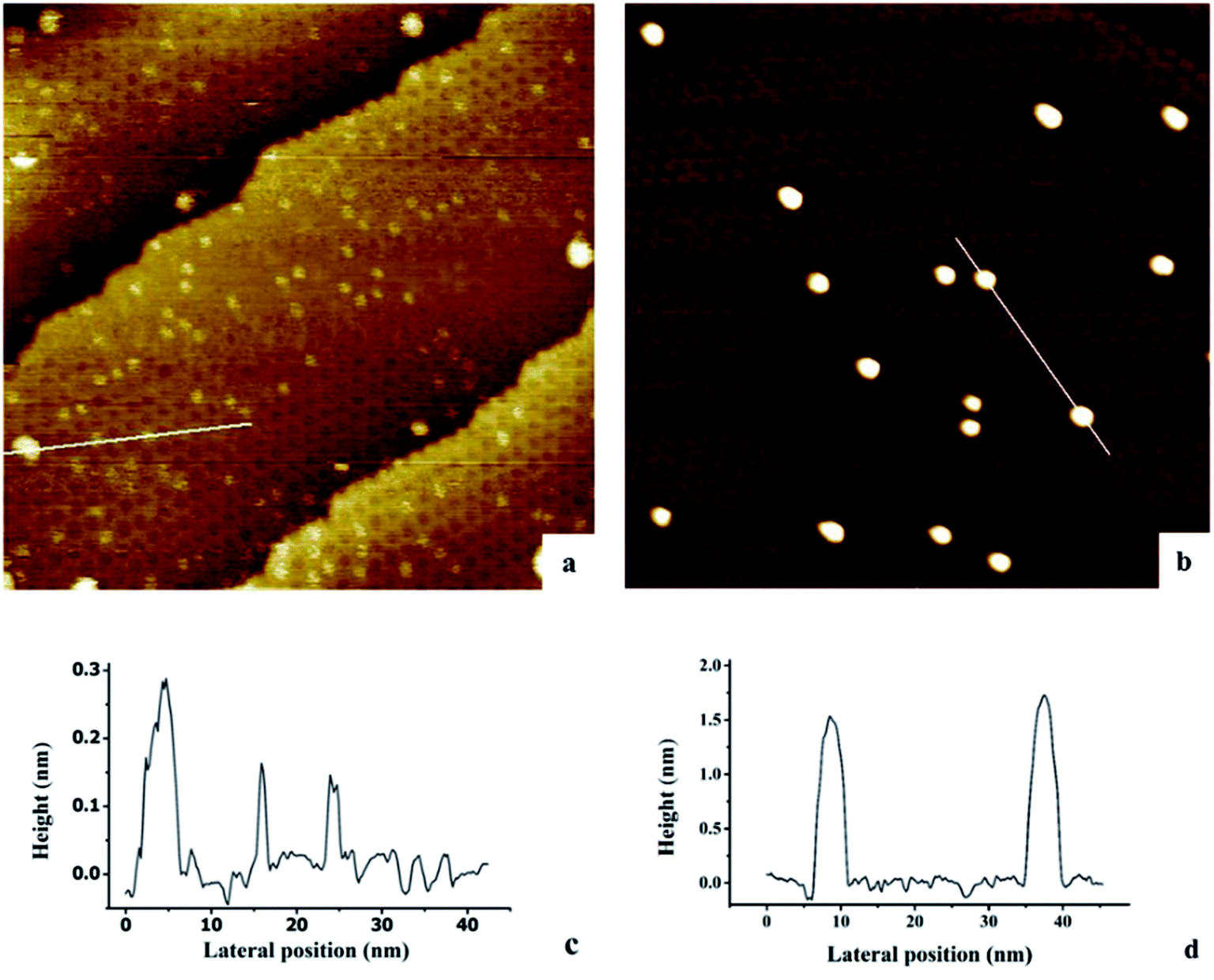 | ||
| Fig. 1 Scanning tunneling microscopy analysis of Au and Ni deposited and imaged at room temperature on h-BN/Rh(111). Tunneling parameters: Vsample = 1.0 V, Itunneling = 0.1 nA, 100 nm × 100 nm. (a) 0.1 ML Au/h-BN/Rh(111); (b) 0.1 ML Ni/h-BN/Rh(111); (c) height profile of the line in (a); (d) height profile of the line in (b). | ||
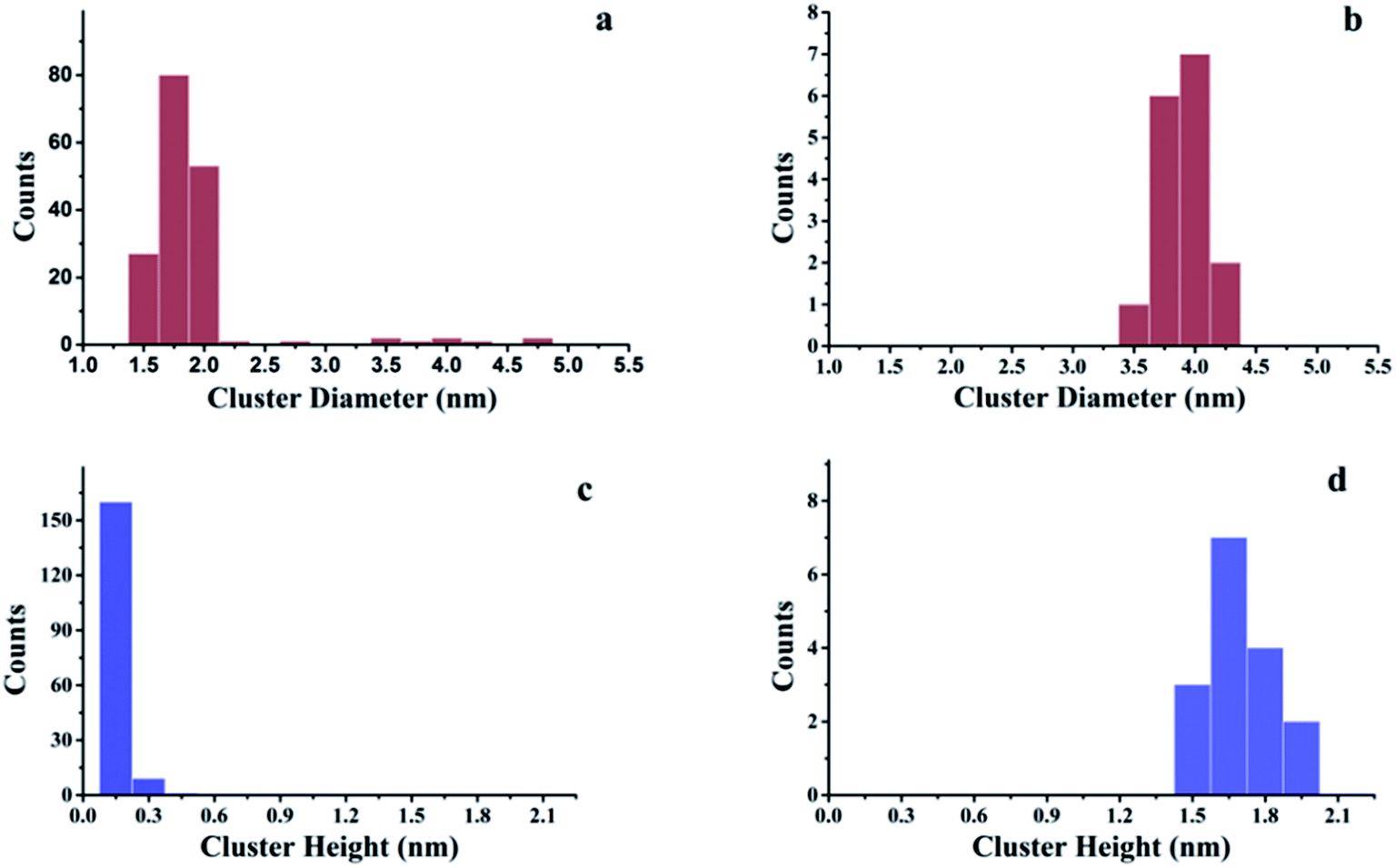 | ||
| Fig. 2 Histograms of cluster diameters and heights for Au and Ni deposited at room temperature on h-BN/Rh(111). (a) Histogram of cluster diameters for 0.1 ML Au/h-BN/Rh(111); (b) histogram of cluster diameters for 0.1 ML Ni/h-BN/Rh(111); (c) histogram of cluster heights for 0.1 ML Au/h-BN/Rh(111); (d) histogram of cluster heights for 0.1 ML Ni/h-BN/Rh(111). All these histograms correspond to the STM images in Fig. 1. | ||
Hexagonal boron nitride forms well-ordered and uniform monolayers on transition metals, for instance, Cu(111),65 Ru(0001)58 and Pd(111).66 It has been reported by Corso et al.57 that h-BN could form a highly corrugated “nanomesh” structure on Rh(111). The STM images and the splitting of the σ bands can be elucidated by a highly corrugated single-layer structure.49,50 In this single-layer model, a “pores-wires” structure could be assigned with “pores” formed when h-BN stayed closer to the Rh surface, while the “wires” formed when h-BN stayed away from the Rh substrate. Although there is only a weak interaction between the Au and the “wires” part of h-BN single-layer, the Rh atoms beneath the “pores” alter the density of states of h-BN, and Au atoms adsorb strongly to the “pores” part, specifically, the B atoms in the “pores”.54,55 Moreover, the Au atoms are considerably charged. Such a strong interaction between the Au and the substrate explains the small diffusion constant of Au clusters on h-BN/Rh(111) substrate. Furthermore, Xu46 has reported that the additional growth of a single-layer Au cluster beyond 30 atoms in one pore is thermodynamically unfavorable. The cluster would transit into double layers, surmounting the pore barriers at room temperature. That is why the sizes of double layer Au nanoclusters observed by STM are larger than the size of the pores. Note that at 0.1 ML dosage, the average number of Au atoms at each pore is 144 × 0.1 ≈ 14, and thus it is reasonable to conclude that the majority of Au clusters under 0.1 ML deposition coverage are template grown in single layer height (consistent to our experiment: 94% are composed of single layers). In addition, the Bader charge analysis for the clusters indicated that the average charge per Au atom would decrease as the sizes of the clusters increase. Although no explanation has yet been ventured for the large diffusivity of Ni on h-BN/Rh(111) experimentally and computationally, it would be reasonable to predict such a behavior, considering its weak bonding to the substrate, higher surface free energy (2.080 J m−2 for Ni vs. 1.333 J m−2 for Au67,68), as well as its strong cohesive energy compared to Au (534 kJ mol−1 for Ni vs. 452 kJ mol−1 for Au69).
In order to investigate the growth of Au–Ni clusters on h-BN/Rh(111), STM experiments were applied by varying deposition sequences. Fig. 3 compares the morphology difference of Au–Ni clusters (depositing Ni first) with those monometallic counterpart Ni on h-BN/Rh(111), and Fig. 4 compares the morphology difference of Ni–Au clusters (depositing Au first) with pure Au.
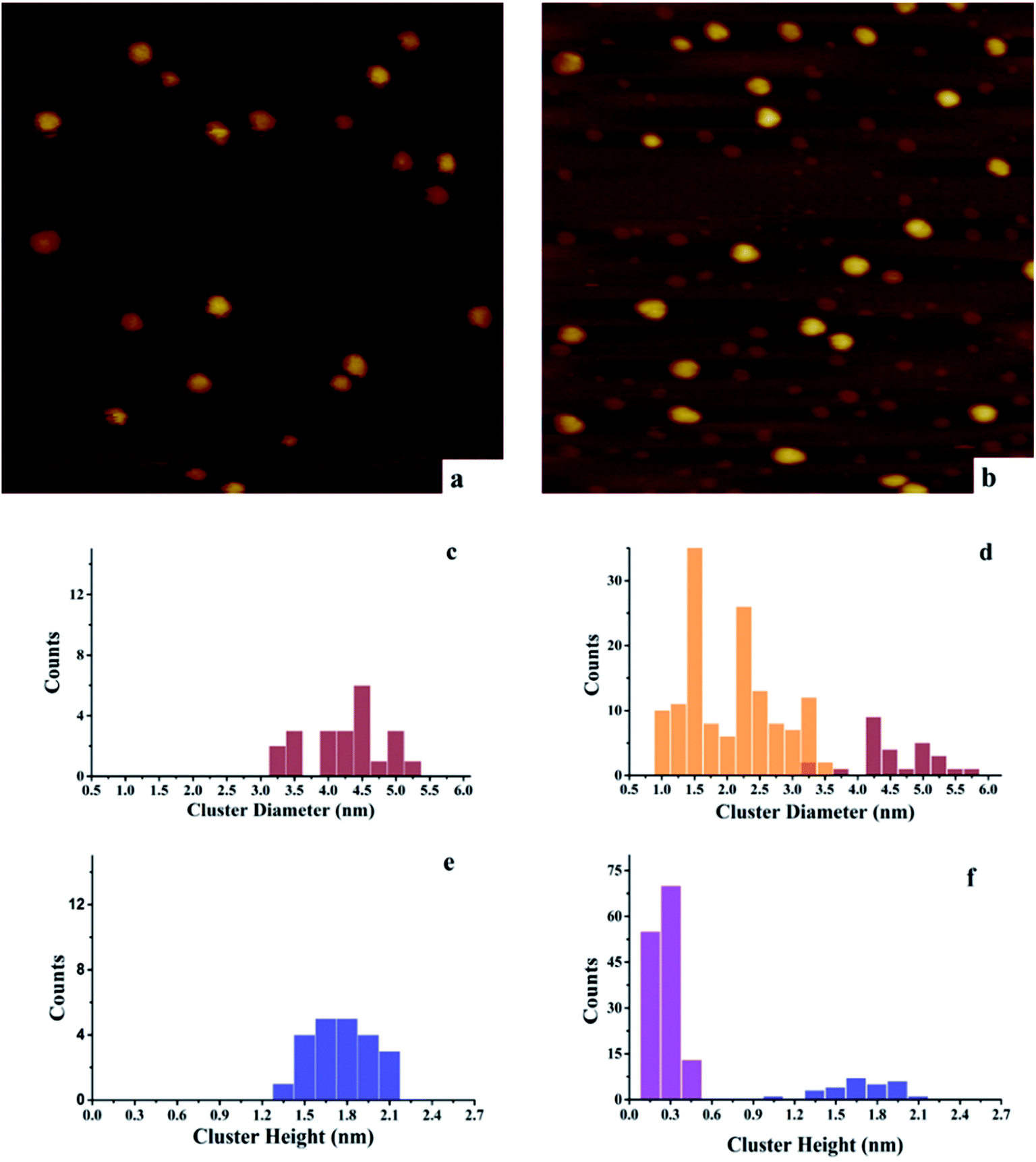 | ||
| Fig. 3 Scanning tunneling microscopy analysis of Ni and Au–Ni clusters deposited and imaged at room temperature on h-BN/Rh(111). Tunneling parameters: Vsample = 1.0 V, Itunneling = 0.1 nA. (a) 0.15 ML Ni/h-BN/Rh(111), 100 nm × 100 nm; (b) 0.15 ML Au/0.15 ML Ni/h-BN/Rh(111), (Ni deposited first), 100 nm × 100 nm; (c) histogram of cluster diameters for 0.15 ML Ni/h-BN/Rh(111) in (a); (d) histogram of cluster diameters for 0.15 ML Au/0.15 ML Ni/h-BN/Rh(111) in (b); (e) histogram of cluster heights for 0.15 ML Ni/h-BN/Rh(111) in (a); (f) histogram of cluster heights for 0.15 ML Au/0.15 ML Ni/h-BN/Rh(111) in (b). | ||
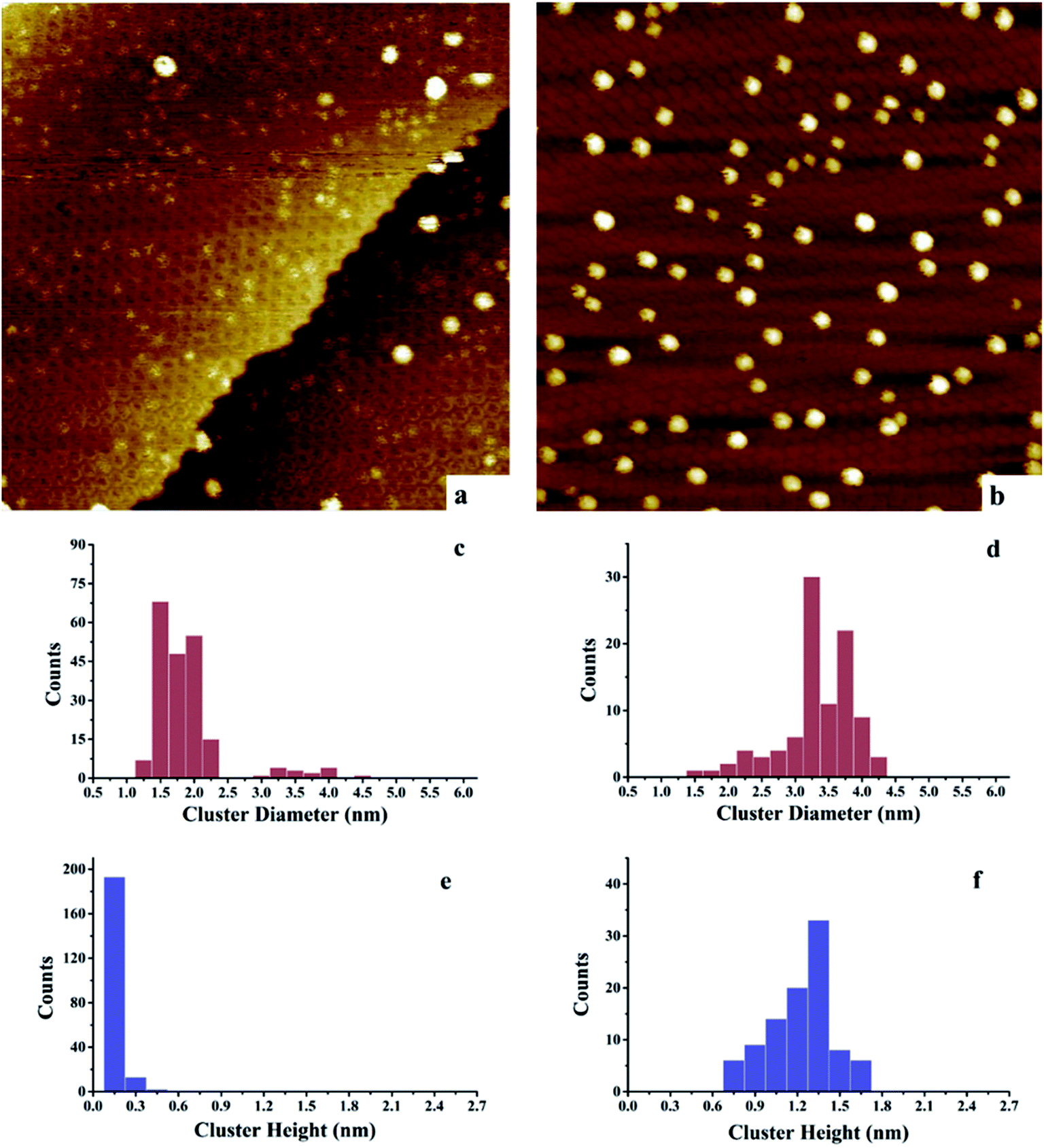 | ||
| Fig. 4 Scanning tunneling microscopy analysis of Au and Ni–Au clusters deposited and imaged at room temperature on h-BN/Rh(111). Tunneling parameters: Vsample = 1.0 V, Itunneling = 38 pA. (a) 0.15 ML Au/h-BN/Rh(111), 100 nm × 100 nm; (b) 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111), (Au deposited first), 100 nm × 100 nm; (c) histogram of cluster diameters for 0.15 ML Au/h-BN/Rh(111) in (a); (d) histogram of cluster diameters for 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111) in (b); (e) histogram of cluster heights for 0.15 ML Au/h-BN/Rh(111) in (a); (f) histogram of cluster heights for 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111) in (b). | ||
After deposition of 0.15 ML Au on 0.15 ML Ni/h-BN/Rh(111), the cluster density increases dramatically from 2.2 × 1011 to 1.65 × 1012 clusters cm−2 (Fig. 3). Due to the significant difference in cluster size between pure 0.15 ML Au (Fig. 4a) and pure 0.15 ML Ni (Fig. 3a) on h-BN/Rh(111), pure Au clusters and Au–Ni clusters can be clearly distinguished as shown in Fig. 3b. Pure Au clusters are all smaller than 0.45 nm (triple layers) in heights with 99% consisting of single or double layers (Fig. 4a and e). In contrast, all monometallic Ni clusters have heights larger than 1 nm with an average value of 1.76 ± 0.4 nm (Fig. 3a and e). Moreover, the average diameter of monometallic Ni clusters is 4.27 ± 0.1 nm, which is 2.26 times larger than the average diameter of pure Au clusters 1.89 ± 0.14 nm. Therefore, it can be concluded from Fig. 3b that the substantial increase of the density after the deposition of 0.15 ML Au on 0.15 ML Ni/h-BN/Rh(111) is mainly due to the growth of monometallic Au clusters on the substrate (corresponding to the orange and purple bins in Fig. 3d and f). The small diffusion length of Au on h-BN/Rh(111) and the large inter-cluster distances for Ni clusters both contribute to the increase of cluster density. No significant expansion of the pre-grown Ni clusters is observed after the deposition of Au (average 4.59 ± 0.1 nm in diameter and 1.69 ± 0.4 nm in height). It can be concluded that almost all Au clusters nucleate and grow between the Ni clusters. Further TPD and AES results will confirm the explanation of the STM results.
However, for the Ni–Au clusters with reversed deposition sequence, a novel growth mode is observed for the Ni–Au bimetallic clusters (Fig. 4). Before depositing 0.15 ML Ni on 0.15 ML Au/h-BN/Rh(111), 99% of Au clusters are template grown in the pore parts (Fig. 4a), with 0.15 or 0.30 nm (Fig. 4e) in height (single or double layers). After Ni is deposited, the average cluster height increases from 0.15 ± 0.02 nm to 1.22 ± 0.3 nm with the minimum cluster height greater than 0.7 nm (Fig. 4f). This suggests that no monometallic Au clusters are left unaffected; in other words, all the Au clusters are covered by Ni. The average cluster diameter also increases from 1.89 ± 0.28 nm to 3.33 ± 0.35 nm (Fig. 4c and d). In addition, compared to pure Ni on h-BN/Rh(111) at the same deposition coverage (Fig. 3a), the cluster density increases from 2.2 × 1011 clusters cm−2 to 1.02 × 1012 clusters cm−2 for such a reversed deposition sequence. All of these phenomena indicate that Ni atoms cover all the pre-deposited Au clusters and the Ni–Au bimetallic clusters are effectively formed. In this process, the pre-existing charged Au clusters act as nucleation sites (seeding effect) for the growth of subsequently dosed Ni, and the Ni atoms enrich on the surface of the Ni–Au bimetallic clusters. On comparing the cluster density of 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111) to that of pure 0.15 ML Au/h-BN/Rh(111), we find that cluster density decreases from 2.08 × 1012 clusters cm−2 to 1.02 × 1012 clusters cm−2, caused mainly by the coalescence of bimetallic clusters. STM characterization demonstrates that bimetallic Ni–Au clusters can be efficiently formed by template growth of Au first on h-BN/Rh(111), and then Ni, whereas the reversed deposition sequence leads to an ineffective bimetallic formation.
The preferential growth of Ni on the surface of Au clusters differs from Ni–Au bimetallic studies carried out by Bao's group with 6H–SiC(0001) substrate64 and Chen's group using TiO2 substrate.63 Both studies showed preferential growth of Au on the surface of the existing Ni clusters due to the relatively high surface free energies of Au (2.080J m−2 for Ni and 1.333J m−2 for Au).67,68 However in our case, the support effect played a more important role compared to the surface energy of the nanoclusters, and the h-BN/Rh(111) support provided a novel approach in the formation of Ni–Au bimetallic clusters with Ni enriched on the cluster surface.
3.2. Temperature-programmed desorption and Auger electron spectroscopy
The surface composition as well as the chemical activity of the Ni–Au bimetallic clusters supported on h-BN/Rh(111), were probed by CO TPD experiments since the adsorption of CO on metal surfaces is a well-understood process.70 At room temperature, CO does not bind to the Au surfaces (no peak for 0.15 ML Au on h-BN/Rh(111) as shown in Fig. 5a). All peaks in Fig. 5a come from CO desorption on Ni surfaces. For 0.15 ML Ni deposited on h-BN/Rh(111), the desorption occurs at 404 K and 634 K separately. The low temperature peak (LTP) is attributed to molecular desorption; the high temperature peak (HTP) is attributed to associated desorption.71,72 The deposition and deposition sequence of Au have little effect on the associated CO desorption peak on Ni, and hence only the LTP will be discussed. After depositing 0.15 ML Au on 0.15 ML Ni/h-BN/Rh(111), only a slightly decrease in LTP can be observed. Although Au has a lower surface energy compared to Ni and tends to cover the Ni cluster surfaces, the adsorption of CO would introduce the diffusion of Ni atoms to the surface of the clusters.63 The density functional theory (DFT) calculation shows that in the presence of CO, the Ni1Au12 system energy will be ∼1.26 eV lower when the Ni atom is on the surface and binds to CO compared to Ni atom inside the cluster with CO binding to the Au surface. In addition, the molecular dynamics (MD) simulation illustrates that the adsorption of CO on Au atoms weakens the Ni–Au bonding and thus increases the diffusivity of Ni atom to the surface. Similar CO-induced diffusion mechanism has also been reported in the case of Au–Rh, Au–Pt and Au–Pd core–shell nanoclusters.73–75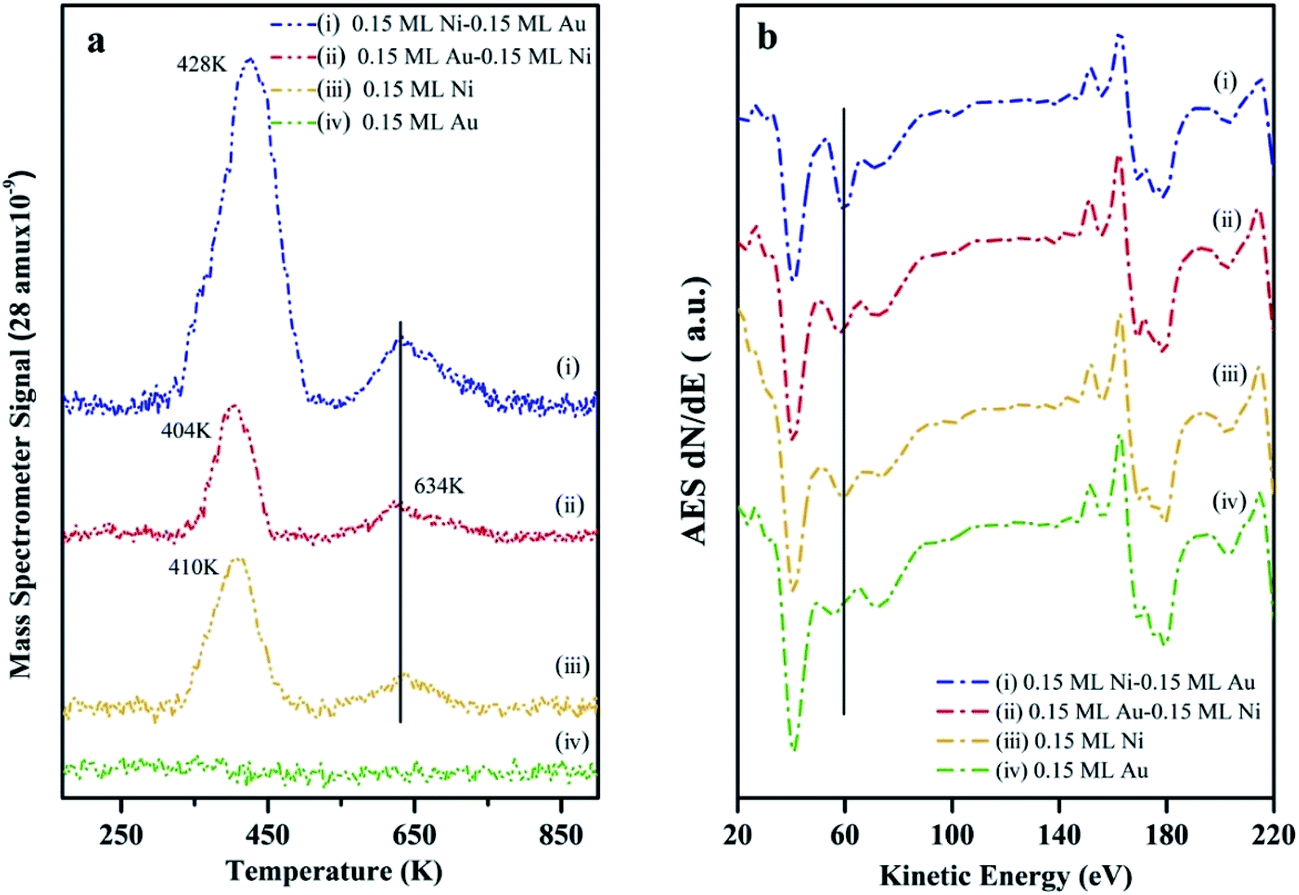 | ||
| Fig. 5 (a) CO temperature-programmed desorption at CO saturation coverage and (b) Auger electron spectroscopy for (i) 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111), (Au deposited first); (ii) 0.15 ML Au/0.15 ML Ni/h-BN/Rh(111), (Ni deposited first); (iii) 0.15 ML Ni/h-BN/Rh(111) and (iv) 0.15 ML Au/h-BN/Rh(111). All TPD and AES spectra have been adjusted to the same scale for direct comparison. | ||
In addition, a slight shift of the LTP peak from 410 K to 404 K can be observed after depositing 0.15 ML Au on 0.15 ML Ni/h-BN/Rh(111) surface. The addition of Au atoms on Ni surfaces leads to the formation of the surface alloy.12 This will decrease the density of state of Ni and shift up its d band center to the Fermi level.76 Therefore, the charge transfer from the Ni to the antibonding state of CO molecules will decrease and thus lead to a reduced binding of the CO molecules to the surface. Another DFT study also shows a 0.2–0.5 eV decrease in adsorption energy with the presence of Au on Ni(111) surface.77 However, after switching the deposition sequence, the LTP peak changes to 428 K for 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111) (Au deposited first). Such a noteworthy difference in peak positions reveals the important role of the h-BN/Rh(111) substrate. As mentioned before, Au atoms are considerably charged after Au is deposited onto the h-BN/Rh(111) substrate. These charged Au atoms not only behave as nucleation sites for the growth of the subsequently dosed Ni, but also transfer electrons to the Ni and change the local density of state on the Ni surface. Molecular orbitals near the Fermi level are very sensitive to any perturbation of the electronic structure of the substrate, since minute changes in the positions of these electronic states compared to the Fermi energy would change their coupling to the molecular states. The presence of the charged Au and the donation of electrons to the Ni atoms significantly perturb the surface density of states near Fermi energy. In this case, Au atoms can also be treated as the promoter that facilitates the charge transfer from the Ni atoms into the antibonding states of the CO molecules. Therefore, the metal–CO bond can be enhanced and this shifts up the desorption temperature (increasing from 410 K for 0.15 ML pure Ni to 428 K for 0.15 ML Ni/0.15 ML Au).78 Another possible consideration could be the size effect of these clusters. It is generally believed that small cluster would have a stronger binding energy to CO compared to its bulk counterpart since there would be more under-coordinated surface sites (steps and kink sites) for smaller nanoclusters. However, in our case (average diameter 3.33 ± 0.35 nm for 0.15 ML Ni/0.15 ML Au; average diameter 4.27 ± 0.1 nm for monometallic Ni clusters), the adsorption energies for CO are almost the same for such large particle sizes (>3 nm) according to Notker's DFT calculation results.79 Thus, the up-shift of the LTP is mainly attributed to the charge transfer between Au and Ni atoms.
In addition, the peak intensity for 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111) increases dramatically compared to that for 0.15 ML Ni/h-BN/Rh(111), further confirming the template growth of the bimetallic Ni–Au clusters and the increase of the surface coverage. The AES data (Fig. 5b) also show the same trend as the 61 eV peak, which can be assigned for Ni element, illustrating a larger feature for 0.15 ML Ni/0.15 ML Au/h-BN/Rh(111) as compared to 0.15 ML Ni/h-BN/Rh(111) peak (peak shifts are hard to be identified in AES, only the intensities are discussed in AES section). Thus, results of TPD and AES confirm previous interpretation of STM results.
4. Conclusions
Ni, Au and Ni–Au bimetallic clusters were prepared on h-BN/Rh(111) template substrate. Au formed mainly single layer clusters on h-BN/Rh(111) substrate at 0.15 ML surface coverage, while Ni formed large clusters at 0.15 ML surface coverage. Bimetallic Ni–Au clusters were prepared by either depositing Ni or Au first. By sequential deposition of Ni first and then Au, most of the Au clusters nucleated and grew between the Ni clusters. Bimetallic clusters could not be successfully formed. However by sequential deposition of Au followed by Ni, bimetallic Ni–Au clusters were effectively formed as the charged Au clusters, which were template grown on h-BN/Rh(111), worked as nucleation sites for the growth of the subsequently dosed Ni. Further TPD and AES experiments confirmed such an interpretation of the STM results. In addition, TPD results illustrated the perturbation of the electronic structure of Ni clusters with the presence of Au atoms. By changing the deposition sequence, Au atoms would either “poison” or “promote” the catalytic activity of Ni clusters. All these results suggested that the h-BN/Rh(111) substrate represented a unique candidate for supporting Ni–Au bimetallic clusters and even other nano-clusters in further catalytic reactions.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the Department of Energy (DE-SC0001058) for supporting this work, and the Robert A. Welch Foundation is also acknowledged.References
- M. Chen and D. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- G. C. Bond, P. A. Sermon, G. Webb, D. A. Buchanan and P. B. Wells, J. Chem. Soc., Chem. Commun., 1973, 444b–445b RSC.
- Y. Gault, J. Chem. Soc., Chem. Commun., 1973, 478–479 RSC.
- S. Overbury, V. Schwartz, D. R. Mullins, W. Yan and S. Dai, J. Catal., 2006, 241, 56–65 CrossRef CAS.
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS PubMed.
- M. Chen and D. W. Goodman, Chem. Soc. Rev., 2008, 37, 1860–1870 RSC.
- N. Lopez, T. Janssens, B. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- M. C. Kung, R. J. Davis and H. H. Kung, J. Phys. Chem. C, 2007, 111, 11767–11775 CAS.
- J. R. Rostrup-Nielsen, in Catalysis, Springer, 1984, pp. 1–117 Search PubMed.
- M. Bradford and M. Vannice, Catal. Rev., 1999, 41, 1–42 CAS.
- F. Besenbacher, I. Chorkendorff, B. Clausen, B. Hammer, A. Molenbroek, J. K. Nørskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS PubMed.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. Clausen, L. Nielsen, A. Molenbroek and J. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- J. H. Sinfelt, Acc. Chem. Res., 1977, 10, 15–20 CrossRef CAS.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS PubMed.
- J. H. Sinfelt, Bimetallic catalysts: discoveries, concepts, and applications, Wiley-Interscience, 1983 Search PubMed.
- J. G. Chen, C. A. Menning and M. B. Zellner, Surf. Sci. Rep., 2008, 63, 201–254 CrossRef CAS.
- J. Rodriguez, Surf. Sci. Rep., 1996, 24, 223–287 CrossRef CAS.
- C. T. Campbell, Annu. Rev. Phys. Chem., 1990, 41, 775–837 CrossRef CAS.
- J. A. Rodriguez and D. W. Goodman, J. Phys. Chem., 1991, 95, 4196–4206 CrossRef CAS.
- B. D. Chandler, C. G. Long, J. D. Gilbertson, C. J. Pursell, G. Vijayaraghavan and K. J. Stevenson, J. Phys. Chem. C, 2010, 114, 11498–11508 CAS.
- S. Zhou, Z. Ma, H. Yin, Z. Wu, B. Eichhorn, S. H. Overbury and S. Dai, J. Phys. Chem. C, 2009, 113, 5758–5765 CAS.
- H. Reichert, A. Schöps, I. Ramsteiner, V. Bugaev, O. Shchyglo, A. Udyansky, H. Dosch, M. Asta, R. Drautz and V. Honkimäki, Phys. Rev. Lett., 2005, 95, 235703 CrossRef CAS PubMed.
- H. Okamoto and T. Massalski, J. Phase Equilib., 1991, 12, 148–168 CrossRef CAS.
- L. P. Nielsen, F. Besenbacher, I. Stensgaard, E. Lægsgaard, C. Engdahl, P. Stoltze, K. W. Jacobsen and J. K. Nørskov, Phys. Rev. Lett., 1993, 71, 754 CrossRef CAS PubMed.
- P. M. Holmblad, J. H. Larsen, I. Chorkendorff, L. P. Nielsen, F. Besenbacher, I. Stensgaard, E. Lægsgaard, P. Kratzer, B. Hammer and J. K. Nørskov, Catal. Lett., 1996, 40, 131–135 CrossRef CAS.
- A. M. Molenbroek, J. K. Nørskov and B. S. Clausen, J. Phys. Chem. B, 2001, 105, 5450–5458 CrossRef CAS.
- D. L. Lahr and S. T. Ceyer, J. Am. Chem. Soc., 2006, 128, 1800–1801 CrossRef CAS PubMed.
- E. K. Vestergaard, R. T. Vang, J. Knudsen, T. M. Pedersen, T. An, E. Lægsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2005, 95, 126101 CrossRef PubMed.
- G.-C. Wang, J. Jiao and X.-H. Bu, J. Phys. Chem. C, 2007, 111, 12335–12339 CAS.
- P. M. Holmblad, J. H. Larsen and I. Chorkendorff, J. Chem. Phys., 1996, 104, 7289–7295 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- K. Okazaki, S. Ichikawa, Y. Maeda, M. Haruta and M. Kohyama, Appl. Catal., A, 2005, 291, 45–54 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- D. Goodman, Catal. Lett., 2005, 99, 1–4 CrossRef CAS.
- N. Spiridis, R. Socha, B. Handke, J. Haber, M. Szczepanik and J. Korecki, Catal. Today, 2011, 169, 24–28 CrossRef CAS.
- M. Amft and N. V. Skorodumova, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 195443 CrossRef.
- J. Guzman and B. C. Gates, J. Am. Chem. Soc., 2004, 126, 2672–2673 CrossRef CAS PubMed.
- G. Pacchioni, Phys. Chem. Chem. Phys., 2013, 15, 1737–1757 RSC.
- C. R. Henry, Catal. Lett., 2015, 145, 731–749 CrossRef CAS.
- S. Bleikamp, P. J. Feibelman and T. Michely, Phys. Rev. Lett., 2006, 97, 215501 CrossRef PubMed.
- I. Brihuega, C. H. Michaelis, J. Zhang, S. Bose, V. Sessi, J. Honolka, M. A. Schneider, A. Enders and K. Kern, Surf. Sci., 2008, 602, L95–L99 CrossRef CAS.
- B. Wang and M.-L. Bocquet, J. Phys. Chem. Lett., 2011, 2, 2341–2345 CrossRef CAS.
- M. C. Patterson, B. F. Habenicht, R. L. Kurtz, L. Liu, Y. Xu and P. T. Sprunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 205423 CrossRef.
- W. C. McKee, M. C. Patterson, D. Huang, J. R. Frick, R. L. Kurtz, P. Sprunger, L. Liu and Y. Xu, J. Phys. Chem. C, 2016, 120, 10909–10918 CAS.
- J. Zhang, V. Sessi, C. Michaelis, I. Brihuega, J. Honolka, K. Kern, R. Skomski, X. Chen, G. Rojas and A. Enders, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 165430 CrossRef.
- R. Laskowski, P. Blaha, T. Gallauner and K. Schwarz, Phys. Rev. Lett., 2007, 98, 106802 CrossRef PubMed.
- R. Laskowski and P. Blaha, J. Phys.: Condens. Matter, 2008, 20, 064207 CrossRef CAS PubMed.
- A. P. T. Farkas, P. Török, F. Solymosi, J. Kiss and Z. Kónya, Appl. Surf. Sci., 2015, 354, 367–372 CrossRef CAS.
- L. Molina and B. Hammer, Phys. Rev. Lett., 2003, 90, 206102 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- H. P. Koch, R. Laskowski, P. Blaha and K. Schwarz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245410 CrossRef.
- H. P. Koch, R. Laskowski, P. Blaha and K. Schwarz, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 155404 CrossRef.
- M. L. Ng, A. Preobrajenski, A. Vinogradov and N. Mårtensson, Surf. Sci., 2008, 602, 1250–1255 CrossRef CAS.
- M. Corso, W. Auwärter, M. Muntwiler, A. Tamai, T. Greber and J. Osterwalder, Science, 2004, 303, 217–220 CrossRef CAS PubMed.
- A. Goriachko, Y. He, M. Knapp, H. Over, M. Corso, T. Brugger, S. Berner, J. Osterwalder and T. Greber, Langmuir, 2007, 23, 2928–2931 CrossRef CAS PubMed.
- L. Liu, Z. Zhou, Q. Guo, Z. Yan, Y. Yao and D. W. Goodman, Surf. Sci., 2011, 605, L47–L50 CrossRef CAS.
- Z. Zhou, F. Gao and D. W. Goodman, Surf. Sci., 2010, 604, L31–L38 CrossRef CAS.
- S. Berner, M. Corso, R. Widmer, O. Groening, R. Laskowski, P. Blaha, K. Schwarz, A. Goriachko, H. Over and S. Gsell, Angew. Chem., Int. Ed., 2007, 46, 5115–5119 CrossRef CAS PubMed.
- A. Winkler and J. Yates Jr, J. Vac. Sci. Technol., A, 1988, 6, 2929–2932 CAS.
- S. A. Tenney, W. He, C. C. Roberts, J. S. Ratliff, S. I. Shah, G. S. Shafai, V. Turkowski, T. S. Rahman and D. A. Chen, J. Phys. Chem. C, 2011, 115, 11112–11123 CAS.
- Z.-J. Wang, Q. Fu, Z. Wang and X. Bao, Surf. Sci., 2012, 606, 1313–1322 CrossRef CAS.
- A. Preobrajenski, A. Vinogradov and N. Mårtensson, Surf. Sci., 2005, 582, 21–30 CrossRef CAS.
- M. Morscher, M. Corso, T. Greber and J. Osterwalder, Surf. Sci., 2006, 600, 3280–3284 CrossRef CAS.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- W. Tyson and W. Miller, Surf. Sci., 1977, 62, 267–276 CrossRef CAS.
- M. Turchanin and P. Agraval, Powder Metall. Met. Ceram., 2008, 47, 26–39 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- H. Madden and G. Ertl, Surf. Sci., 1973, 35, 211–226 CrossRef CAS.
- W. Erley and H. Wagner, Surf. Sci., 1978, 74, 333–341 CrossRef CAS.
- J. Kiss, L. Óvári, A. Oszkó, G. Pótári, M. Tóth, K. Baán and A. Erdóhelyi, Catal. Today, 2012, 181, 163–170 CrossRef CAS.
- S. A. Tenney, J. S. Ratliff, C. C. Roberts, W. He, S. C. Ammal, A. Heyden and D. A. Chen, J. Phys. Chem. C, 2010, 114, 21652–21663 CAS.
- F. Gao, Y. Wang and D. W. Goodman, J. Phys. Chem. C, 2010, 114, 4036–4043 CAS.
- K. Termentzidis, J. Hafner and F. Mittendorfer, J. Phys.: Condens. Matter, 2006, 18, 10825 CrossRef CAS.
- K. Termentzidis and J. Hafner, J. Phys.: Condens. Matter, 2007, 19, 246219 CrossRef PubMed.
- P. J. Feibelman and D. Hamann, Surf. Sci., 1985, 149, 48–66 CrossRef CAS.
- I. V. Yudanov, A. Genest, S. Schauermann, H.-J. Freund and N. Rösch, Nano Lett., 2012, 12, 2134–2139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08880a |
| This journal is © The Royal Society of Chemistry 2017 |