
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel information fusion strategy based on a regularized framework for identifying disease-related microRNAs
Li Peng ab,
Manman Peng*a,
Bo Liaoa,
Qiu Xiaoa,
Wei Liuc,
Guohua Huangd and
Keqin Lie
ab,
Manman Peng*a,
Bo Liaoa,
Qiu Xiaoa,
Wei Liuc,
Guohua Huangd and
Keqin Lie
aCollege of Information Science and Engineering, Hunan University, Changsha, Hunan 410082, China. E-mail: plpeng@hnu.edu.cn; pengmanman@hnu.edu.cn
bCollege of Computer Science and Engineering, Hunan University of Science and Technology, Xiangtan, Hunan 411201, China
cCollege of Information Engineering, XiangTan University, Xiangtan, Hunan 411105, China
dCollege of Information Engineering, Shaoyang University, Shaoyang, Hunan 422000, China
eDepartment of Computer Science, State University of New York, New Paltz, New York 12561, USA
First published on 15th September 2017
Abstract
Abnormal microRNA (miRNA) expression can induce various complex human diseases. Thus, revealing the underlying relationship between miRNA and human diseases contributes to the early diagnosis and treatment of diseases. Utilizing a computational approach in selecting the most likely miRNA candidates related to a given disease for further biological experimental validation can save time and manpower costs. In this study, we propose a novel information fusion strategy called RLSSLP, which is based on a regularized framework, for discovering the underlying associations between miRNAs and diseases. RLSSLP integrates two submodels to construct effective prediction frameworks and quantify the similarities between miRNAs and diseases by fully using multiple omics data, which include verified associations, particularly miRNA–disease, miRNA–gene, and weighted gene–gene network associations. The 10-fold cross-validation and case studies for lung cancer, hepatocellular carcinoma and breast cancer indicate that RLSSLP performs well in predicting miRNA–disease interactions.
Introduction
MiRNAs are a set of small non-protein-coding RNAs and approximately 22 nt long.1,2 MiRNAs are involved in many crucial pathological and biological processes. Thus, abnormal miRNA expression can induce various complex human diseases, including cancer.3,4 Revealing the underlying relationships between miRNA and human diseases contributes to the early diagnosis and treatment of diseases. However, traditional experimental methods for detecting disease-related miRNAs require huge amounts of time and manpower. Utilizing computational approaches in selecting miRNA candidates that are likely related to a particular disease for further biological experimental validation is more efficient than traditional approaches and costs less.In recent years, many efforts have been extensively exerted to investigate the associations between miRNAs and diseases. Some network-based methods that predict miRNA–disease interactions are based on the hypothesis that similar miRNAs are likely to relate to similar diseases, and vice versa. Jiang et al.5 first proposed a similarity-based approach that measures miRNA functional similarity based on the common sets of their associated target gene and identify disease-related miRNAs based on hypergeometric distributions. Shi et al.6 investigated the relationships of miRNA–target and disease–gene and constructed a bipartite network for discovering the miRNA regulation of disease gene. However, due to the false-positive rate in target predictions, the accuracy of the above methods is often negatively affected. Xuan et al.7 proposed a new method called HDMP, which sorts the most likely miRNA candidates related to diseases according to the weighted k most similar neighbor. Chen et al.8 presented “RWRMDA: predicting novel human microRNA–disease associations”, which is a predictive approach wherein the algorithm of random walk with restart is applied to construct a global network for capturing underlying miRNA–disease associations. Li et al.9 presented a new computational method based on the algorithm of matrix completion to recover the associations score of each miRNA–disease pair. However, these methods cannot be used to predict diseases with no known related miRNAs.
Some classical machine learning methods were also utilized for mining the relationship between miRNAs and diseases. Jiang et al.10 advanced a computational approach based on Naïve Bayes for discovering disease-associated miRNAs. Xu et al.11 presented a method, which applied support vector machine in distinguishing associated or nonassociated miRNAs for particular diseases. However, one weakness of these approaches is that the negative samples utilized in these machine-learning methods are practically hard to obtain because verified nonassociations between miRNAs and diseases cannot be found exactly in any existing database.
By incorporating experimentally verified miRNA–disease associations and diverse similarity information based on miRNAs and diseases into a heterogeneous graph, Chen et al.12 advanced a novel approach named HGIMDA to infer underlying relationship between miRNAs and diseases. You et al.13 presented a path-based prediction method, named PBMDA, which applied depth-first search algorithm in the integrated heterogeneous graph to capture the potential miRNA–disease associations. Chen et al.14 developed a new method named WBSMDA, which predicted miRNA–disease interactions based on the framework of within and between score. They also proposed a computational approach named SDMMDA15 based on super-disease and super miRNA to predict underlying miRNA–disease interactions. “RBMMMDA: predicting multiple types of disease-microRNA associations”, which proposed by Chen et al.,16 is the first model that can predict not only whether there is a link between each miRNA and disease pairs, but also the corresponding type of association.
Zou et al.17 presented a method called KATZ for predicting miRNA–disease interactions. In this method, social network analysis methods are adopted to construct miRNA–disease association networks. However, although KATZ has excellent performance, its capability to spare known associations is relatively poor. Chen et al.18 presented a semi-supervised approach, called RLSMDA, for exposing unknown miRNA–disease interactions on the basis of regularized least squares. Luo et al.19 utilized heterogeneous omics data and adopted Kronecker regularized least-squares framework to identify potential disease-related miRNAs. However, cross-validation performance of these methods is not so good.
Overall, the aforementioned approaches have the following limitations: some methods cannot predict diseases without known related miRNAs, some methods require negative samples that are practically hard to obtain, and exhibit predictive performance that requires further improvement.
To overcome the above challenges, we proposed a novel information fusion strategy called RLSSLP, which is based on a regularized framework, for discovering underlying associations between miRNAs and diseases. RLSSLP comprehensively measures the similarity for miRNAs and diseases by fully using the multiple omics data, which include known miRNA–disease interactions, gene–gene networks, and the experimentally verified association data of miRNA–gene from three different databases. RLSSLP adopts the eigenvalue transformation technique to reduce computational time and memory requirement, as well as utilizes integrated regularized framework based on regularized least squares (RLS)18,19,29 and semi-supervised link prediction (SLP)20,30 to prioritize disease-related miRNAs.
The main contributions of this study are as follows:
(1) RLSSLP does not need negative training samples, which are practically hard to obtain.
(2) Various omics data can be fully utilized in RLSSLP and are beneficial for comprehensive evaluation of the similarities between miRNAs and diseases. MiRNA–gene association data are obtained from three different experimentally verified databases, which help reduce influence of false-positive rate on the performance of the miRNA–target prediction process.
(3) Eigenvalue transformation technique has been adopted to reduce computational time and memory requirement for the storage and calculation between the similarities matrices during the Kronecker operation.
(4) Two submodels are combined in RLSSLP to enhance predictive performance.
Materials and methods
Data preparation
The standard dataset used in our study include data on known miRNA–disease associations, disease similarities, and relevant information about miRNA similarities. MiRNA–disease associations are retrieved from HMDD v2.0.21 After duplicated associations are filtered, the data contain 5424 experimentally verified interactions, which consist of 495 miRNAs and 378 diseases. Disease directed acyclic graph (DAG), which is used to calculate disease semantic similarities, is derived from the MeSH database (available from: https://www.nlm.nih.gov/mesh/). We obtain miRNA target genes from the miRTarBase v4.5,22 TarBase v6.0,23 and miRecords v4.0 (ref. 24) to obtain more accurate and comprehensive information. The probabilistic functional gene network is obtained from HumanNet25 (available from: http://www.functionalnet.org/humannet/).Problem description
The problem can be regard as predicting novel interactions in a miRNA–disease association network. Formally, Xm = {m1,m2,…mnm} and Xd = {d1,d2,…dnd} represent the sets of miRNA and disease nodes, respectively. The known miRNA–disease association network is characterized as nm × nd adjacency matrix pre. If miRNA i interacts with disease j, preij is 1; otherwise, 0. In this study, we calculate the prediction score of each unknown miRNA–disease pair though a computational approach and recover the underlying association between miRNAs and diseases.Disease similarity calculation
Accumulating findings show that miRNAs with similar functions tend to regulate similar diseases. In this work, the method for estimating disease semantic similarities was based on the strategy of Wang et al.26In MeSH database, diseases can be expressed into a DAG. Formally, disease i can be denoted as DAG(i) = (i,Ti,Ei), where Ti is the disease set containing i itself and all its ancestors, and Ei represents the sets of corresponding links of disease i. The semantic contribution of ancestor node t to disease i is as follows:
 | (1) |
The semantic similarity score between disease i and j is denoted as follows:
 | (2) |
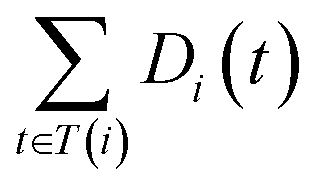 and
and 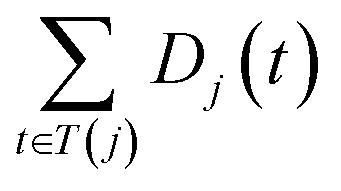 are the semantic value of disease i and disease j, respectively. As shown in eqn (2), the two diseases share a common part of DAG, the semantic similarity score is high, and the two diseases are increasingly similar.
are the semantic value of disease i and disease j, respectively. As shown in eqn (2), the two diseases share a common part of DAG, the semantic similarity score is high, and the two diseases are increasingly similar.
MiRNA similarity calculation
The more the two miRNAs share target genes, the more similar they are. In this work, we measure the similarities for miRNA according to an experimentally validated miRNA–target gene relationship and probabilistic functional gene network.First, we extract the common miRNA–target gene set from three experimentally valid databases mentioned above. The gene–gene relationship network can be acquired from HumanNet, in which the closeness of the link between each pair of genes is measured by associated log-likelihood scores. SL(ei,ej)stands for the associated log-likelihood scores between genes ei and ej. Second, we normalize SL(ei,ej) and obtain the normalized similarity SLnorm(ei,ej) between genes ei and ej as follows:
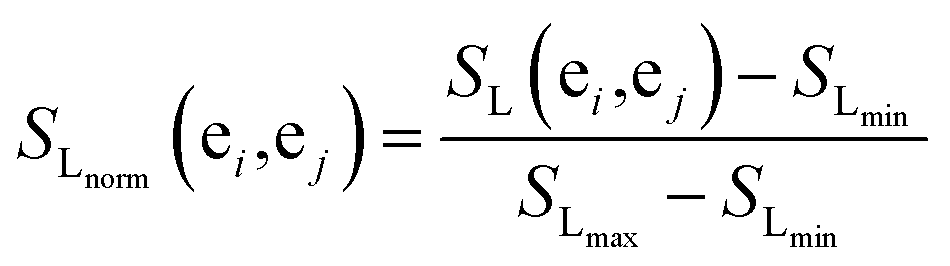 | (3) |
The similarity between genes et and gene set E = {et1,et2,…etk} is provided as follows:
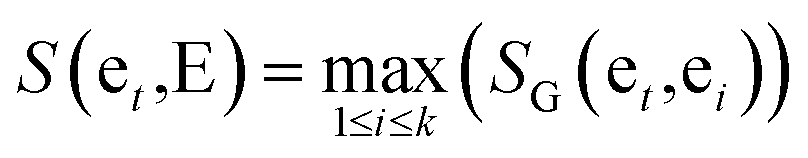 | (4) |
Finally, basing on a best matching average (BMA) strategy,27,28 we calculate the functional similarity between miRNAs i and j according to common genes.
 | (5) |
Methods for miRNA–disease interaction prediction
In our study, we apply an information fusion strategy and combine RLS and SLP to establish a prediction model. The overall flowchart of RLSSLP is shown in Fig. 1.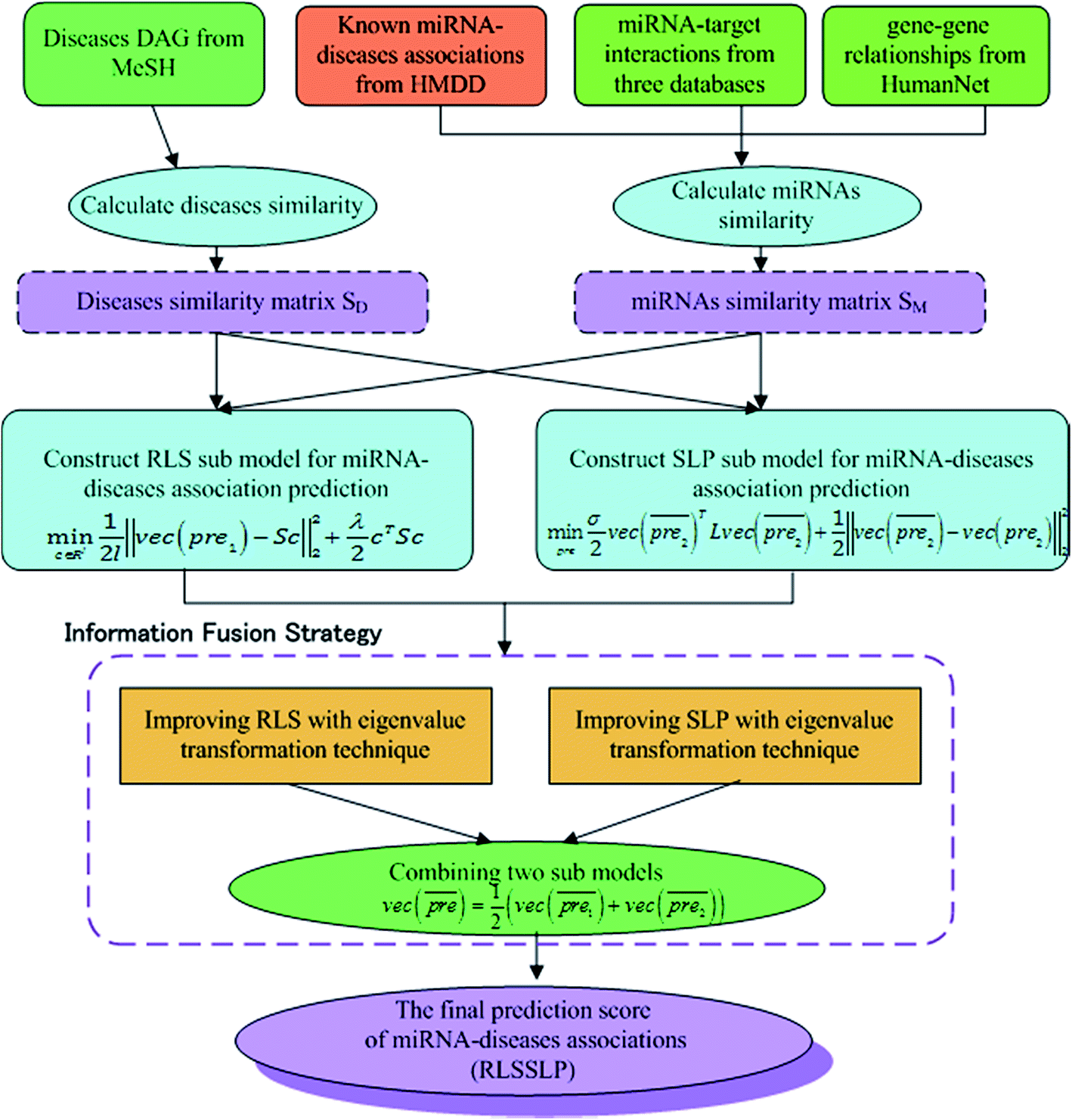 | ||
| Fig. 1 The overall flowchart of RLSSLP. | ||
 | (6) |
In this study, λ is a regularization parameter. Kernel matrix S is defined as S = SM ⊗ SD, which represents the Kronecker product of miRNA similarity matrix SM and disease similarity matrix SD. Through the derivative of c, the optimal solution of c is c = (S + σI)−1vec(pre), where σ = λI. I is the identity matrix. The prediction association score matrix  is calculated as follows:
is calculated as follows:
 | (7) |
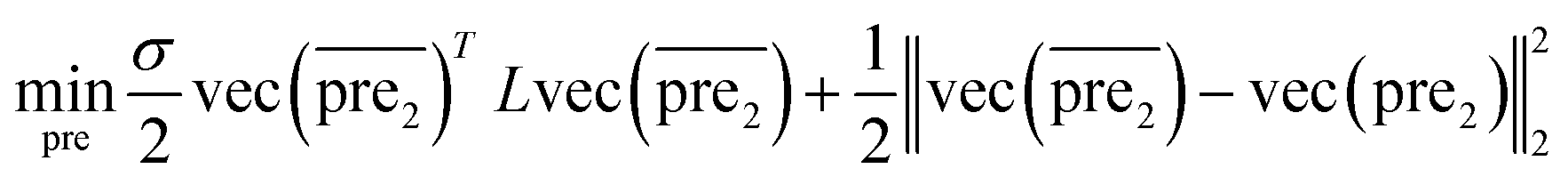 | (8) |
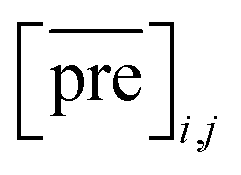 and
and 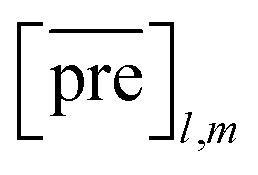 for the two pairs (for instance, the association between miRNA i–disease j and miRNA l–disease m) should be close to each other if some significant similarities between the two pairs are present. The second term is a regularization term and represents the loss function that fits the prediction matrix
for the two pairs (for instance, the association between miRNA i–disease j and miRNA l–disease m) should be close to each other if some significant similarities between the two pairs are present. The second term is a regularization term and represents the loss function that fits the prediction matrix  to known miRNA–disease association matrix pre2. Parameter σ represents the regularization parameter to balance the two terms. The Laplacian matrix L is denoted as L = I − SM ⊗ SD. Specifically, L can be denoted as
to known miRNA–disease association matrix pre2. Parameter σ represents the regularization parameter to balance the two terms. The Laplacian matrix L is denoted as L = I − SM ⊗ SD. Specifically, L can be denoted as
 | (9) |
 and
and 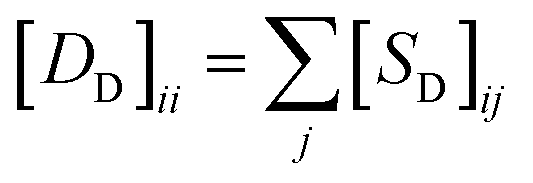 , respectively.
, respectively.Therefore, the prediction score matrix 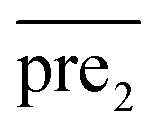 is calculated as follows:
is calculated as follows:
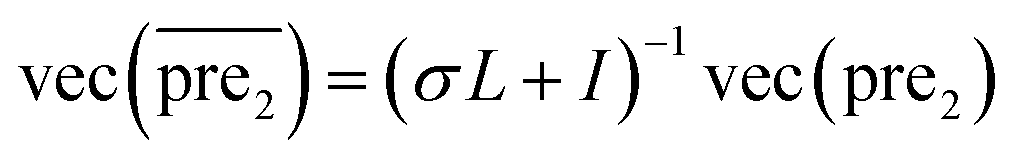 | (10) |
Information fusion strategy (RLSSLP)
In general, we can directly use conjugate gradient-based method30 to solve eqn (7) and (10). However, an unavoidable limitation exists due to the large memory requirements and computational time for storing the matrices, which include SM, SD, and
and 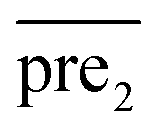 during the Kronecker operation. Therefore, we perform the eigenvalue transformation technique31 to improve the calculation efficiency and further extend its application on large-size miRNA–disease association network.
during the Kronecker operation. Therefore, we perform the eigenvalue transformation technique31 to improve the calculation efficiency and further extend its application on large-size miRNA–disease association network.
 | (11) |
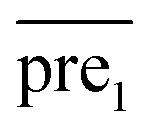 after eigenvalue transformation:
after eigenvalue transformation:
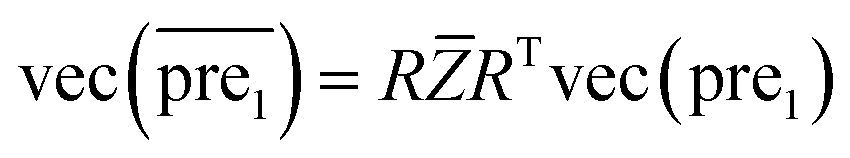 | (12) |
![[Z with combining macron]](https://www.rsc.org/images/entities/i_char_005a_0304.gif) is a diagonal matrix and its diagonal elements are []ii = λiα/(λiα + σ).
is a diagonal matrix and its diagonal elements are []ii = λiα/(λiα + σ).![[S with combining macron]](https://www.rsc.org/images/entities/i_char_0053_0304.gif) =
= ![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) ΛT as the Eigen decomposition of matrix for SLP. Then eqn (10) can be rewritten as:
ΛT as the Eigen decomposition of matrix for SLP. Then eqn (10) can be rewritten as:
 | (13) |
.Similar to RLS, after the transformation of the eigenvalue, the new prediction matrix 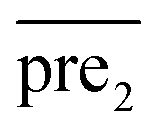 in SLP is as follows:
in SLP is as follows:
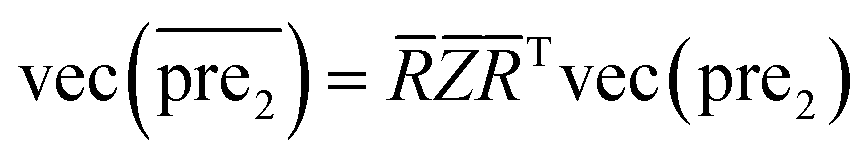 | (14) |
]ii = 1/(1 + σ − σλiα) is a diagonal matrix and its diagonal elements are []ii = 1/(1 + σ − σλiα).
Step3: combining the two submodels
Finally, we combine the new prediction score matrix and
and  after the eigenvalue transformation. The prediction matrix is obtained as follows:
after the eigenvalue transformation. The prediction matrix is obtained as follows:
 | (15) |
The more miRNA i strongly associates with disease j, the higher the corresponding prediction score  is.
is.
The eigen decompositions of similarity matrix SM and SD are one of the key technique in this paper to efficiently compute the inverse matrix on the eqn (6) and (8) that involves Kronecker operators. By applying this technique, the time complexity of the RLS sub-model is reduced from O((nm × nd)3) to O((nm)3 + (nd)3), in which nm and nd represent the number of miRNAs and diseases, respectively. Moreover, the time complexity of the SLP sub-model is O((nm)3 + (nd)3 + nm·nd), more detail can be found from ref. 14. Thus, the total time complexity is O((nm)3 + (nd)3 + nm·nd).
Results
Performance evaluation of RLSSLP
In this section, we implement a 10-fold cross-validation on the standard dataset to evaluate the performance of RLSSLP and other compared methods. In the experiment, both known and unknown miRNA–disease interactions are randomly divided into 10 subsets of equal sizes. In each repetition of the method, one subset of known miRNA–disease interactions and one of unknown interactions are selected (their scores in the adjacency matrix pre were set to 0) as test sets. We use the remaining nine subsets as training sets to recover the prediction score matrix. The value of area under the curve (AUC receiver operating characteristic (ROC)) is used as the main quantitative index for performance evaluation.Basing on the RLS and SLP models, we apply information fusion strategy and eigenvalue transformation technique to construct a global prediction framework to uncover potential miRNA–disease interactions. We evaluate the predictive performance of RLSSLP by considering the following aspects: (1) RLSSLP with information fusion strategy and eigenvalue transformation technique, (2) RLSSLP with RLS model only, and (3) RLSSLP with SLP model only. The ROC curves and average AUC values of RLSSLP at different situations mentioned above are displayed in Fig. 2.
 | ||
| Fig. 2 ROC curve and average AUC value of RLSSLP in different situations. (1) RLSSLP after information fusion strategy, (2) RLS only, (3) SLP only. | ||
As illustrated in Fig. 1, RLSSLP exhibited desirable predictive performance and achieved an AUC value of 0.9265. The AUC values for the RLS model and SLP models are 0.8992 and 0.8735, respectively. The RLSSLP framework of the combined information increases the AUC value, which is 2.73% and 5.30% higher compare with those of the RLS and SLP models, respectively. Evidently, the information fusion strategy and eigenvalue transformation technique enhance the predictive ability of RLSSLP.
Effect of parameter on RLSSLP performance
Parameters σ and α are introduced in the RLSSLP method. Parameter σ is the regularization parameter for the RLS and SLP models and balances the first and second terms in each regularization framework. Parameter α is an eigenvalue exponent and influences the improvement performance attributed to the application of the eigenvalue transformation technique. In this study, we set σ to 0.05 for the RLS model and 0.01 for SLP model in a noninformative manner according to a previous study.32 To explore the influence of parameter α on predictive performance, we fix regularization parameter σ and vary the value of α from 0 to 2 with an interval of 0.1 in the 10-fold cross-validation experiments. Fig. 3 shows the average AUC obtained from RLSSLP at different values of α. When we set the value of α from 0 to 2, the AUC value fluctuates between 0.6987 and 0.9450. When α = 1.3, RLSSLP has an optimal AUC value. More remarkably, when α = 1, the algorithm is equal to the original algorithm without applying the eigenvalue transformation technique.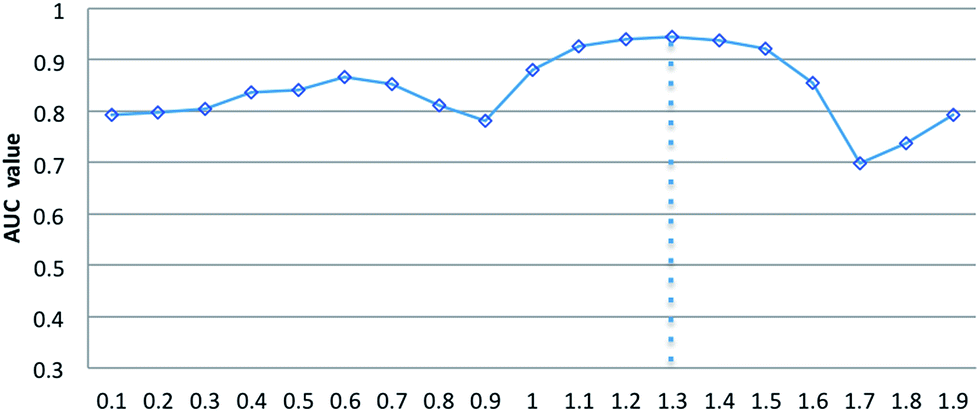 | ||
| Fig. 3 The effect of parameters α on the RLSSLP. | ||
Comparison with other methods
To the best of our knowledge, RWRMDA, RLSMDA, KATZ, HDMP, and KRLSM are the representative computer methods for the prediction of miRNA–disease associations. Given that RWRMDA and HDMP cannot predict diseases with no related miRNAs, we compared RLSSLP with RLSMDA, KATZ, and KRLSM.We implement a 10-fold cross-validation for RLSSLP and three other methods. The optimal parameters of RLSMDA, KATZ, and KRLSM are selected as described in literature. The ROC curves and AUC values of the four methods are shown in Fig. 4. The average AUC values of RLSSLP, RLSMDA, KATZ, and KRLSM are 0.9250, 0.8547, 0.9081, and 0.8324, respectively. RLSSLP has the best prediction result, and its average AUC value is increased and is 7.03%, 1.69%, and 9.26% higher than the other three approaches. The comparison results demonstrate that RLSSLP performs better than RLSMDA, KATZ, and KRLSM during the 10-fold cross-validation.
 | ||
| Fig. 4 Comparison among RLSSLP, RLSMDA, KATZ, and KRLSM with respect to their ROC curves and average AUC values obtained through the 10-fold cross-validation. | ||
Case studies
Few miRNAs play a crucial regulatory role in various human cancers. Case studies for breast, hepatocellular carcinoma (HCC), and lung cancer were implemented to evaluate the capability of RLSSLP to discover disease-related miRNA candidates. All the known miRNA–diseases interactions in the standard dataset were assigned as training sets, and the remaining unknown interactions served as testing sets. The predictive disease-related miRNAs were confirmed by public databases dbDEMC33 and miRCancer.34Lung cancer is one of the primary cancers that kill thousands of people annually. Early diagnosis and intervention can improve the low survival rate of patients with lung cancer. Many researchers reported that miRNAs, such as let-7e, mir-21, mir-25, mir-223, and mir-486, are potential premonitory biomarkers for lung cancer.35 In particular, Mir-145 inhibits tumor cell proliferation and is known to act as a tumor suppressor. Meanwhile, miRNA-192, miRNA-200c, and mir-21 are overexpressed during the progression of lung neoplasms.36 The top 20 potential miRNA candidates associated with lung cancer and predicted by RLSSLP are listed in Table 1. Of these candidates, 17 are verified by the dbDEMC and miRCancer databases to be associated with lung neoplasms. Meanwhile, three are not verified on these two databases, although we find that mir-296 suppresses cell viability in lung cancer37 (PMID: 26549165).
| Rank | miRNA name | Evidences |
|---|---|---|
| 1 | hsa-mir-708 | dbDEMC |
| 2 | hsa-mir-149 | dbDEMC |
| 3 | hsa-mir-625 | dbDEMC |
| 4 | hsa-mir-429 | miRCancer, dbDEMC |
| 5 | hsa-mir-296 | Unconfirmed |
| 6 | hsa-mir-302b | miRCancer, dbDEMC |
| 7 | hsa-mir-520b | dbDEMC |
| 8 | hsa-mir-92b | dbDEMC |
| 9 | hsa-mir-193b | dbDEMC |
| 10 | hsa-mir-378a | Unconfirmed |
| 11 | hsa-mir-20b | dbDEMC |
| 12 | hsa-mir-204 | dbDEMC |
| 13 | hsa-mir-302c | dbDEMC |
| 14 | hsa-mir-151a | Unconfirmed |
| 15 | hsa-mir-345 | dbDEMC |
| 16 | hsa-mir-367 | dbDEMC |
| 17 | hsa-mir-302d | dbDEMC |
| 18 | hsa-mir-99a | dbDEMC |
| 19 | hsa-mir-139 | dbDEMC |
| 20 | hsa-mir-211 | dbDEMC |
Meanwhile, HCC is the most common form of liver cancer. Analyzing miRNA expression data in cancerous liver tissues and normal tissues may facilitate the discovery of novel miRNA biomarkers and may assist the early detection of HCC cancer state. For instance, the expression levels of mir-125a, let-7e, mir-99b, and mir-195 are lower in HCC neoplasm tissues compared with those in normal liver tissues.38 Mir-92, mir-20, mir-100, mir-10a, mir-122, and mir-222 are more overexpressed in HCC tumor tissues compared with those in nontumor liver tissues.39 The top 20 potential HCC-related miRNAs predicted by RLSSLP and their confirmed interactions are listed in Table 2. All these miRNA candidates are associated with HCC. The associations are verified in the dbDEMC and miRCancer databases.
| Rank | miRNA name | Evidences |
|---|---|---|
| 1 | hsa-mir-185 | miRCancer, dbDEMC |
| 2 | hsa-mir-302d | dbDEMC |
| 3 | hsa-mir-135b | dbDEMC |
| 4 | hsa-mir-520h | dbDEMC |
| 5 | hsa-mir-302a | dbDEMC |
| 6 | hsa-mir-429 | miRCancer, dbDEMC |
| 7 | hsa-mir-367 | miRCancer, dbDEMC |
| 8 | hsa-mir-204 | miRCancer, dbDEMC |
| 9 | hsa-mir-638 | miRCancer, dbDEMC |
| 10 | hsa-mir-708 | miRCancer |
| 11 | hsa-mir-149 | miRCancer, dbDEMC |
| 12 | hsa-mir-215 | miRCancer, dbDEMC |
| 13 | hsa-mir-331 | miRCancer, dbDEMC |
| 14 | hsa-mir-625 | miRCancer |
| 15 | hsa-mir-186 | dbDEMC |
| 16 | hsa-mir-371a | miRCancer, dbDEMC |
| 17 | hsa-mir-211 | miRCancer, dbDEMC |
| 18 | hsa-mir-95 | miRCancer, dbDEMC |
| 19 | hsa-mir-194 | miRCancer, dbDEMC |
| 20 | hsa-mir-30e | miRCancer, dbDEMC |
Breast cancer is another type of cancer that seriously affects people's health, especially women. Few miRNAs are involved in the regulation of some critical processes in breast cancer progression, such as proliferation and apoptosis of breast neoplasm cell. For instance, mir-99a, mir-24, mir-101, mir-152, mir96, and the let-7 family are involved in the development of breast cancer.40,41 The top 20 potential breast neoplasm-related miRNAs predicted by RLSSLP and their confirmed interactions are listed in Table 3. Among these candidates, 18 were verified on dbDEMC and miRCancer databases, and only 2 miRNAs were unconfirmed. However, ref. 42 (PMID: 26657485) proved that mir-142 inhibits the invasiveness of human breast neoplasm cell. In addition, ref. 43 (PMID: 28121348) investigated that mir-542 regulates the proliferation and invasion of breast tumor cell.
| Rank | miRNA name | Evidences |
|---|---|---|
| 1 | hsa-mir-186 | dbDEMC |
| 2 | hsa-mir-330 | dbDEMC |
| 3 | hsa-mir-130a | miRCancer, dbDEMC |
| 4 | hsa-mir-185 | miRCancer, dbDEMC |
| 5 | hsa-mir-449a | dbDEMC |
| 6 | hsa-mir-99a | dbDEMC |
| 7 | hsa-mir-106a | miRCancer, dbDEMC |
| 8 | hsa-mir-95 | dbDEMC |
| 9 | hsa-mir-142 | Unconfirmed |
| 10 | hsa-mir-449b | dbDEMC |
| 11 | hsa-mir-650 | dbDEMC |
| 12 | hsa-mir-574 | miRCancer |
| 13 | hsa-mir-98 | miRCancer, dbDEMC |
| 14 | hsa-mir-376a | dbDEMC |
| 15 | hsa-mir-130b | dbDEMC |
| 16 | hsa-mir-381 | miRCancer, dbDEMC |
| 17 | hsa-mir-32 | dbDEMC |
| 18 | hsa-mir-99b | dbDEMC |
| 19 | hsa-mir-542 | Unconfirmed |
| 20 | hsa-mir-487b | dbDEMC |
Finally, we implement another experiment on an isolated disease (diseases without known related miRNAs) to demonstrate the strength of our method. We remove known verified miRNAs related to three diseases discussed above and predict potential miRNA candidates associated with a particular disease by only using similarity information and associations of other diseases. Consequently, the average AUC value of RLSSLP for the prediction of isolated diseases is 0.8175. Fig. 5 displays the predicted results of breast cancer, colonic cancer, and lung cancer.
 | ||
| Fig. 5 Network of top 40 miRNA candidates predicted by RLSSLP to be related to isolated diseases, namely, breast cancer, colonic cancer, and lung cancer. | ||
The results of these case studies further illustrate that RLSSLP exhibits good performance in identifying underlying disease-related miRNAs.
Discussion
In this study, we have presented a novel information fusion strategy called RLSSLP, which is based on regularized framework, for discovering underlying associations between miRNAs and diseases. RLSSLP can predict isolated diseases and does not require negative training samples. The results of the 10-fold cross-validation and case studies for lung cancer, HCC, and breast cancer indicate that RLSSLP performs well in predicting miRNA–disease interactions. The AUC value obtained through cross-validation also demonstrated that RLSSLP performs better compared with other state-of-art approaches.The favorable performance of RLSSLP can be mainly attributed to the following aspects. First, RLSSLP is a comprehensive prediction approach, which fuses various omics data that include the verified associations of miRNA–diseases, miRNAs–gene, and weighted gene–gene network. Second, RLSSLP combines two submodels to construct a more effective prediction framework for predicting miRNA–disease associations. Third, in RLSSLP, eigenvalue transformation technique can be used to improve the efficiency of the calculations.
Inevitably, the current version of RLSSLP has limitations. First, a more comprehensive similarity measurement for evaluating similarities for miRNAs and diseases can be adopted in the algorithm to improve the performance of RLSSLP. Second, the optimal value of parameter α can be obtained in a more satisfactory way. Finally, in RLSSLP, miRNA similarity measurement is based on miRNA-target associations. The number of known verified miRNA-target associations affects the prediction accuracy. The more the number of experimental validated miRNA targets, the more accurate the prediction is. Hence, the performance of the RLSSLP could be improved by obtaining more miRNA-target associations in the future. Nevertheless, RLSSLP exhibited good performance and can thus be considered a useful bioinformatics tool for biomedical research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is in part supported by National Natural Science Foundation of China (61702179, 61672356, 61572188), by Hunan Natural Science Foundation (2017JJ2239, 2016JJ2058), by Scientific Research Fund of Hunan Provincial Education Department (15B216), and by the Science and Technology Plan Project of Changsha (k1509003-11).References
- V. Ambros, Nature, 2004, 431, 350–355 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- P. Paul, A. Chakraborty, D. Sarkar, M. Langthasa, M. Rahman, M. Bari, R. Singha, A. K. Malakar and S. Chakraborty, J. Cell. Physiol., 2017, 9999 Search PubMed , 1–12.
- A. Ganju, S. Khan, B. B. Hafeez, S. W. Behrman, M. M. Yal lapu, S. C. Chauhan and M. Jaggi, Drug Discov. Today, 2017, 22, 424–432 CrossRef CAS PubMed.
- Q. Jiang, Y. Hao, G. Wang, L. Juan, T. Zhang, M. Teng, Y. Liu and Y. Wang, BMC Syst. Biol., 2010, 4, S2 CrossRef PubMed.
- H. Shi, J. Xu, G. Zhang, L. Xu, C. Li, L. Wang, Z. Zhao, W. Jiang, Z. Guo and X. Li, BMC Syst. Biol., 2013, 7, 101 CrossRef PubMed.
- P. Xuan, K. Han, M. Guo, Y. Guo, J. Li, J. Ding, Y. Liu, Q. Dai, J. Li and Z. Teng, et al., PLos One, 2013, 8, e70204 CAS.
- H. Chen and Z. Zhang, Sci. World J., 2013, 10, 204658 Search PubMed.
- J. Q. Li, Z. H. Rong, X. Chen, G. Y. Yan and Z. H. You, Oncotarget, 2017, 8, 21187 Search PubMed.
- Q. Jiang, G. Wang and Y. Wang, 2010 3rd International Conference On, 2010, vol. 6, pp. 2270–2274 Search PubMed.
- J. Xu, C. X. Li, J. Y. Lv, Y. S. Li, Y. Xiao, T. T. Shao, X. Huo, X. Li, Y. Zou and Q. L. Han, Mol. Cancer Ther., 2011, 10, 1857–1866 CrossRef CAS PubMed.
- X. Chen, C. C. Yan, X. Zhang, Z. H. You, Y. A. Huang and G. Y. Yan, Oncotarget, 2016, 7, 65257–65269 Search PubMed.
- Z. H. You, Z. A. Huang, Z. Zhu, G. Y. Yan, Z. W. Li, Z. Wen and X. Chen, PLoS Comput. Biol., 2017, 13, e1005455 Search PubMed.
- X. Chen, C. C. Yan, X. Zhang, Z. H. You, L. Deng, Y. Liu, Y. Zhang and Q. Dai, Sci. Rep., 2016, 6, 21106 CrossRef CAS PubMed.
- X. Chen, Z. C. Jiang, D. Xie, D. S. Huang, Q. Zhao, G. Y. Yan and Z. H. You, Mol. BioSyst., 2017, 13, 1202–1212 RSC.
- X. Chen, C. C. Yan, X. Zhang, Z. Li, L. Deng, Y. Zhang and Q. Dai, Sci. Rep., 2015, 5, 13877 CrossRef PubMed.
- Q. Zou, J. Li, L. Song, X. Zeng and G. Wang, Briefings Funct. Genomics, 2015, elv024 CrossRef PubMed.
- X. Chen and G. Y. Yan, Sci. Rep., 2014, 4, 5501 CrossRef CAS PubMed.
- J. Luo, Q. Xiao, C. Liang, C. Liang and P. Ding, IEEE Access, 2017, 5, 2503–2513 CrossRef.
- R. Raymond and H. Kashima, Mach Learn Discov., 2010, pp. 131–147 Search PubMed.
- Y. Li, C. Qiu, J. Tu, B. Geng, J. Yang, T. Jiang and Q. Cui, Nucleic Acids Res., 2014, 42, D1070–D1074 CrossRef CAS PubMed.
- S. D. Hsu, Y. T. Tseng, S. Shrestha, Y. L. Lin, A. Khaleel, C. H. Chou, C. F. Chu, H. Y. Huang, C. M. Lin and S. Y. Ho, et al., Nucleic Acids Res., 2014, 42, D78–D85 CrossRef CAS PubMed.
- T. Vergoulis, I. S. Vlachos, P. Alexiou, G. Georgakilas, M. Maragkakis, M. Reczko, S. Gerangelos, N. Koziris, T. Dalamagas and A. G. Hatzigeorgiou, Nucleic Acids Res., 2011, 40, D222–D229 CrossRef PubMed.
- F. Xiao, Z. Zuo, G. Cai, S. Kang, X. Gao and T. Li, Nucleic Acids Res., 2008, 37, D105–D110 CrossRef PubMed.
- I. Lee, U. M. Blom, P. I. Wang, J. E. Shim and E. M. Marcotte, Genome Res., 2011, 21, 1109–1121 CrossRef CAS PubMed.
- D. Wang, J. Wang, M. Lu, F. Song and Q. Cui, Bioinformatics, 2010, 26, 1644–1650 CrossRef CAS PubMed.
- A. Schlicker, F. S. Domingues, J. Rahnenfuhrer and T. Lengauer, BMC Bioinf., 2006, 7, 302 CrossRef PubMed.
- R. Rifkin and A. Klautau, J. Mach. Learn. Res., 2004, 5, 101–141 Search PubMed.
- Z. Xia, L. Y. Wu, X. Zhou and S. T. C. Wong, BMC Syst. Biol., 2010, 4, S6 CrossRef PubMed.
- H. Kashima, T. Kato, Y. Yamanishi, M. Sugiyama and K. Tsuda, in SDM, 2009, pp. 1100–1111 Search PubMed.
- Q. Kuang, X. Xu, R. Li, Y. Dong, Y. Li, Z. Huang, Y. Li and M. Li, Sci. Rep., 2015, 5, 13867 CrossRef PubMed.
- T. vanLaarhoven, S. B. Nabuurs and E. Marchiori, Bioinformatics, 2011, 27, 3036–3043 CrossRef CAS PubMed.
- Z. Yang, F. Ren, C. Liu, S. He, G. Sun, Q. Gao, L. Yao, Y. Zhang, R. Miao, Y. Cao, Y. Zhao, Y. Zhong and H. Zhao, BMC Genom., 2010, 11, S5 CrossRef CAS PubMed.
- B. Xie, Q. Ding, H. Han and D. Wu, Bioinformatics, 2013, 29, 638–644 CrossRef CAS PubMed.
- P. Leidinger, A. Keller and E. Meese, Front. Genet., 2012, 2, 104 Search PubMed.
- H. J. Yan, J. Y. Ma, L. Wang and W. Gu, Med. Sci. Mon., 2015, 21, 722 CrossRef PubMed.
- C. Xu, S. Li, T. Chen, H. Hu, C. Ding, Z. Xu, J. Chen, Z. Liu, Z. Lei and H. T. Zhang, et al., Oncol. Rep., 2016, 35, 497–503 CrossRef CAS PubMed.
- J. Tang, L. Li, W. Huang, C. Sui, Y. Yang, X. Lin, G. Hou, X. Chen, J. Fu and S. Yuan, et al., Canc. Lett., 2015, 364, 33–43 CrossRef CAS PubMed.
- H. S. Jung, Y. R. Seo, Y. M. Yang, J. H. Koo, J. An, S. J. Lee, K. M. Kim and S. G. Kim, Cell. Signal., 2014, 26, 1456–1465 CrossRef CAS PubMed.
- M. V. Iorio, M. Ferracin, C. G. Liu, A. Veronese, R. Spizzo, S. Sabbioni, E. Magri, M. Pedriali, M. Fabbri and M. Campiglio, Cancer Res., 2005, 65, 7065–7070 CrossRef CAS PubMed.
- L. Ma, G. Li, Z. Wu and G. Meng, Med. Oncol., 2014, 31, 773 CrossRef PubMed.
- A. Schwickert, E. Weghake, K. Brüggemann, A. Engbers, B. F. Brinkmann, B. Kemper, J. Seggewiß, C. Stock, K. Ebnet and L. Kiesel, et al., PLoS One, 2015, 10, e0143993 Search PubMed.
- H. X. Wu, G. M. Wang, X. Lu and L. Zhang, Eur. Rev. Med. Pharmacol. Sci., 2017, 21, 108–114 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |