
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic degradation of norfloxacin on different TiO2−X polymorphs under visible light in water†
Hai Yang ab,
Liangyong Meib,
Pengcheng Wangb,
Joseph Genereuxb,
Yinsheng Wangb,
Bing Yia,
Chaktong Aua,
Limin Danga and
Pingyun Feng*b
ab,
Liangyong Meib,
Pengcheng Wangb,
Joseph Genereuxb,
Yinsheng Wangb,
Bing Yia,
Chaktong Aua,
Limin Danga and
Pingyun Feng*b
aCollege of Chemistry and Chemical Engineering, Hunan Institute of Engineering, Xiangtan 411104, P. R. China
bDepartment of Chemistry, University of California, Riverside, California 92521, USA. E-mail: pingyun.feng@ucr.edu
First published on 26th September 2017
Abstract
Reduced TiO2 (TiO2−X) materials with different crystallographic structures were prepared and characterized. Cat.I-A, Cat.II-R, Cat.III-B are TiO2−X with anatase, rutile and brookite structures, respectively, while the Cat.IV-A&R series are materials with anatase and rutile phases mixed in different ratios. All samples exhibit efficient photocatalytic activity for the degradation of norfloxacin (Nor) under visible light, and Cat.IV-A&R-4 is the best among the samples studied. Our results show that the photocatalytic activity is governed by different factors such as the specific surface area of the catalysts as well as the concentrations of Ti3+ and the density of oxygen vacancies in the photocatalytic materials. Mechanistic study of the materials demonstrates that photohole (h+) transfer contributes more to Nor degradation than reaction with ˙OH radicals and the other reactive oxygen species (ROSs). Intermediate species were characterized by HPLC-TOF-HRMS and HPLC-MS/MS to construct a general transformation mechanism of Nor on the family of TiO2−X under visible light. The study shows that Nor adsorption onto TiO2−X occurs by its heteroatoms followed by cleavage of its piperazine ring and hydroxylation of its quinolone ring under the attack of h+ and ˙OH radicals. The study could assist the further search for efficient photocatalytic materials for the degradation of organic pollutants.
1. Introduction
Owing to its abundance, low toxicity, chemical and thermal stability, and resistance to photocorrosion, TiO2 has been extensively studied as a catalyst for different photocatalytic reactions,1–4 and used for paints,5 coatings,6 supports for drug delivery,7 angiogenic agents8 and so on. Crystalline TiO2 itself has limited practical applications as a photocatalytic material because its activity is driven by ultraviolet (UV) light. Nonetheless, it was demonstrated that partially reduced TiO2 with Ti3+ and/or oxygen vacancies can be an efficient photocatalytic material under visible light irradiation.9–12 In the presence of Ti3+ and/or oxygen vacancies, the photo response of TiO2−X can be extended from the UV to the visible light region, leading to visible-light photocatalytic activity not only for the generation of hydrogen from water but also for the degradation of different organic pollutants.13–17The most stable polymorphs of TiO2 are three, viz. anatase, rutile and brookite.18,19 Although rutile is thermodynamically the most stable, anatase is often dominant in nanocrystalline TiO2 because of its lower surface energy. In numerous studies, anatase was found to be photocatalytically more active than rutile.20 Brookite is stable at relatively lower temperature and is commonly obtained together with the other polymorphs.21 Recently, strategies have been developed to synthesize TiO2−X with different morphologies and phases. It was found that TiO2−X crystallinity and morphology can have a dramatic impact on photocatalytic properties. For example, TiO2−X in the form of anatase,14,22,23 rutile,13,17,24–26 and brookite27,28 were studied as photocatalytic materials for water splitting under visible light, which is the most widely studied application. The use of titanium oxide, especially anatase, has also been studied for hydrogen generation by methanol photoreforming.29 However, few systematic studies on TiO2−X isoforms for photocatalytic the degradation of organic pollutants in water have been reported, nor have the degradation mechanisms been assigned.30–34 It would be of great scientific interest and practical importance to find out the differences among the various TiO2−X in terms of reaction kinetics and mechanism for the degradation of organic pollutants in water.
Norfloxacin (Nor) is a fluoroquinolone antibiotic widely used for respiratory and bacterial infections.35–37 Because of its low metabolic rate and poor biodegradability, fluoroquinolone antibiotics are detected not only in effluents of wastewater treatment plant, but also in surface water and other environmental matrixes.38–40 The persistence of fluoroquinolone antibiotics in aquatic environment in particular can lead to antibiotic resistance.41–43 Almost 70% of Nor was left in the sludge of biological treatment plants.41,44 As a result, pathogens become increasingly resistant to the drugs,43,45,46 posing a great threat to aquatic and terrestrial organisms as well as to humans. Recently, systems of photocatalytic degradation under UV light,47–49 nanoscale zero-valent iron with H2O2,50 electro-Fenton treatment,51 thermally activated persulfate,52 and gamma-ray irradiation53 were deployed to remove Nor. However, these approaches suffer from drawbacks such as high energy consumption, low degradation efficiency and being environmentally unfriendly.49,54 The development of efficient photocatalytic materials for the degradation of Nor still remains as an important issue and challenge.
In this study, a series of TiO2−X with different crystalline phases and Ti3+ concentrations were synthesized and studied for the degradation of Nor under visible light irradiation in water. The degradation kinetics and the roles of reactive oxygen species (ROSs) were elucidated. The mechanistic steps of Nor degradation on the TiO2−X were explored based on both theoretical calculations and experimental results.
2. Experimental
2.1 Chemicals
Nor (Sigma-Aldrich) was used as received (≥98%). 5,5-Dimethyl-1-pyrrolidine N-oxide (DMPO, 97%) was supplied by Sigma-Aldrich (Saint Louis, MO, USA). Water of HPLC grade was obtained using a Millipore Milli-Q system in which a xenon arc lamp at 172 nm was used to provide constant illumination to keep the total organic carbon (TOC) concentration of water below 13 μg L−1. Formic acid (88%) and tert-butyl alcohol (99.5%) were purchased from Fisher Chemical and J.T. Baker, respectively. Acetonitrile for mobile phase was also purchased from Fisher Chemical. The syringe filters with MCE membrane having pore size of 0.22 μm were from CELLTREAT Scientific. Other reagents were all analytical grade.2.2 Preparation of different TiO2−X polymorphs
All catalysts were prepared following procedures published elsewhere,13,17,23,27 and the details are provided as ESI.†2.3 Characterization of different TiO2−X polymorphs
Powder X-ray diffraction data were collected on a Bruker D8-Advance powder diffractometer operating at 40 kV and 40 mA (Cu Kα radiation, λ = 1.5406 Å). The EPR spectra were obtained on a Bruker EMX/plus spectrometer (Germany), with a resonance frequency of 9.363 GHz, microwave power of 20.0 mW, modulation frequency of 100 kHz, modulation amplitude of 1.0 G, sweep width of 800 Gauss, center field of 3400 Gauss, time constant of 40.96 ms, sweep time of 81.92 s, and receiver gain of 1.0 × 103. DMPO (100 mM) was used as the spin-trap agent. The UV-visible absorption spectra were recorded on a Shimadzu UV-3101 PC UV-Vis-NIR spectrophotometer operating in the diffuse mode with the application of Kubelkae–Munk equation. BET measurements were performed using an ASAP 2020 (Micromeritics) equipment.2.4 Photocatalytic degradation experiments
The experiments of Nor degradation were performed in a photochemical reactor (Perfect, Beijing, Perfect Technology Co., LTD). The light was from a 300 W Xe lamp (350–780 nm, operated at 10 A) installed with a 400 nm cut-on filter (Newport Corp.) that was housed on top of a 150 mL beaker. The distance between the light and reactor was 15 cm and the light intensity (>400 nm) was 14 mW cm−2. Prior to illumination, a mixture of Nor (100 μM L−1) and TiO2−X (0.100 g) was stirred in dark for 30 min to achieve adsorption–desorption equilibrium. Then, the light was turned on and the solution was irradiated under constant stirring. The reaction solution was sampled (2 mL) at fixed time intervals, and analyzed (after going through a 0.22 μm filter) by HPLC, HPLC/TOF/HRMS and HPLC-MS/MS techniques. Upon termination of the experiment, the reaction solution was subject to TOC measurement after centrifugation for the removal of solid substances. All experiments were performed at room temperature (21 ± 1 °C) and the pH values were adjusted to 7.0. Each batch experiment was performed in triplicate. In most cases, the error was less than 5%.2.5 Analysis
The photocatalytic degradation kinetics of Nor was studied at 25 °C using an Agilent 1260 HPLC (Kromasil C18 column, 150 × 4.6 mm, particle size of 5.0 μm) instrument. The mobile phase was 60% CH3CN and 40% H2O (containing 0.3% HCOOH). All of the solutions were filtered with a Water Associates (Milford; MA, USA) 0.45 μm filter before analysis. The injection volume was 20 μL. The detection wavelength for all compounds was 276 nm,47 and the flow rate of mobile phase was 0.5 mL min−1.To confirm the generation of radicals, EPR investigation was conducted using DMPO as a spin-trapping agent over a Bruker instrument (EMX EPR/X-band, Germany). Samples were collected at designated time and subject to centrifugation (8000 rpm min−1) for 5 min. After that, the solution was transferred into a 100 μL capillary tube which was then fixed in the cavity of the EPR spectrometer.
The photocatalytic degradation intermediates were analyzed first over an Agilent 1200 Infinity LC system coupled with a quadrupole time-of-flight high resolution mass spectrometer (Q-TOF HRMS, Triple TOF 6210, AB SCIEX, USA). The injection volume was 10 μL. Chromatographic separation was performed using a Thermo BDS Hypersil C18 column (2.1 × 100 mm, particle size of 2.4 μm, Thermo Fisher Scientific, Waltham, MA) maintained at 30 °C at a flow rate of 300 μL min−1. The mobile phase was composed of A (water containing 0.3% formic acid) and B (acetonitrile). The A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B proportion was changed linearly from 90:10 (v/v) to 10:90 in 20 min, and then returned to 90:10 in 5 min and held for 5 min for re-equilibration. Mass spectrometric analysis was performed in positive-ion mode using an electrospray ionization (ESI†) source. The mass range was m/z 50–800. The operation parameters are: ion source gas I, 55 (arbitrary units); gas II, 55 (arbitrary units); source temperature, 550 °C; ion spray voltage floating, −4500 V; and declustering potential, −80 V. Water signals were recorded for the subtraction of sample background.
:B proportion was changed linearly from 90:10 (v/v) to 10:90 in 20 min, and then returned to 90:10 in 5 min and held for 5 min for re-equilibration. Mass spectrometric analysis was performed in positive-ion mode using an electrospray ionization (ESI†) source. The mass range was m/z 50–800. The operation parameters are: ion source gas I, 55 (arbitrary units); gas II, 55 (arbitrary units); source temperature, 550 °C; ion spray voltage floating, −4500 V; and declustering potential, −80 V. Water signals were recorded for the subtraction of sample background.
To separate and purify the degradation intermediates, Agilent 1260 HPLC was employed as semi-preparative HPLC to obtain the individual intermediates. The separation was performed at fixed flow rate of 0.5 mL min−1 with linearly changing the ratio of A (water containing 0.3% formic acid) and B (acetonitrile) from 90:10 (v/v) to 10:90 in 30 min, and then returning to 90:10 in 10 min. The injection volume was 100 μL. Once the peaks of degradation intermediates were recorded, the sample was retained for future HPLC-MS/MS analysis over a LTQ linear ion trap mass spectrometer (Thermo Electron, San Jose, CA, USA) with Agilent Zorbax SB C18 column (0.5 × 250 mm, particle size of 5 μm size). The mobile phase was composed of C (water containing 0.1% formic acid) and D (acetonitrile containing 0.1% formic acid). The flow rate was 8 μL min−1, and the gradient profile for HPLC-MS/MS analysis in terms of solution D was from 10% to 90% in 30 min. The temperature for the ion-transport tube was maintained at 300 °C. An electrospray interface was used for MS and MS-MS measurements in positive ionization mode, and scan acquisition was between m/z 50 and 500. The collision energy was varied according to requirement, and spray voltage was 5.5 keV.
TOC contents of samples were measured on a Shimadzu TOC-500 analyzer with catalytic oxidation on a Pt set at 680 °C. Triplicate analyses were performed for each sample.
2.6 Theoretical calculation for PCs and FEDs
Molecular orbital calculations were performed using Gaussian 03 program (Gaussian, Inc.) at the single determinant (HF/3-21) level. The optimal conformation having a minimum energy was obtained at the B3LYP/6-31G* level. The (FEDHOMO2 + FEDLUMO2) and 2FEDHOMO2 values were acquired to predict the initial state of hydroxyl radicals (˙OH) and photoholes (h+) interaction with Nor, respectively. Also, the point charges (PCs) were calculated by means of natural bond orbital (NBO) to predict the adsorption orientation of Nor molecules on the surface of TiO2−X.3. Results and discussion
3.1 Characterization of different TiO2−X
As shown in Fig. 1, Cat.I-A displays XRD peaks at 2θ = 25.4°, 37.9°, 48.1° and 53.1° and these peaks match well with the structure of anatase. For Cat.II-R, the XRD pattern matches well with rutile structure. According to JCPDS card no. 29–1360, Cat.III-B is a brookite with no other phases.27 As for the four Cat.IV-A&R samples, the XRD results indicate the presence of both anatase and rutile phases.13 We used the WR = AR/(0.884AA + AR) equation to estimate the weight percentage of rutile (WR) in the mixture, with AA and AR corresponding to the integrated intensity of the rutile (110) and anatase (101) peak.24 The results indicate that WR for Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4 is 88.9%, 82.4%, 24.7% and 5.3%, respectively. And correspondingly the anatase weight percentage is 11.1%, 17.6%, 75.3% and 94.7%.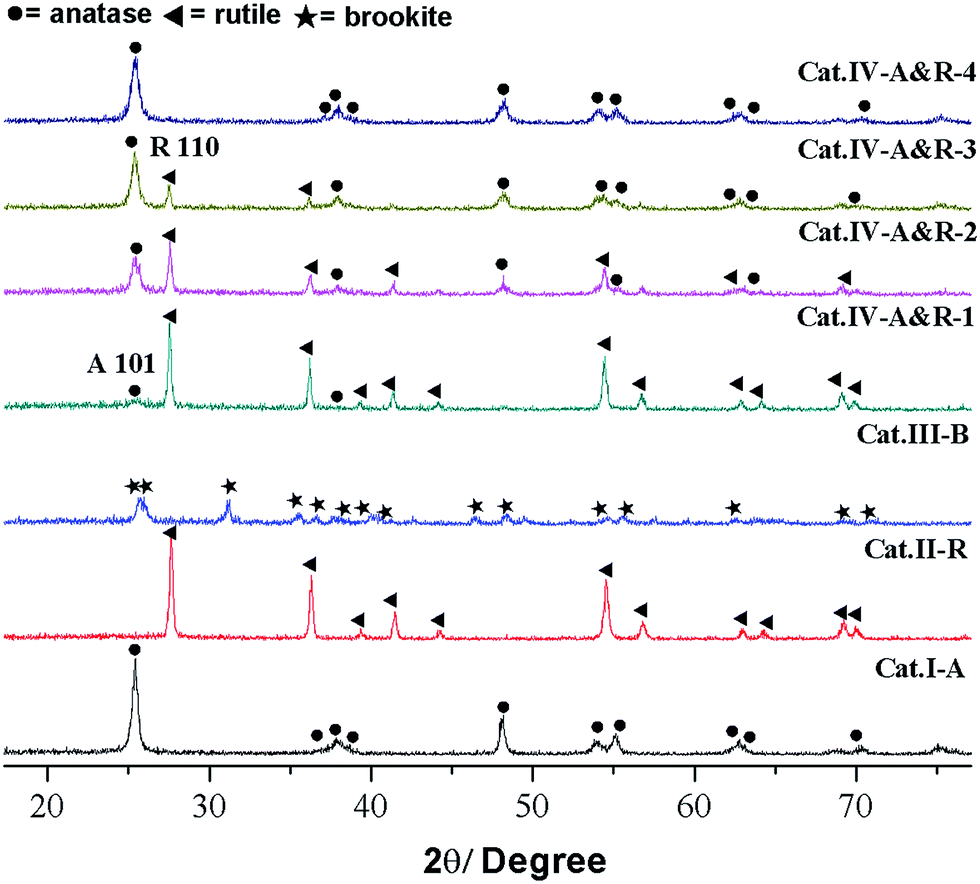 | ||
| Fig. 1 XRD patterns of different TiO2−X, ● = anatase; ◀ = rutile; ★ = brookite. | ||
The Ti3+ concentration of the samples was estimated by EPR measurements (Fig. 2). All samples display strong EPR signals with the Landé g factor values (g-values) equal to 2.003, 1.971, 1.984, and 1.971 for Cat.I-A, Cat.II-R, Cat.III-B and Cat.IV-A&R series samples, respectively. Different g-values obtained indicate that there is no Ti3+ but rather oxygen vacancies in Cat.I-A,23 whereas Ti3+ is present in the other six samples. The EPR signals at g-value of 2.003 is caused by electrons trapped on surface oxygen vacancies, while the representative signals of Ti3+ appears at g ≈ 1.98.27 Quantitative determination of Ti3+ concentration was achieved by numerical double integration of the EPR spectra. The standard EPR calibration curve was prepared by measuring a series of frozen aqueous solution of Cu2+ (shown in Fig. S1†). Referring to the calibration curve, the Ti3+ concentration of Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4 is 4.95, 4.34, 16.70, 25.50, 4.50 and 0.30 μmol g−1, respectively (Table 1), giving a decreasing order of Cat.IV-A&R-2 > Cat.IV-A&R-1 > Cat.II-R > Cat.IV-A&R-3 > Cat.III-B > Cat.IV-A&R-4. The specific surface area of Cat.I-A, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4 is 263.95, 5.50, 5.20, 11.72, 18.96, 27.86, and 56.83 m2 g−1, respectively, giving a decreasing order of Cat.I-A > Cat.IV-A&R-4 > Cat.IV-A&R-3 > Cat.IV-A&R-2 > Cat.IV-A&R-1 > Cat.II-R ≈ Cat.III-B. It should be noted that there are significant variations in the particle size, which are 10, 160, 130, 80 and 60 nm for Cat.I-A, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4, respectively.
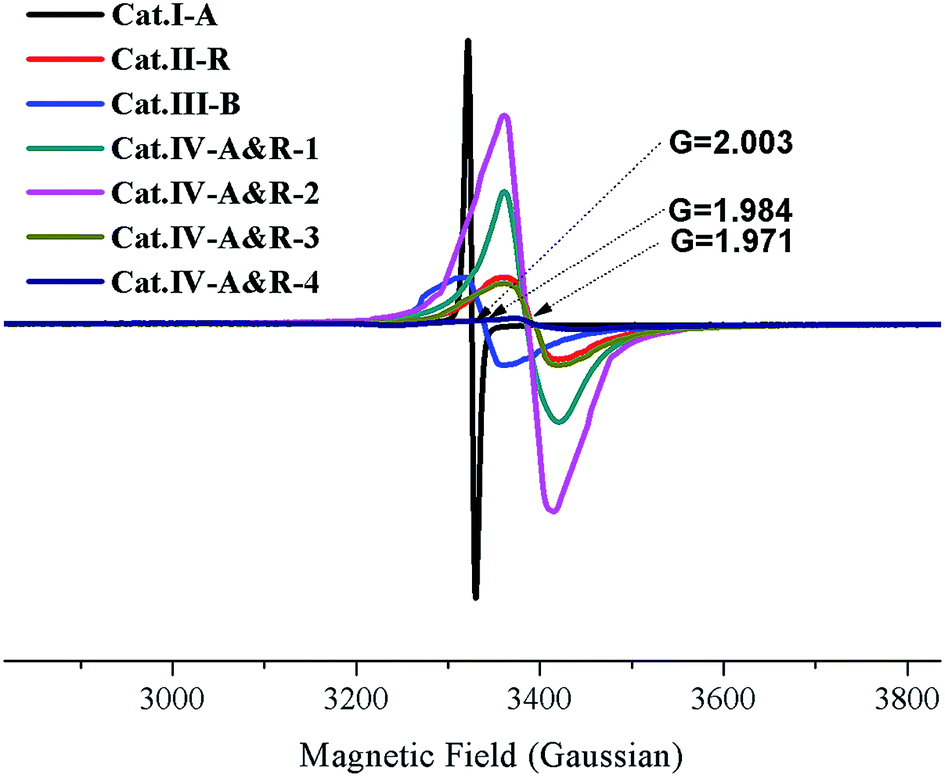 | ||
| Fig. 2 EPR spectra of different TiO2−X. | ||
| Cat. | Crystallinity | Ti3+ concentration (μmol g−1) | Oxygen vacancy | Surface area (m2 g−1) | Particle size (nm) | ||
|---|---|---|---|---|---|---|---|
| Ana.% | Rut.% | Bro.% | |||||
| a Ref. 19.b Ref. 13.c Ref. 23, platelike features crystal.d Ref. 9 and 20.e Tetragonal bipyramid crystal, the angle between the edges of prismatic and pyramidal facets is 53.6. | |||||||
| Cat.I-A | 100 | 0 | 0 | — | Non stoichiometric | 263.95a | 10a |
| Cat.II-R | 0 | 100 | 0 | 4.95 | — | 5.5b | ≈1500 × 700 × 700e |
| Cat.III-B | 0 | 0 | 100 | 4.34 | — | 5.2 | ≈400 × 180 × 10c |
| Cat.IV-A&R-1 | 11.1 | 88.9 | 0 | 16.7 | — | 11.72d | 160d |
| Cat.IV-A&R-2 | 17.6 | 82.4 | 0 | 25.5 | — | 18.96d | 130d |
| Cat.IV-A&R-3 | 75.3 | 24.7 | 0 | 4.5 | — | 27.86d | 80d |
| Cat.IV-A&R-4 | 94.7 | 5.3 | 0 | 0.3 | — | 56.83d | 60d |
3.2 Degradation kinetics of nor on TiO2−X under visible light
The application of the seven catalysts for the degradation of Nor under visible light (>400 nm) irradiation was performed, and the results are presented in Fig. 3. Without a catalyst or with P25 TiO2, there is no degradation of Nor, whereas in the presence of the catalysts, Nor degradation is substantial. All these TiO2−X catalysts enable degradation close or equal to 100% within 240 min except for Cat.II-R. The plots of −ln(C/C0) vs. time display a degradation rate of pseudo-first order (Fig. S2†),55,56 and the rate constants for Cat.I-A, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4 are 0.0156, 0.0059, 0.0059, 0.0093, 0.0146, 0.0210 and 0.0361 min−1, respectively; giving a decreasing order of Cat.IV-A&R-4 > Cat.IV-A&R-3 > Cat.I-A > Cat.IV-A&R-2 > Cat.IV-A&R-1 > Cat.II-R ≈ Cat.III-B, which does not follow the decreasing order of Ti3+ concentration, which is Cat.IV-A&R-2 > Cat.IV-A&R-1 > Cat.II-R > Cat.IV-A&R-3 > Cat.III-B > Cat.IV-A&R-4. It is noted that Cat.I-A, which is the highest in specific surface area and with oxygen vacancies rather than Ti3+, is moderate in activity. While despite lowest in Ti3+ concentration (not counting Cat.I-A), Cat.IV-A&R-4 is photocatalytically the most active one for the degradation of Nor.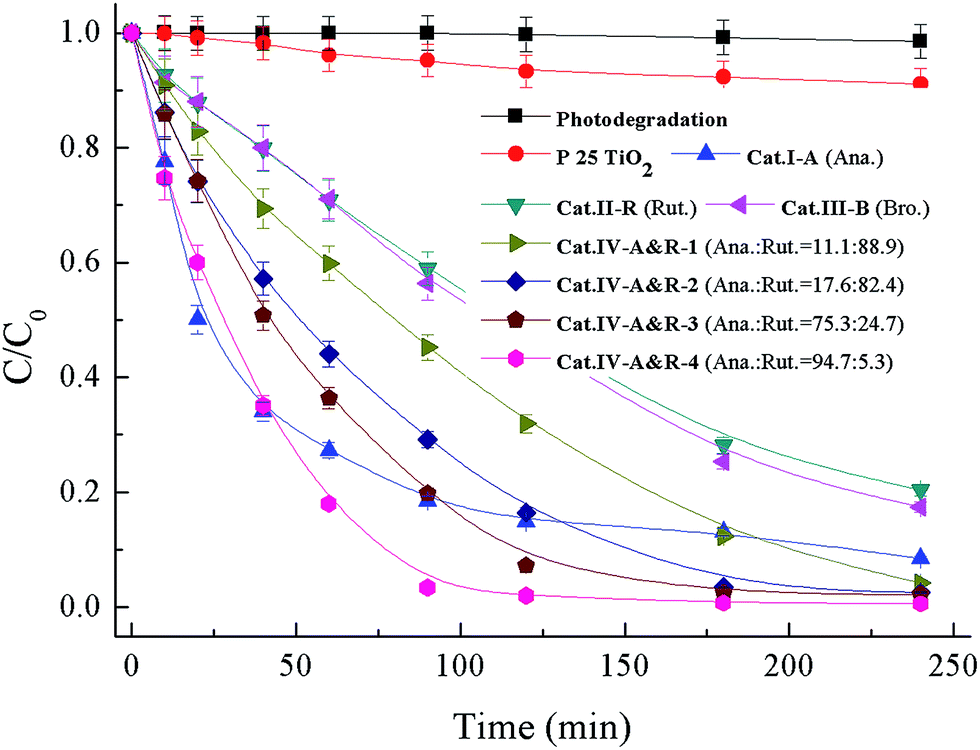 | ||
| Fig. 3 Degradation curves of Nor in the system of 0.1 g L−1 TiO2−X under visible light (>400 nm). Anatase = Ana.; rutile = Rut.; brookite = Bro. | ||
It is envisaged that the adsorption capacity of Nor on catalyst surface could have an influence on reaction activity. The adsorption isotherms of the seven catalysts are displayed in Fig. S3,† showing an adsorption capacity follows a decreasing order of Cat.I-A > Cat.IV-A&R-4 > Cat.IV-A&R-3 > Cat.IV-A&R-2 > Cat.III-B > Cat.II-R > Cat.IV-A&R-1, which is somewhat different from that of degradation rate constants. In addition, Cat.IV-A&R-1, Cat.II-R, and Cat.III-B are relatively high in Ti3+ concentrations, however these samples show relatively low degradation rates. In this special case of using TiO2−X for Nor degradation, Cat.IV-A&R-4, which is low in Ti3+ concentration and second in Nor adsorption capacity, exhibits highest photocatalytic activity. The results suggest that the degradation efficiency is a synergetic effect of a number of factors.
We then turned our attention to the contribution of ROSs such as ˙OH and photogenerated holes (h+) and electrons (e−) (Table 2). Formic acid (FA) is considered a scavenger for h+ and ˙OH, while tert-butanol (t-BuOH) is an excellent quencher of ˙OH as illustrated by eqn (1).57,58 FA under neutral and acidic conditions is strongly adsorbed on the surface of TiO2, consuming h+ (eqn (2)) and eliminating or intercepting the generation of ˙OH (eqn (3)).When 0.1 g of FA is introduced to the solution as a diagnostic tool for suppressing the h+ and ˙OH species, the photocatalytic degradation of Nor is inhibited significantly (Table 2). The rate constants, respectively, decrease from 0.0156, 0.0059, 0.0059, 0.0093, 0.0146, 0.0210 and 0.0361 min−1 to 0.0021, 0.0008, 0.0010, 0.0019, 0.0017, 0.0024 and 0.0035 min−1 for Cat.I-A, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4, suggesting that 87%, 86%, 83%, 80%, 88%, 89% and 90% of the photocatalytic degradation efficiency may be caused by h+ and ˙OH. On the contrary, when 6 mL t-BuOH is added, the degradation rate constants change only slightly to 0.0142, 0.0042, 0.0047, 0.0074, 0.0138, 0.0195 and 0.0238 min−1, indicating that 9%, 29%, 20%, 20%, 5%, 7% and 34% of photocatalytic degradation efficiency is due to ˙OH for Cat.I-A, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4, respectively. Taking away the contribution of ˙OH, that of h+ is 78%, 57%, 63%, 60%, 83%, 82% and 56% for Cat.I-A, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4, respectively. And the rest, viz. 13%, 14%, 17%, 20%, 12%, 11% and 10% for Cat.I-A, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4, could be collectively due to the other ROSs. The results confirm that over the TiO2−X, the h+ species play an important role in the degradation while ˙OH only takes a minor role. Such a phenomenon is significantly different from that of P25 TiO2 for the degradation of organic pollutants under UV light,59,60 in which ˙OH plays a decisive role while h+ and the other ROSs have a minor effect.
| Cat. | Rate constant (min−1) | Degradation contribution% | ||||
|---|---|---|---|---|---|---|
| No scavenger | FA | t-BuOH | h+ | ˙OH radicals | Other ROSs | |
| Cat.I-A | 0.0156 | 0.0021 | 0.0142 | 78 | 9 | 13 |
| Cat.II-R | 0.0059 | 0.0008 | 0.0042 | 57 | 29 | 14 |
| Cat.III-B | 0.0059 | 0.0010 | 0.0047 | 63 | 20 | 17 |
| Cat.IV-A&R-1 | 0.0093 | 0.0019 | 0.0074 | 60 | 20 | 20 |
| Cat.IV-A&R-2 | 0.0146 | 0.0017 | 0.0138 | 83 | 5 | 12 |
| Cat.IV-A&R-3 | 0.0210 | 0.0024 | 0.0195 | 82 | 7 | 11 |
| Cat.IV-A&R-4 | 0.0361 | 0.0035 | 0.0238 | 56 | 34 | 10 |
It is envisaged that if h+ has a major role, a catalyst of higher specific surface area should perform better. However, it is noted that despite Cat.IV-A&R-4 is the second highest in specific surface area, the contribution of h+ (56%) is the lowest. This phenomenon could be related to the high adsorption capacity of Cat.IV-A&R-4 which is about 40 μmol g−1, the second highest among the seven catalysts (Fig. S3†). In such a case, the surplus h+ reacts with H2O to produce ˙OH (eqn (4)), and then the attack of ˙OH on Nor results in its relatively more contribution (34%) during the photocatalytic degradation process. It is noted that Cat.I-A (one with no Ti3+ but only oxygen vacancies), which has the highest specific surface area and decent h+ concentration among the studied samples, shows only mediocre activity. It is hence deduced that oxygen vacancies possess lower activity than Ti3+ under visible light. This could be caused by the higher Ti3+ concentration, which contributed positively to the donor density, and the higher donor density could lead to better electrons–hole separation as we observed previously.61
| ˙OH + t-BuOH → ˙CH2C(CH3)2OH + H2O | (1) |
| HCOOH + 2h+ → CO2 + 2H+ | (2) |
| HCOOH + ˙OH → HCO3− + H2O | (3) |
| h+ + H2O → ˙OH + ˙H | (4) |
EPR spectroscopy using DMPO as a spin-trapping agent can be employed to monitor the behavior of ROSs during photocatalytic processes.62–65 ROSs such as ˙OH and ˙O2− react with DMPO as illustrated in Scheme S1.† Simulated with Win EPR acquisition software, DMPO-OH signals (four lines, 1:2:2:1) of the Cat.IV-A&R-1/visible light system can be identified based on hyperfine splitting constants (aH = aN = 14.7 G) with no detection of other ROSs such as DMPO-OOH (Fig. 4a). The DMPO-OH signals become stronger with reaction time, and are the strongest at 10 min. Further increase of reaction time results in gradual decline and final disappearance of the DMPO-OH signals. The results indicate that ˙OH generation is due to visible light irradiation of TiO2−X, most plausibly from photo activated e− and h+ generation at Ti3+ and oxygen vacancies. Furthermore, there is no detection of ˙O2− signals due to the rapid conversion of DMPO-OOH to DMPO-OH (Scheme S1†).61
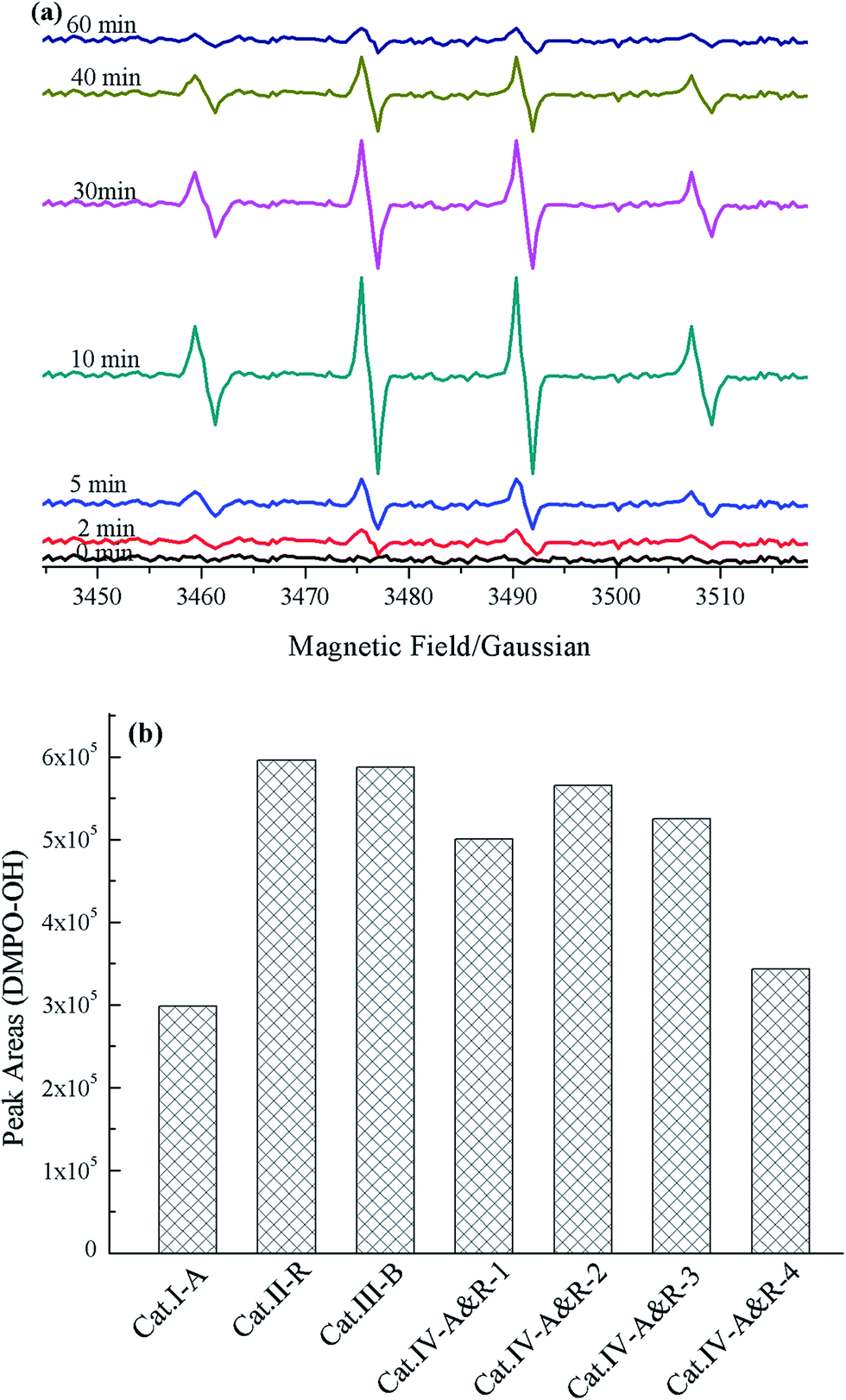 | ||
| Fig. 4 (a) EPR signals versus reaction time of the Cat.IV-A&R-1 system, and (b) intensity of DMPO-OH peaks of the catalytic systems acquired at 10 min. | ||
To evaluate the generation efficiency of ˙OH of the different TiO2−X, the DMPO-OH signals collected at 10 min was subject to integration and the results were compared (Fig. 4b). To our surprise, there is no direct relationship between ˙OH concentration and the presence of Ti3+ or oxygen vacancies. Despite different in Ti3+ concentration, Cat.II-R, Cat.III-B, Cat.IV-A&R-1, Cat.IV-A&R-2 and Cat.IV-A&R-3 are similar in terms of ˙OH concentration. Furthermore, Cat.I-A and Cat.IV-A&R-4 which are high in concentration of oxygen vacancies and Ti3+, respectively, are low in ˙OH concentration. It is clear that the concentrations of Ti3+ and oxygen vacancies are not the only factor to afford the generation of ˙OH. More importantly, it is also indicated that ˙OH doesn't play a decisive role in the degradation of Nor during TiO2−X/visible light system.
We come to a similar conclusion when we compared the TOC values of the reaction solutions after 4 h of photodegradation over the different TiO2−X (Fig. 5). Among the seven catalysts, Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4 show higher ability for Nor mineralization. Compared to a TOC value of 19.62 mg L−1 for an initial Nor amount of 100 μmol L−1, the TOC values over Cat.IV-A&R-2, Cat.IV-A&R-3 and Cat.IV-A&R-4 are much smaller: 10.18, 10.59, and 9.93 mg L−1, respectively. The TOC value over Cat.II-R, Cat.III-B, and Cat.IV-A&R-1 is 16.93, 14.31, and 14.67 mg L−1. The results again indicate that the photocatalytic activity cannot be directly related to Ti3+ concentration of the catalysts. A TOC value of 19.33 mg L−1 is observed over Cat.I-A after 4 h of degradation reaction. The result is inconsistent with the fact that Cat.I-A shows moderate activity for Nor degradation. However, as L-ascorbic acid is employed in Cat.I-A preparation, the high TOC value could represent residual L-ascorbic acid in Cat.I-A.
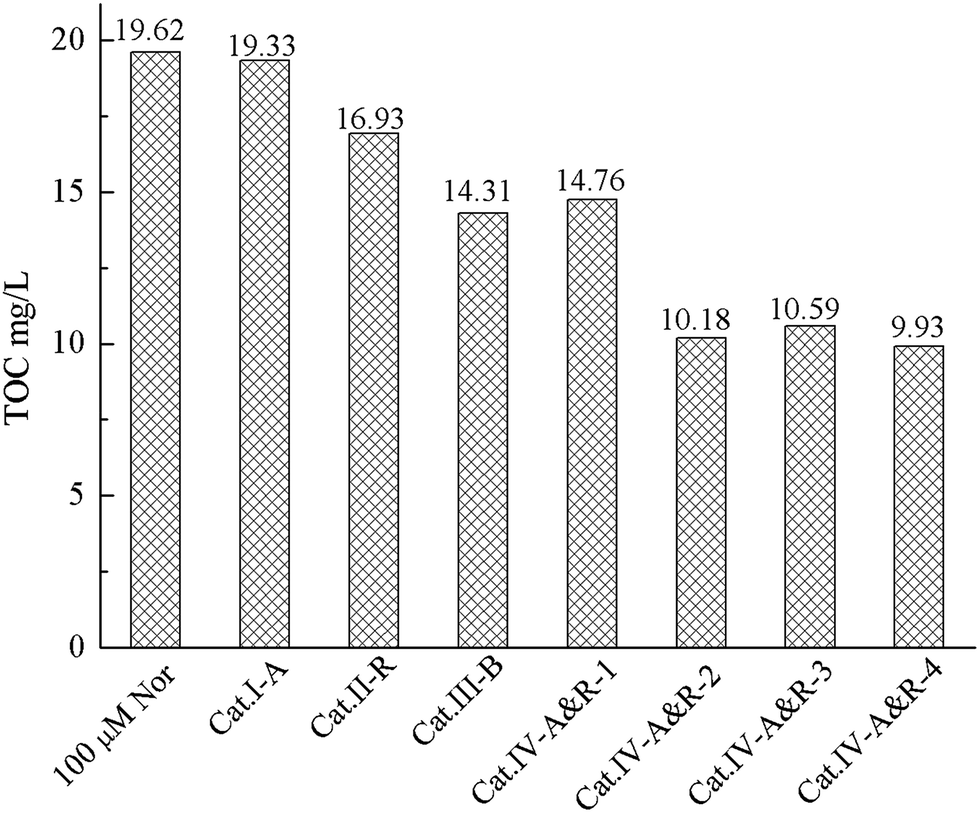 | ||
| Fig. 5 Comparison of TOC values after 4 h of photocatalytic degradation. | ||
3.3 Intermediates and pathways for nor degradation on TiO2−X under visible light
We used the HPLC-TOF-HRMS technique to identify the degradation intermediates and to elucidate the reaction mechanism of Nor degradation on the TiO2−X samples. As shown in Table 3, 19 degradation intermediates, namely, P1 to P19, were detected. Not all, but most of these are detected over each individual catalyst. Briefly, P9, P14 and P18 are not detected over Cat.I-A; P2, P7, P10, P12, P14 and P15 are not detected over Cat.II-R; and P2, P7, P10, P12 and P15 are not detected over Cat.III-B. Furthermore, P2, P7, P9, P10, P12 and P15 are not detected over Cat.IV-A&R-1; P7, P9, P10, P12 and P15 are not detected over Cat.IV-A&R-2; P4, P7, P12, P14, P15 and P18 are not detected over Cat.IV-A&R-3; and P4, P7, P14, P10, P15 and P18 are not detected over Cat.IV-A&R-4. It is apparent that the reaction mechanisms are somewhat different over the seven catalysts.| DIa | HRMS | Formula | Different catalytic system | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cat.I-A | Cat.II-R | Cat.III-B | Cat.VI-A&R-1 | Cat.VI-A&R-2 | Cat.VI-A&R-3 | Cat.VI-A&R-4 | |||
| a Degradation Intermediates. | |||||||||
| P1 | 350.1273 | C16H19N3O6 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P2 | 171.0482 | C7H7FN2O2 | ✓ | — | — | — | ✓ | ✓ | ✓ |
| P3 | 223.0395 | C10H7FN2O3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P4 | 332.1168 | C16H17N3O5 | ✓ | ✓ | ✓ | ✓ | ✓ | — | — |
| P5 | 279.1066 | C14H15FN2O3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P6 | 322.1485 | C16H20FN3O3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P7 | 273.0808 | C11H13FN2O5 | ✓ | — | — | — | — | — | — |
| P8 | 261.0797 | C13H12N2O4 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P9 | 205.0898 | C11H12N2O2 | — | ✓ | ✓ | — | — | ✓ | ✓ |
| P10 | 223.0762 | C11H11FN2O2 | ✓ | — | — | — | — | ✓ | ✓ |
| P11 | 125.0576 | C6H8N2O | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P12 | 327.0914 | C14H15FN2O6 | ✓ | — | — | — | — | — | ✓ |
| P13 | 352.1287 | C16H18FN3O5 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P14 | 336.1324 | C16H18FN3O4 | — | — | ✓ | ✓ | ✓ | — | — |
| P15 | 244.2311 | C10H14FN3O3 | ✓ | — | — | — | — | — | — |
| P16 | 251.0866 | C12H11FN2O3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P17 | 233.0847 | C12H12N2O3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| P18 | 205.0890 | C10H8N2O3 | — | ✓ | ✓ | ✓ | ✓ | — | — |
| P19 | 312.1486 | C14H18FN3O4 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
The HPLC-TOF-HRMS technique can only offer molecular weights of species based on which one can propose molecular formulas but not molecular structures. To achieve the latter, we purified the purified main degradation intermediates by semi-preparative HPLC and analyzed them by HPLC-MS/MS. The results are summarized in Table 4 while the tandem mass spectra are shown in Fig. S4.† On the basis of the data, we come to the following understandings:
| DIa | RTb (min) | HRMSc | Formula | MS/MS fragmentsd | Proposed structure | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| [M + H]+ | [M + H–NH3]+ | [M + H–H2O]+ | [M + H–C2H5N]+ | [M + H–CO2]+ | Others | |||||
| a Degradation intermediates.b Retention time during HPLC/TOF/HRMS.c High resolution mass spectrometry, data obtained by HPLC/TOF/HRMS.d Data obtained by HPLC-MS/MS after separation and purification of degradation intermediates by semi-preparative HPLC. | ||||||||||
| Nor | 7.50 | 320.1466 | C16H18FN3O3 | 320.01 | 303.03 | 302.02 | 277.03 | 276.04 | 232.99 | — |
| P1 | 1.35 | 350.1273 | C16H19N3O6 | 350.23 | — | 322.29 | — | — | — | 1-Ethyl-2,5,6-trihydroxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid |
| P2 | 14.47 | 171.0482 | C7H7FN2O2 | 170.99 | 154.05 | 153.06 | — | 126.94 | 143.03, 135.08, 125.03 | 2,4-Diamino-5-fluorobenzoic acid |
| P3 | 12.45 | 223.0395 | C10H7FN2O3 | 223.13 | 206.18 | 205.08 | — | 179.08 | 165.07, 148.99, 139.04 | 7-Amino-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P4 | 1.42 | 332.1168 | C16H17N3O5 | 332.23 | — | 314.24 | — | — | 304.23, 299.98, 262.18 | 1-Ethyl-6-hydroxy-4-oxo-7-(3-oxopiperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid |
| P5 | 17.72 | 279.1066 | C14H15FN2O3 | 279.14 | — | 261.15 | — | 235.15 | 251.18, 233.09, 209.19, 204.91, 149.02 | 1-Ethyl-7-(ethylamino)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P6 | 1.62 | 322.1485 | C16H20FN3O3 | 322.22 | 305.25 | 304.26 | — | 278.11 | 289.01, 277.27, 276.23, 275.10 | 7-((2-Aminoethyl)(ethyl)amino)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P7 | 1.85 | 273.0808 | C11H13FN2O5 | 273.09 | 256.17 | 255.25 | — | — | 254.13, 241.24, 240.29 | 7-Amino-1-ethyl-6-fluoro-2,3,5,8-tetrahydroxy-2,3-dihydroquinolin-4(1H)-one |
| P8 | 14.23 | 261.0797 | C13H12N2O4 | 261.03 | — | 243.10 | — | — | 246.99, 229.07, 223.16, 215.10, 185.04 | 7-(Divinylamino)-2,5,6-trihydroxyquinolin-4(1H)-one |
| P9 | 13.25 | 205.0898 | C11H12N2O2 | 205.01 | 188.07 | 187.05 | — | — | 177.04, 172.99, 171.96, 169.08, 158.03 | 7-Amino-1-ethyl-6-hydroxyquinolin-4(1H)-one |
| P10 | 11.43 | 223.0762 | C11H11FN2O2 | 223.02 | 206.14 | 205.05 | — | — | 195.06, 193.08, 179.05 | 7-Amino-1-ethyl-6-fluoro-2-hydroxyquinolin-4(1H)-one |
| P11 | 16.34 | 125.0576 | C6H8N2O | 124.95 | 108.27 | 106.96 | — | — | — | 2,4-Diaminophenol |
| P12 | 1.84 | 327.0914 | C14H15FN2O6 | 326.99 | — | 309.09 | — | — | — | 1-Ethyl-7-(ethylamino)-6-fluoro-2,5,8-trihydroxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P13 | 1.23 | 352.1287 | C16H18FN3O5 | 352.17 | 335.44 | 334.46 | 309.13 | 308.40 | 319.38, 315.40 | 1-Ethyl-6-fluoro-2,5-dihydroxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid |
| P14 | 1.62 | 336.1324 | C16H18FN3O4 | 336.15 | 319.26 | 318.28 | 293.18 | 292.20 | 308.16, 303.11, 301.02, 283.16, 261.19 | 1-Ethyl-6-fluoro-2-hydroxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid |
| P15 | 1.92 | 244.2311 | C10H14FN3O3 | 244.23 | 227.14 | 226.07 | — | — | 216.93, 198.93, 197.06 | 3-Amino-5-((2-aminoethyl)(vinyl)amino)-6-fluorobenzene-1,2,4-triol |
| P16 | 18.04 | 251.0866 | C12H11FN2O3 | 250.95 | 234.04 | 233.00 | — | 216.04 | 223.02, 219.02 | 7-Amino-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P17 | 18.67 | 233.0847 | C12H12N2O3 | 233.27 | 216.08 | 215.14 | — | 189.14 | 205.18, 149.06 | 7-Amino-1-ethyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P18 | 17.34 | 205.0890 | C10H8N2O3 | 204.99 | 188.07 | 187.06 | — | 160.99 | 172.99, 158.04 | 7-Amino-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
| P19 | 10.11 | 312.1486 | C14H18FN3O4 | 312.07 | 295.29 | 294.31 | 269.45 | — | 255.08, 174.98 | 2-(6-(Ethylamino)-3-fluoro-2-hydroxy-4-(piperazin-1-yl)phenyl)-2-oxoacetic acid |
The parent molecule of Nor with C16H18FN3O3 formula and an m/z of 320.1466 for [M + H]+ can be cleaved to yield the fragment ions of m/z 303.03, 302.02, 277.03, and 276.04, which are attributed to the losses of NH3, H2O, –C2H5N and CO2, respectively. The ion of m/z 232.99 is attributed to arise from the further loss of a CO2 from the ion of m/z 277.03.
With C16H19N3O6 formula and an m/z of 350.1273 for the [M + H]+ ion, P1 is assigned to 1-ethyl-2,5,6-trihydroxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid. Nor is trihydroxylated to give this production which the F atom is substituted with a hydroxyl group together with the addition of two hydroxyl groups to the quinoline ring. Aside from the precursor ion, the MS/MS of the [M + H]+ ion of P1 displays a fragment ion emanating from the neutral loss of an H2O molecule.
With C7H7FN2O2 formula and an m/z of 171.0482 for [M + H]+ ion, P2 can be assigned to 2,4-diamino-5-fluorobenzoic acid. The fragment ions with m/z 154.05, 153.06 and 126.94 arise from the neutral losses of NH3, H2O, and CO2 respectively. The specie with m/z 143.03 and 125.03 could be produced from the eliminations of CO and [CO + H2O], respectively.
With C10H7FN2O3 formula and an m/z of 223.0395 for the [M + H]+ ion, P3 can be assigned to 7-amino-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid. The fragment ions of m/z 206.18, 205.08 and 179.08 are attributed to the losses of NH3, H2O, and CO2, respectively.
Unlike the parent molecule, P4 with C16H17N3O5 formula and an m/z of 332.1168 for the [M + H]+ ion does not display the combined neutral loss of –C2H5N, indicating the change of piperazine ring in the molecule. The [M + H–C2H4]+ ion observed in the MS/MS nonetheless suggests that the ethyl group on the N atom of quinolone ring is still present. Importantly, the fragment ion of m/z 262.18, due to the loss of CH2CO from the ion with m/z 304.23, indicates the appearance of carbonyl group.
P5 has C14H15FN2O3 formula with the [M + H]+ ion at m/z 279.1066. The detection of fragment ions of m/z 261.15 and 251.18 suggests the presence of –OH and –C2H4 groups. The fragment ion of m/z 204.91 may be due to the loss of a CO from the ion of m/z 233.09. Therefore, P5 can be ascribed to 1-ethyl-7-(ethylamino)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid.
With C16H20FN3O3 formula and m/z 322.1485 for the [M + H]+ ion, P6 has additional 2 Da added to the parent molecule. The fragment ions of m/z 305.25, 304.26 and 276.23 are attributed to the losses of NH3, H2O and [H2O + C2H4], respectively. The structure of P6 is proposed to be 1-ethyl-7-(ethylamino)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid.
The molecular formula of P7 is C11H13FN2O5, indicating that the F atom is present. Moreover, the fragment ions with m/z 256.17 and 255.25 may be attributed to the loss of NH3 and H2O from the 273.09 ion, respectively. The intermediate is hence ascribed to 7-amino-1-ethyl-6-fluoro-2,3,5,8-tetrahydroxy-2,3-dihydroquinolin-4(1H)-one.
P8 may be 7-(divinylamino)-2,5,6-trihydroxyquinolin-4(1H)-one. There is no signal of [M + H–C2H5N]+, indicating the absence of piperazine ring, and the fragment ion due to the loss of H2O is highly abundant. Moreover, the m/z 233.16 and 215.10 ions may emanate from the loss of a C2H4 from the ions of 261.03 and 243.10, respectively.
No neutral loss of CO2 can be detected in the MS/MS for the [M + H]+ ion of P9, though the fragment ions of m/z 188.07 and 187.05 were observed. The latter were attributed to the losses of NH3 and H2O, respectively. Based on these results, the compound is ascribed to 7-amino-1-ethyl-6-hydroxyquinolin-4(1H)-one.
P10 has C11H11FN2O2 formula and m/z 223.0762 for the [M + H]+ ion. The fragment ions of m/z 206.14, 205.05 and 195.06 are generated through the loss of NH3, H2O and C2H4, respectively. There is no detection of signal that can be assigned to the neutral loss of a CO2. Accordingly, P10 is ascribed to 7-amino-1-ethyl-6-fluoro-2-hydroxyquinolin-4(1H)-one.
P11 possesses C6H8N2O formula and an m/z of 125.0576 for the [M + H]+ ion. The fragment ions of m/z 108.27 and 106.96 in the MS/MS may be assigned to the losses of NH3 and H2O from the parent ions, respectively. There is no detection of any other fragment ions, and thus the compound is ascribed to 2,4-diaminophenol.
P12 can be ascribed to 1-ethyl-7-(ethylamino)-6-fluoro-2,5,8-trihydroxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid. Its fragmentations only suggest the presence of –OH group, and with a molecular formula of C14H15FN2O6.
With C16H18FN3O5 formula and an m/z of 352.1287 for [M + H]+ ion, P13 may be dihydroxylated products. The formation of 335.44, 334.36 and 308.40 ions are due to the losses of NH3, H2O and CO2 group, respectively. The fragmentation of m/z 309.13, assigned to [M + H–C2H5N]+, suggests the presence of piperazine ring as similar as parent molecule. Accordingly, the compound is deduced to be 1-ethyl-6-fluoro-2,5-dihydroxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid.
The MS/MS of the [M + H]+ ion of P14 display fragment ions originating from the neutral losses of NH3, H2O, CO2 and –C2H5N. Moreover, the fragment ion arising from the loss of a C2H4 at m/z 308.16 is also detected. This compound with C16H18FN3O4 formula and an m/z of 336.1324 for the [M + H]+ ion, with a molecular weight increase of 16 Da compared to the parent molecule, may be deduced to be a monohydroxylated product of Nor, though the position for hydroxylation is unclear.
With C10H14FN3O3 formula and an m/z of 244.23 for the [M + H]+ ion, P15 may be ascribed to 3-amino-5-((2-aminoethyl)(vinyl)amino)-6-fluorobenzene-1,2,4-triol. The MS/MS for the compound revealed the presence of fragment ions arising from the losses of NH3, H2O and –C2H5N; however, no loss of CO2 was observed, indicating the cleavage of the quinolone ring.
P16 with C12H11FN2O3 formula and the [M + H]+ ion at m/z 251.0866 may be assigned to be 7-amino-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid. The MS/MS exhibited fragment ions emanating from the neutral losses of H2O, NH3 and CO2.
Interestingly, the MS/MS of the protonated ions of P17 and P18 are very similar, which revealed the neutral losses of NH3, H2O and CO2, but not the neutral loss of –C2H5N, suggesting the disappearance of the piperazine ring. Therefore, the two compounds are ascribed to 7-amino-1-ethyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid and 7-amino-4-oxo-1,4-dihydroquinoline-3-carboxylic acid, respectively.
The MS/MS of the [M + H]+ ions of P19 showed the neutral losses of NH3, H2O and –C2H5N, though no loss of CO2 was observed. The compound may be ascribed to 2-(6-(ethylamino)-3-fluoro-2-hydroxy-4-(piperazin-1-yl)phenyl)-2-oxoacetic acid.
Despite these variations in degradation intermediates across the TiO2−X, we propose a general transformation mechanism for Nor degradation as shown in Fig. 6. We find that, h+ is the species most responsible for Nor decomposition. Following oxidation, a Nor molecule loses its piperazine ring, and the P5 and P6 intermediates are detected across all catalysts. Consequent degradation mechanisms include loss of a carboxyl group, addition of –OH to the quinolone ring, and substitution of the F atom. P19, a product from direct interaction of ˙OH and the quinolone ring, is detected across the catalysts. Also, P4 with the F atom substituted by –OH is observed for each TiO2−X except for Cat.IV-A&R-3 and Cat.IV-A&R-4. Additionally, the monohydroxylated product P14 is detected over Cat.III-B, Cat.IV-A&R-1 and Cat.IV-A&R-2. With prolonged reaction time, the degradation intermediates further react with h+, ˙OH, and other ROSs leading to the mineralization of Nor.
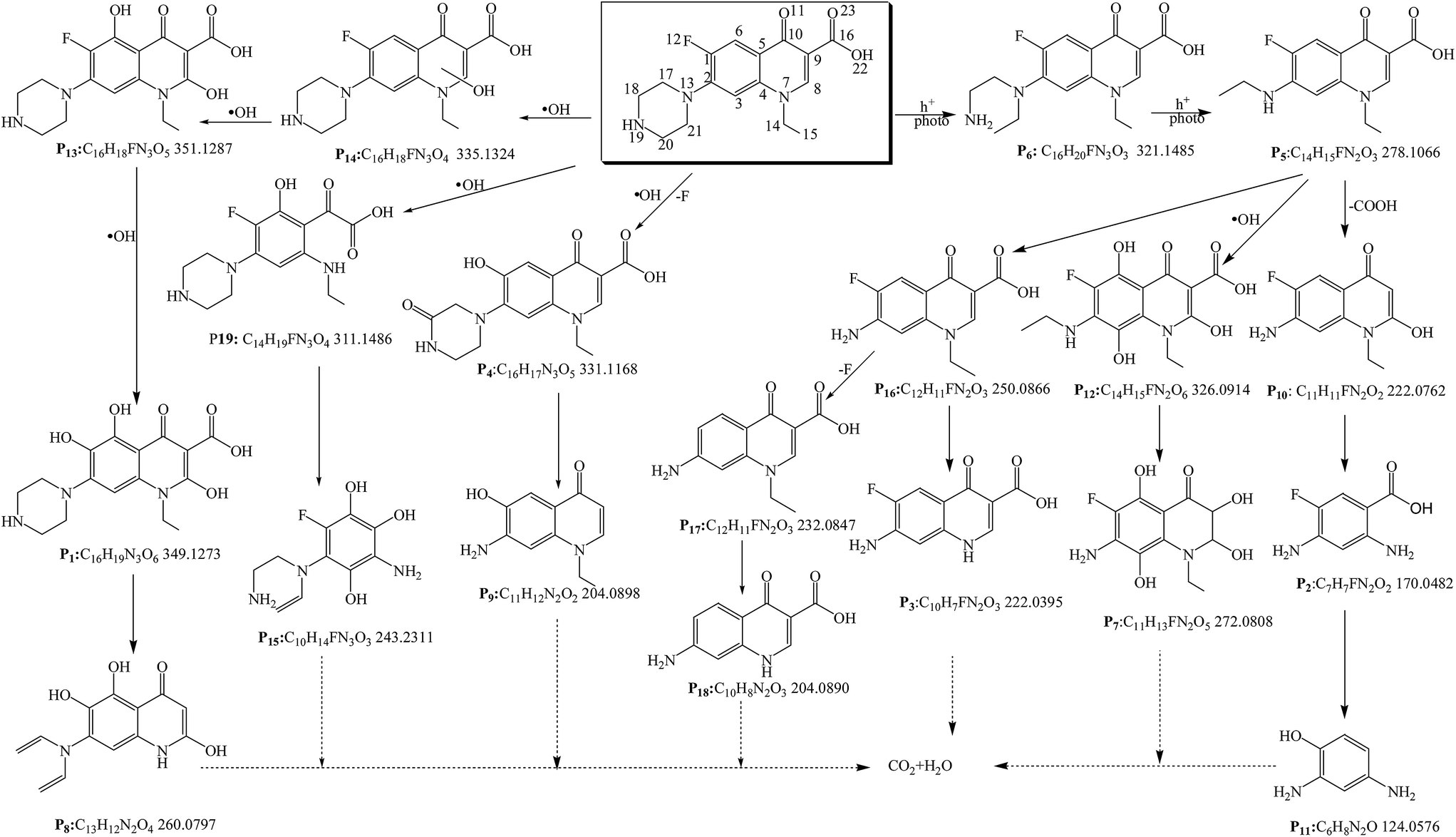 | ||
| Fig. 6 Proposed degradation pathway of Nor on TiO2−X under visible light. | ||
3.4 Mechanism of photocatalytic degradation of nor on TiO2−X
Under visible light, the TiO2−X can be activated to generate photo-excited electrons in the conduction band and hole in the valence band. The experiment shows that the e− reacts with dissolved oxygen and subsequently results in the generation of ˙OH and other ROSs. On the other hand, h+ interacts with H2O to generate ˙OH or reacts with Nor directly as shown in Fig. 7.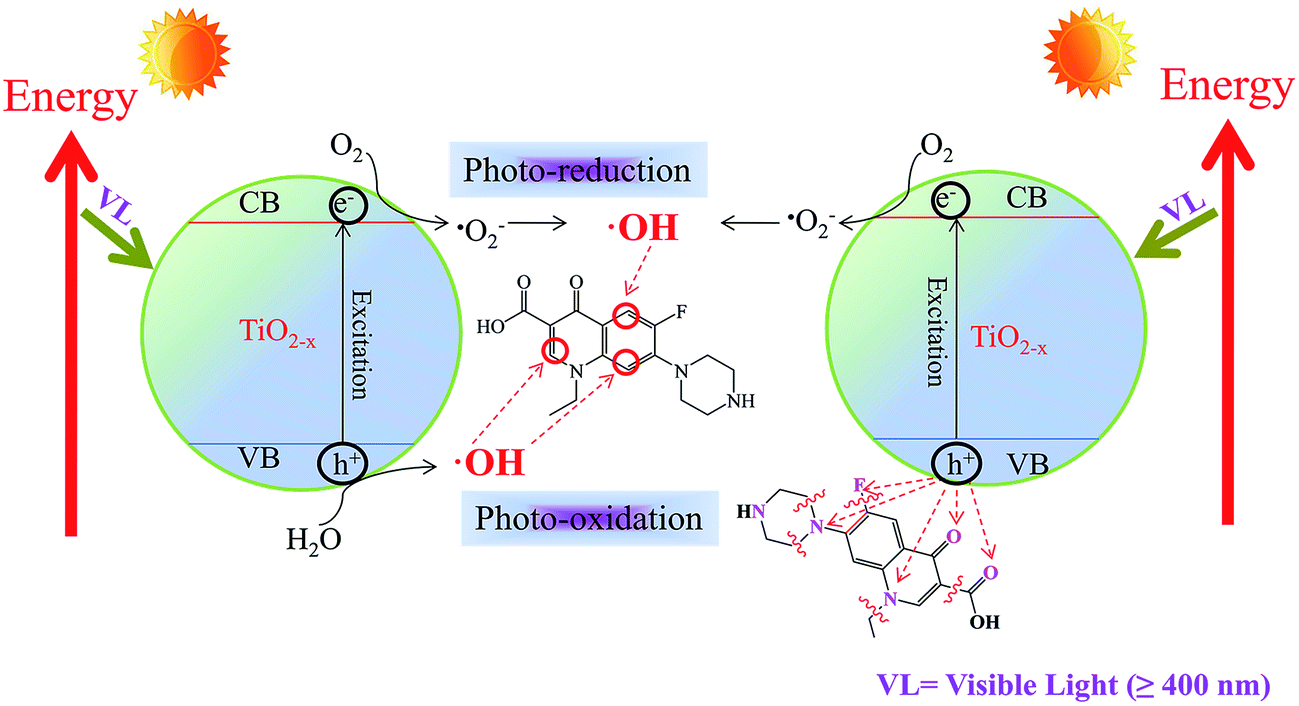 | ||
| Fig. 7 Proposed reaction mechanism between TiO2−X and Nor. | ||
With the help of FEDs values, the active sites of Nor interacting with ROSs can be predicted (Table S1†). According to frontier orbital theory, an electron can be more readily extracted at positions with higher 2FEDHOMO2 values, while ˙OH reaction usually occurs at a position with higher 2FEDHOMO2 + 2FEDLUMO2 value.66,67 C1, C5, N7, O11 and N13 are found to have high 2FEDHOMO2 values, corresponding to 0.2072, 0.2510, 0.1976, 0.1730, and 0.2060. The results indicate that these are positions likely to react with h+. Nonetheless, a Nor molecule has to be on the catalyst surface in order to interact with surface h+. Therefore, we use the point charges to elucidate the adsorption orientation of Nor molecule on the surface of catalyst. The point charges of N7, O11, N13 and O23 are −1.0801, −0.6139, −0.8332 and −0.5985, respectively, suggesting these are the positions likely to have direct contact with h+. Hence, the loss of a piperazine ring or a –COOH group from Nor can be attributed to the direct interaction of Nor with h+ as shown in Fig. 7. In addition, the interaction of Nor with ˙OH results in Nor hydroxylation. Higher 2FEDHOMO2 + 2FEDLUMO2 values are found at C2, C3, C5, C6, C8 and O11. Considering that C2 and C5 are subject to steric hindrance, C3, C6, C8 and O11 are the main positions for ˙OH attack. All catalysts show degradation intermediates that are products of Nor hydroxylation. Further interaction of the intermediates with h+, ˙OH and other ROSs would result in mineralization of Nor.
4. Conclusions
Seven TiO2−X were prepared and studied for the degradation of Nor under visible light. Cat.I-A, Cat.II-R and Cat.III-B have anatase, rutile and brookite structure, respectively. Cat.IV-A&R-1, Cat.IV-A&R-2, Cat.IV-A&R-3, and Cat.IV-A&R-4 are mixtures of anatase and rutile with different ratios. All of these TiO2−X materials exhibit photocatalytic activity under visible light irradiation for Nor mineralization with degradation rate of Cat.IV-A&R-4 > Cat.IV-A&R-3 > Cat.I-A > Cat.IV-A&R-2 > Cat.IV-A&R-1 > Cat.II-R ≈ Cat.III-B. It is observed that photocatalytic activity is influenced by factors such as Ti3+ concentration, oxygen vacancy density and catalyst specific surface area. The contribution of h+ for Nor degradation is significantly higher than that of ˙OH. A mechanism for Nor degradation with TiO2−X under visible light is deduced based on the identified degradation intermediates. It is shown that under the attack of h+ and ˙OH, the loss of piperazine ring and the occurrence of hydroxylation reaction are the major initial steps, and the resulted intermediates further react with ROSs to achieve the mineralization of Nor. The interaction of a Nor molecule with TiO2−X is proposed based on FEDs values and calculations. The adsorption of Nor on the surface of catalyst is likely realized first through the N, O and F atoms on Nor where Nor perhaps reacts with h+. On the other hand, a Nor molecule is likely attacked by ˙OH at the C3, C6 and C8 positions. It is expected that the present results can motivate further studies on TiO2−X for the promotion of their practical application in wastewater treatments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the China Scholarship Council (H. Yang), the National Natural Science Foundation of China (21207034, H. Yang), the Provincial Natural Science Foundation of Hunan (2015JJ3056, H. Yang), and the Scientific Research Fund of Hunan Provincial Education Department (17B061 and 11C0331, H. Yang). This research was also supported by the National Science Foundation (DMR-1506661, P. Feng).References
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- E. J. W. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander-Webber and H. J. Snaith, Nature, 2013, 495, 215–219 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–642 CrossRef CAS PubMed.
- M. Setvin, U. Aschauer, J. Hulva, T. Simschitz, B. Daniel, M. Schmid, A. Selloni and U. Diebold, J. Am. Chem. Soc., 2016, 138, 9565–9571 CrossRef CAS PubMed.
- A. Colombo, F. Gherardi, S. Goidanich, J. K. Delaney, E. R. de la Rie, M. C. Ubaldi, L. Toniolo and R. Simonutti, RSC Adv., 2015, 5, 84879–84888 RSC.
- P. Rattanawaleedirojn, K. Saengkiettiyut, Y. Boonyongmaneerat, S. Sangsuk, N. Promphet and N. Rodthongkum, RSC Adv., 2016, 6, 69261–69269 RSC.
- V. B. Damodaran, D. Bhatnagar, V. Leszczak and K. C. Popat, RSC Adv., 2015, 5, 37149–37171 RSC.
- S. K. Nethi, N. A. Anand P., B. Rico-Oller, A. Rodríguez-Diéguez, S. Gómez-Ruiz and C. R. Patra, Sci. Total Environ., 2017, 599–600, 1263–1274 CrossRef CAS PubMed.
- E. Lira, S. Wendt, P. P. Huo, J. O. Hansen, R. Streber, S. Porsgaard, Y. Y. Wei, R. Bechstein, E. Laegsgaard and F. Besenbacher, J. Am. Chem. Soc., 2011, 133, 6529–6532 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- L. J. Gao, Y. G. Li, J. B. Ren, S. F. Wang, R. N. Wang, G. S. Fu and Y. Hu, Appl. Catal., B, 2017, 202, 127–133 CrossRef CAS.
- X. Y. Pan, M. Q. Yang, X. Z. Fu, N. Zhang and Y. J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- F. Zuo, L. Wang, T. Wu, Z. Y. Zhang, D. Borchardt and P. Y. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- Y. Zhou, C. H. Chen, N. N. Wang, Y. Y. Li and H. M. Ding, J. Phys. Chem. C, 2016, 120, 6116–6124 CAS.
- X. L. Liu, H. M. Zhang, X. D. Yao, T. C. An, P. R. Liu, Y. Wang, F. Peng, A. R. Carroll and H. J. Zhao, Nano Res., 2012, 5, 762–769 CrossRef CAS.
- B. C. Qiu, Y. Zhou, Y. F. Ma, X. L. Yang, W. Q. Sheng, M. Y. Xing and J. L. Zhang, Sci. Rep., 2015, 5, 8591–8597 CrossRef CAS PubMed.
- F. Zuo, K. Bozhilov, R. J. Dillon, L. Wang, P. Smith, X. Zhao, C. Bardeen and P. Y. Feng, Angew. Chem., Int. Ed., 2012, 51, 6223–6226 CrossRef CAS PubMed.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751–6761 CrossRef CAS PubMed.
- G. Cappelletti, C. L. Bianchi and S. Ardizzone, Appl. Catal., B, 2008, 78, 193–201 CrossRef CAS.
- L. J. Liu, H. L. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- L. Li and C. Y. Liu, Eur. J. Inorg. Chem., 2009, 20, 3727–3733 CrossRef.
- J. Z. Chen, L. P. Zhang, Z. H. Lam, H. B. Tao, Z. P. Zeng, H. B. Yang, J. Q. Luo, L. Ma, B. Li, J. F. Zheng, S. P. Jia, Z. J. Wang, Z. P. Zhu and B. Liu, J. Am. Chem. Soc., 2016, 138, 3183–3189 CrossRef CAS PubMed.
- M. W. Shah, Y. Q. Zhu, X. Y. Fan, J. Zhao, Y. X. Li, S. Asim and C. Y. Wang, Sci. Rep., 2015, 5, 15804–15812 CrossRef PubMed.
- F. Zuo, L. Wang and P. Y. Feng, Int. J. Hydrogen Energy, 2014, 39, 711–717 CrossRef CAS.
- Q. Zhu, Y. Peng, L. Lin, C. M. Fan, G. Q. Gao, R. X. Wang and A. W. Xu, J. Mater. Chem. A, 2014, 2, 4429–4437 CAS.
- K. Sasan, F. Zuo, Y. Wang and P. Y. Feng, Nanoscale, 2015, 7, 13369–13372 RSC.
- X. Y. Xin, T. Xu, L. Wang and C. Y. Wang, Sci. Rep., 2016, 6, 23684–23692 CrossRef CAS PubMed.
- G. L. Zhu, T. Q. Lin, X. J. Lu, W. Zhao, C. Y. Yang, Z. Wang, H. Yin, Z. Q. Liu, F. Q. Huang and J. H. Lin, J. Mater. Chem. A, 2013, 1, 9650–9653 CAS.
- B. Rico-Oller, A. Boudjemaa, H. Bahruji, M. Kebir, S. Prashar, K. Bachari, M. Fajardo and S. Gómez-Ruiz, Sci. Total Environ., 2016, 563–564, 921–932 CrossRef CAS PubMed.
- L. H. Li, L. L. Yu, Z. Y. Lin and G. W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 8536–8545 CAS.
- Y. Zhou, Y. C. Liu, P. W. Liu, W. Y. Zhang, M. Y. Xing and J. L. Zhang, Appl. Catal., B, 2015, 170, 66–73 CrossRef.
- B. J. Jiang, Y. Q. Tang, Y. Qu, J. Q. Wang, Y. Xie, C. G. Tian, W. Zhou and H. G. Fu, Nanoscale, 2015, 7, 5035–5045 RSC.
- M. Y. Xing, X. Li and J. L. Zhang, Sci. Rep., 2014, 4, 5493–5500 CrossRef CAS PubMed.
- R. R. Fu, S. M. Gao, H. Xu, Q. Y. Wang, Z. Y. Wang, B. B. Huang and Y. Dai, RSC Adv., 2014, 4, 37061–37069 RSC.
- S. Snowberger, H. Adejumo, K. He, K. P. Mangalgiri, M. Hopanna, A. D. Soares and L. Blaney, Environ. Sci. Technol., 2016, 50, 9533–9542 CrossRef CAS PubMed.
- Y. G. Xu, W. T. Yu, Q. Ma and H. Zhou, Sci. Total Environ., 2015, 530, 191–197 CrossRef PubMed.
- Y. P. Ma, M. Li, M. M. Wu, Z. Li and X. Liu, Sci. Total Environ., 2015, 518, 498–506 CrossRef PubMed.
- N. A. M. Nor, J. Jaafar, A. F. Ismail, M. A. Mohamed, M. A. Rahman, M. H. D. Othman, W. J. Lau and N. Yusof, Desalination, 2016, 391, 89–97 CrossRef CAS.
- X. W. Hao, Y. Cao, L. Zhang, Y. Y. Zhang and J. G. Liu, Environ. Toxicol. Chem., 2015, 34, 2764–2770 CrossRef CAS PubMed.
- H. W. Leung, T. B. Minh, M. B. Murphy, J. C. W. Lam, M. K. So, M. Martin, P. K. S. Lam and B. J. Richardson, Environ. Int., 2012, 42, 1–9 CrossRef CAS PubMed.
- L. Lv, T. Jiang, S. H. Zhang and X. Yu, Environ. Sci. Technol., 2014, 48, 8188–8195 CrossRef CAS PubMed.
- X. M. Wang, B. Li, T. Zhang and X. Y. Li, Desalination, 2015, 370, 7–16 CrossRef CAS.
- P. Liu, H. Zhang, Y. Feng, C. Shen and F. Yang, Desalination, 2015, 371, 134–143 CrossRef CAS.
- B. Li and T. Zhang, Environ. Sci. Technol., 2010, 44, 3468–3473 CrossRef CAS PubMed.
- R. P. Singh, K. V. H. Sastry, P. K. Dubey, R. Agrawal, R. Singh, N. K. Pandey and J. Mohan, Environ. Toxicol. Chem., 2013, 32, 2134–2138 CrossRef CAS PubMed.
- A. Speltini, M. Sturini, F. Maraschi, S. Viti, D. Sbarbada and A. Profumo, J. Chromatogr. A, 2015, 1410, 44–50 CrossRef CAS PubMed.
- T. C. An, H. Yang, W. H. Song, G. Y. Li, H. Y. Luo and W. J. Cooper, J. Phys. Chem. A, 2010, 114, 2569–2575 CrossRef CAS PubMed.
- L. Tang, J. J. Wang, G. M. Zeng, Y. N. Liu, Y. C. Deng, Y. Y. Zhou, J. Tang, J. J. Wang and Z. Guo, J. Hazard. Mater., 2016, 306, 295–304 CrossRef CAS PubMed.
- T. Paul, P. L. Miller and T. J. Strathmann, Environ. Sci. Technol., 2007, 41, 4720–4727 CrossRef CAS PubMed.
- W. J. Zhang, H. Y. Gao, J. J. He, P. Yang, D. S. Wang, T. Ma, H. Xia and X. Z. Xu, Sep. Purif. Technol., 2017, 172, 158–167 CrossRef CAS.
- A. Ozcan, A. A. Ozcan and Y. Demirci, Chem. Eng. J., 2016, 304, 518–526 CrossRef CAS.
- H. G. Guo, N. Y. Gao, Y. Yang and Y. L. Zhang, Chem. Eng. J., 2016, 292, 82–91 CrossRef CAS.
- M. Sayed, J. A. Khan, L. A. Shah, N. S. Shah, H. M. Khan, F. Rehman, A. R. Khan and A. M. Khan, Environ. Sci. Pollut. Res., 2016, 23, 13155–13168 CrossRef CAS PubMed.
- J. M. Wang, H. Lin, W. C. Sun, Y. Xia, J. W. Ma, J. R. Fu, Z. L. Zhang, H. Z. Wu and M. R. Qian, J. Hazard. Mater., 2016, 304, 49–57 CrossRef CAS PubMed.
- T. C. An, H. Yang, G. Y. Li, W. H. Song, W. J. Cooper and X. P. Nie, Appl. Catal., B, 2010, 94, 288–294 CrossRef CAS.
- H. Yang, T. C. An, G. Y. Li, W. H. Song, W. J. Cooper, H. Y. Luo and X. D. Guo, J. Hazard. Mater., 2010, 179, 834–839 CrossRef CAS PubMed.
- R. P. Cavalcante, R. F. Dantas, B. Bayarri, O. Gonzalez, J. Gimenez, S. Esplugas and A. Machulek, Appl. Catal., B, 2016, 194, 111–122 CrossRef CAS.
- T. L. Xu, Y. Cai and K. E. O'shea, Environ. Sci. Technol., 2007, 41, 5471–5477 CrossRef CAS PubMed.
- H. Yang, H. J. Liu, Z. B. Hu, J. W. Liang, H. L. Pang and B. Yi, Chem. Eng. J., 2014, 245, 24–33 CrossRef CAS.
- H. Yang, G. Y. Li, T. C. An, Y. P. Gao and J. M. Fu, Catal. Today, 2010, 153, 200–207 CrossRef CAS.
- C. Y. Mao, F. Zuo, Y. Hou, X. H. Bu and P. Y. Feng, Angew. Chem., Int. Ed., 2014, 53, 10485–10489 CrossRef CAS PubMed.
- K. Sasan, A. G. Kong, Y. Wang, C. Y. Mao, Q. G. Zhai and P. Y. Feng, J. Phys. Chem. C, 2015, 119, 13545–13550 CAS.
- C. Y. Zhu, G. D. Fang, D. D. Dionysiou, C. Liu, J. Gao, W. X. Qin and D. M. Zhou, J. Hazard. Mater., 2016, 316, 232–241 CrossRef CAS PubMed.
- Y. H. Guan, J. Ma, X. C. Li, J. Y. Fang and L. W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- G. Hubner and E. Roduner, J. Mater. Chem., 1999, 9, 409–418 RSC.
- H. Yang, H. Q. Wei, L. T. Hu, H. J. Liu, L. P. Yang, C. T. Au and B. Yi, Chem. Eng. J., 2016, 300, 209–216 CrossRef CAS.
- J. Zeng, H. Yang, J. Y. Deng, H. J. Liu, X. Yi, L. P. Yang and B. Yi, Chem. Eng. J., 2015, 273, 519–526 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09022f |
| This journal is © The Royal Society of Chemistry 2017 |