DOI:
10.1039/C7RA09289J
(Paper)
RSC Adv., 2017,
7, 45269-45273
Pseudo three-component approach to coumarin-annulated azepines: synthesis of coumarin[3,4-b]azepines†
Received
22nd August 2017
, Accepted 15th September 2017
First published on 22nd September 2017
Abstract
A series of coumarin-annulated azepines were synthesized via acid-catalyzed condensation of 3-amino-4-hydroxycoumarin with two equivalents of substituted acetophenones in toluene with moderate to good yields. A plausible mechanism for this pseudo three-component reaction was proposed.
Introduction
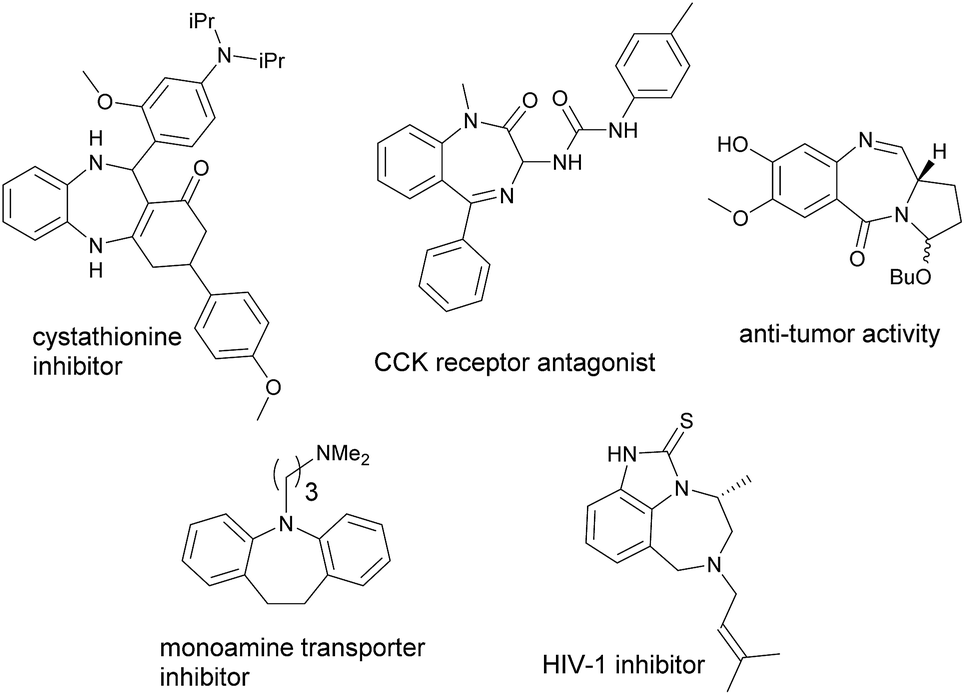
Azepines, unsaturated seven-membered heterocycles with a nitrogen replacing a carbon at one position, are one of the most well-known molecules that exhibit a wide range of therapeutic activities.1 Their activities are generally imparted when the azepines are fused with other molecular scaffolds such as benzene. Apart from this, the annulated 1,4- and 1,5-diazepine derivatives are also found to be the privileged heterocycles with various potent biological properties. These molecular skeletons are frequently seen in many pharmaceuticals2 currently available on the markets. For instance, benzene-annulated azepines (benzazepines) such as benazepril are ACE inhibitors used primarily in the treatment of hypertension;3 fenoldopam is used as an antihypertensive agent;4 lorcaserinis is a weight-loss drug;5 and varenicline is a prescription medication used to treat nicotine addiction.6 Fig. 1 shows structures of some biologically important annulated azepine/diazine derivatives which are the active components of the existing drugs. On the other hand, coumarins represent another important class of heterocycles that have wide applications in the areas of medicinal chemistry7 and functional materials.8 Similar to that of azepines, when coumarins are fused with other molecular scaffolds, the resulting compounds may also exhibit unique or even unprecedented properties. For example, phenanthridine-annulated coumarins have been found to possess negative thermochromic properties;9 pyran-annulated coumarins have been reported to exhibit molecular switching properties;10 pyrrole-annulated coumarins have been demonstrated to possess redox-switching properties.11
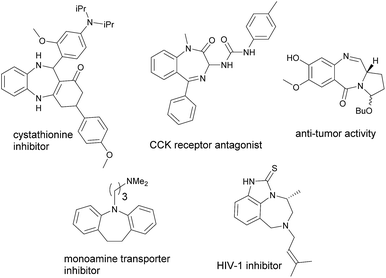 |
| | Fig. 1 Structures of some biologically active azepine/diazine derivatives. | |
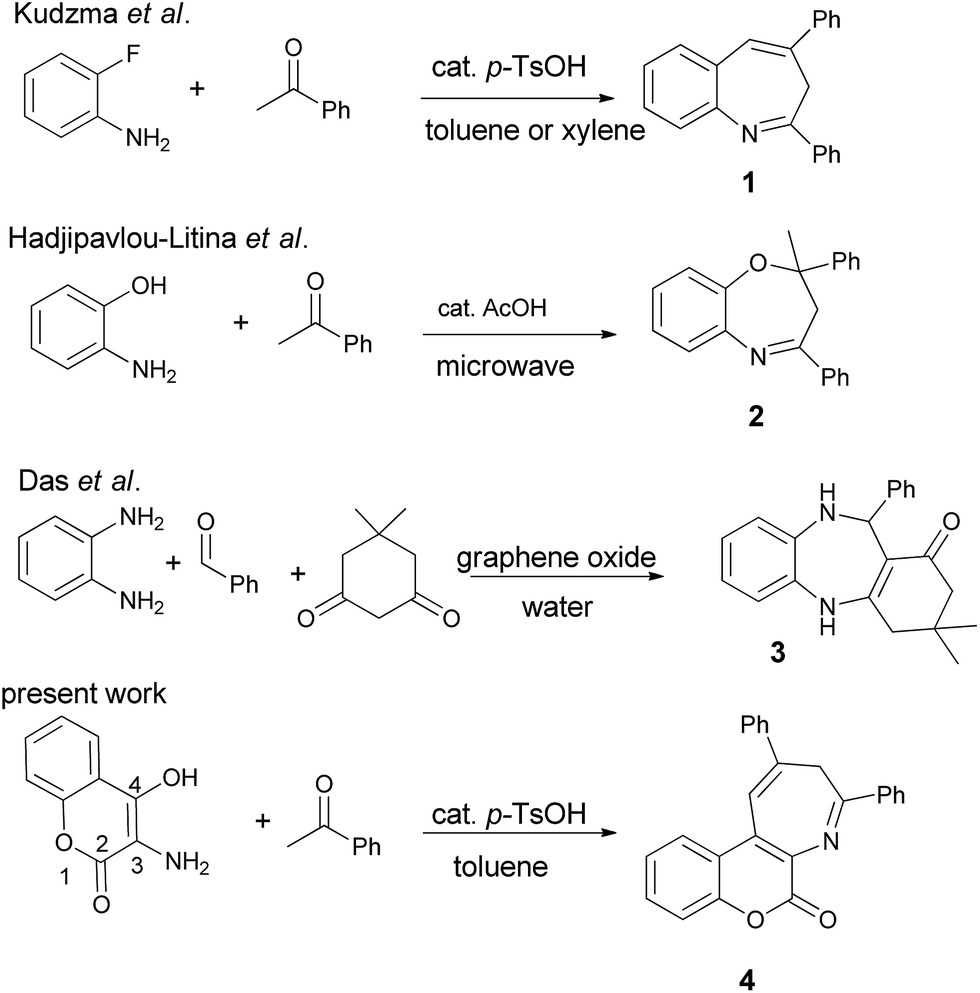
While various efforts have been focused on the preparation of benzazepines and coumarin-fused heterocycles, the synthesis of coumarin-annulated azepines have not been well-explored in the literature.12 Scheme 1 outlines the previous preparation of benzazepines 1 and 2 via acid-catalyzed condensation of acetophenone with 2-fluoroaniline in toluene under reflux conditions and with 2-aminophenol under microwave irradiation conditions, respectively. The fluorine atom of 2-fluoroaniline serves as a leaving group in the synthesis of 2,4-diphenyl-3H-1-benzazepine 1,13 whereas the hydroxyl group of 2-aminophenol functions as a nucleophile in the pseudo three-component synthesis of diphenyloxazepine 2.14 Recently graphene nanosheets have also been used to prepare dibenzo[1,4]diazepine 3 in water.15 Nevertheless, none of the reported methods has focused on the preparation of coumarin[3,4-b]azepines. In our continuing efforts to develop new methodology for the preparation of coumarin-fused heterocycles16 and to subsequently investigate their potential biological or functional properties, we made an attempt to synthesize the coumarin-fused azepine derivatives in a multi-component manner. Herein we report the acid-catalyzed pseudo three-component synthesis of coumarin[3,4-b]azepines by sequential condensations of 3-amino-4-hydroxycoumarin with two equiv. of substituted acetophenones. For present methodology, the substrate scope is investigated and a possible reaction mechanism is deduced.
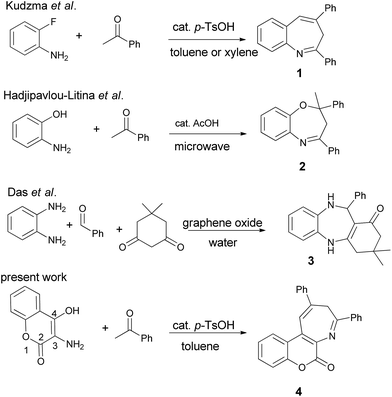 |
| | Scheme 1 Synthetic methods for benzazepines and their derivatives. | |
Results and discussion
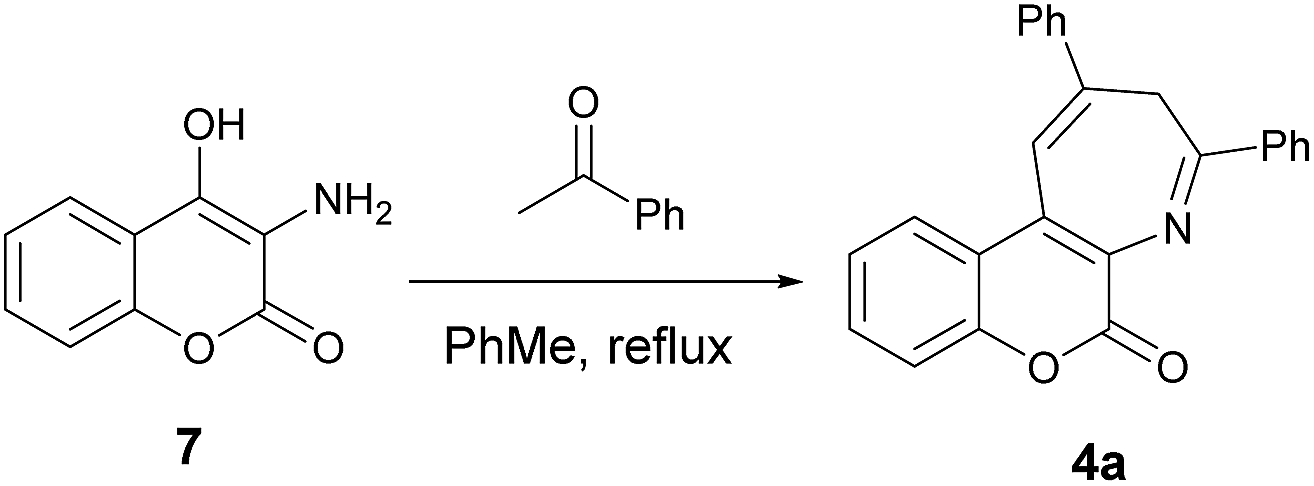
The proposed synthesis of coumarin[3,4-b]azepine 4a is shown in Scheme 2. It started with nitration of commercially available 4-hydroxycoumarin (5) with sodium nitrite and nitric acid in acetic acid to give 3-hydroxy-4-nitrocoumarin17 (6) and followed by the Pd/C reduction under hydrogen atmosphere to yield the 3-amino-4-hydroxycoumarin (7).18 Final refluxing of aminocoumarin 7 with two equivalent of acetophenone in the presence of one equivalent of p-TsOH in toluene for 12 h afforded the coumarin[3,4-b]azepine 4a. To our delight, the pseudo three-component approach proved to be successful. The molecular structure of 4a was elucidated by spectroscopic data as well as by the ORTEP crystal analysis.19 In the 1H NMR spectra of 4a, two characteristic broad singlet absorption peaks at the chemical shift of 2.19 and 4.99 ppm were observed, which were assigned to the methylene protons flanked by the two phenyl rings in the azepine moiety. A similar observation has been previously reported in the literature for 2,4-diaryl-3H-1-benzazepines.20 Since the yield of the target compound 4a was found to be mere 32%, the optimizations of reaction conditions were then under taken. Table 1 lists the results of optimization of reaction parameters for 4a. The combination of p-TsOH and molecular sieves (4 Å) was found to be the best reaction conditions. As the loading of p-TsOH decreased from 1.0 to 0.5 equiv. (entries 1 and 2), the yield of 4a increased from 32 to 57%. The highest yield (80%) was recorded when 0.2 equiv. of p-TsOH was employed (entry 3). Among the Lewis acids screened, only Yb(OTf)3 showed considerable activity (entry 6, 64%) and the rest of them were found be below average (entries 4 and 8). These results prompted us to employ the conditions of 0.2 equiv. of p-TsOH (entry 3) in the subsequent reactions reported in the present study.
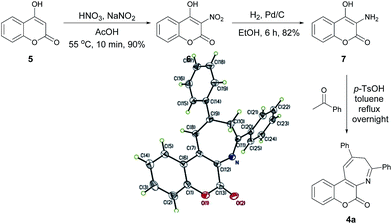 |
| | Scheme 2 Preparation of compound 4a and its ORTEP crystal structure.19 | |
Table 1 Optimization of reaction parameters for 4aa
|
| Entry |
Acid |
Equiv. |
Time (h) |
Yieldb (%) |
| In entries 3 to 8, 4 Å MS was added. Isolated yield. No reaction. |
| 1 |
p-TsOH |
1.0 |
12 |
32 |
| 2 |
p-TsOH |
0.5 |
12 |
57 |
| 3 |
p-TsOH |
0.2 |
6 |
80 |
| 4 |
FeCl3 |
0.2 |
12 |
nrc |
| 5 |
AlCl3 |
0.2 |
12 |
16 |
| 6 |
Yb(OTf)3 |
0.2 |
10 |
64 |
| 7 |
SnCl4 |
0.2 |
12 |
nrc |
| 8 |
ZnCl2 |
0.2 |
12 |
42 |
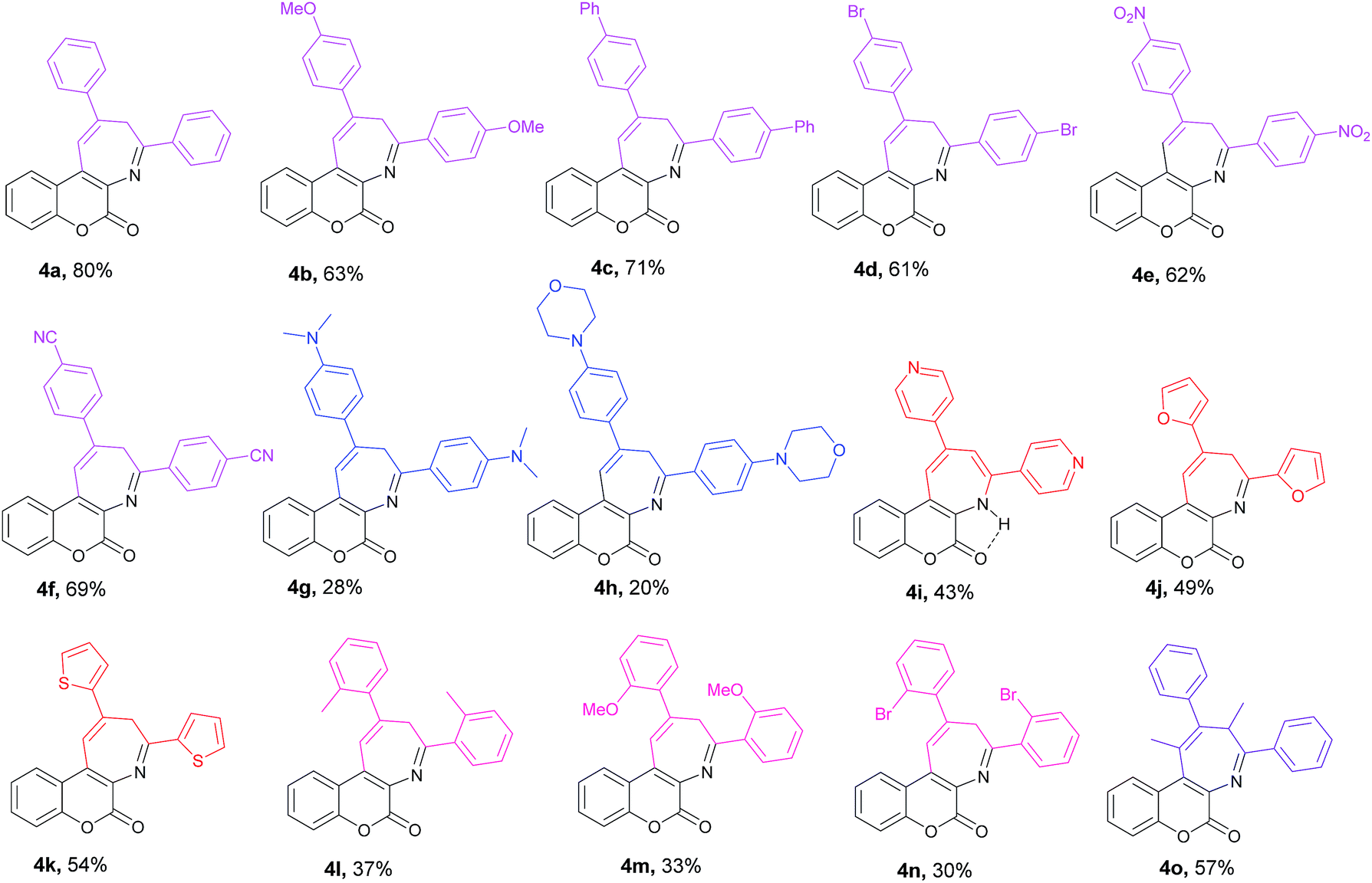
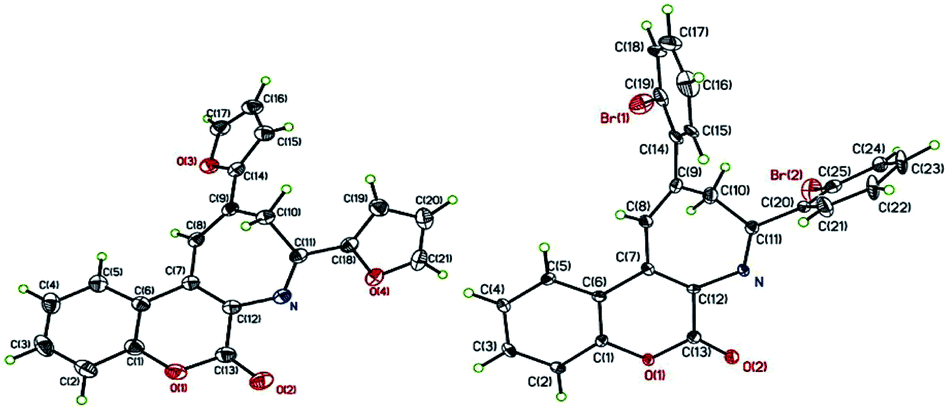
After successfully realizing the preparation of coumarin-annulated azepine 4a, we then turned our attention to investigate the scope and limitation of this pseudo three-component reaction. A series of acetophenones bearing different substituents at either ortho or para position of the aromatic ring were examined. Fig. 2 lists the structures and yields of the prepared coumarin[3,4-b]azepines 4a–o. The results suggested that substrates with either an electron-donating group such as OMe and phenyl or an electron-withdrawing group such as Br, NO2, and CN substituted at the para position of acetophenone all gave the corresponding products 4b–f with yields between 60–70%, indicating that the inductive effect of the para-substituents has little influence on the reaction yield. Nevertheless, both para-amino substituted acetophenones such as 1-(4-(dimethylamino)phenyl)ethan-1-one and 1-(4-morpholinophenyl)ethan-1-one gave the respective products 4g and 6h with low yields. This observation may be attributed to the strong resonance effect of p-amino group rendering 3-acetophenone less susceptible to nucleophilic attack by aminocoumarin. For compounds containing hetero aryl groups like 4-acetylpyridine, 2-acetylfuran, and 2-acetylthiophene, they can all serve as substrates for this reaction to afford the corresponding azepines 4i–k in moderate yields. The substrates bearing methyl, methoxy, or bromo at the ortho position at the phenyl ring of acetophenone generally suffered for low yields, presumably due to the steric hindrance caused by the ortho substituent. Consequently, the even bulkier substrates such as o-nitroacetophenone and 1-(naphthalen-2-yl)ethan-1-one failed to give any of the expected azepines. When propiophenone was employed as the aromatic ketone substrate, the azepine 4o was obtained in 57% yield. This observation suggests that the substrate scope of this reaction is not limited to methyl ketones only. Nevertheless, when the ketone substrate was further extended from propiophenone to isopropyl phenyl ketone, no expected product was observed. Fig. 3 shows the X-ray crystal structures of 4j and 4n.19 It is noteworthy that the prepared azepines all exist in the imine forms on the azepine ring, except for compound 4i. In proton NMR spectra of 4i, the observation of an intramolecular hydrogen bonding absorption peak at 12.14 ppm along with two small doublets (J = 1.6 Hz) olefinic hydrogen absorptions at 8.24 and 8.22 ppm clearly indicates that 4i exists in the enamine form. The reason why only 4i exists in the enamine form in solution is currently under investigation.
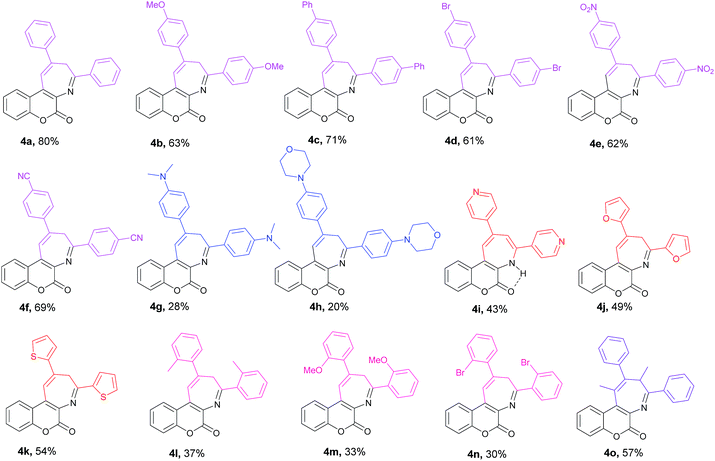 |
| | Fig. 2 Structures of the prepared coumarin[3,4-b]azepines 4a–o. | |
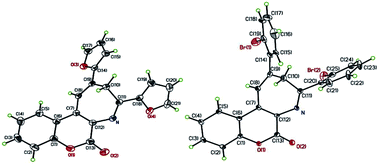 |
| | Fig. 3 ORTEP crystal structures of 4j (left) and 4n (right). | |
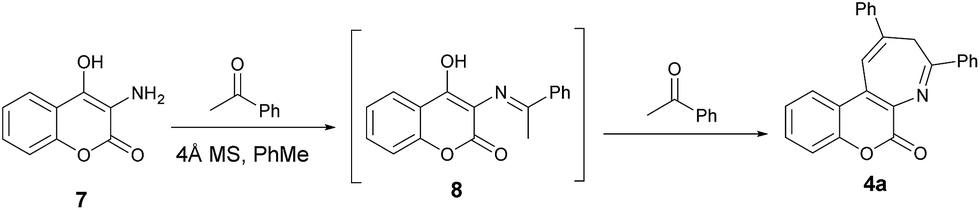
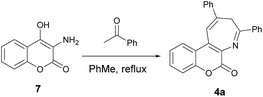
In order to gain more insights into the mechanism of this pseudo three-component reaction, we performed an experiment by introducing just one equiv. of acetophenone to react with aminocoumarin 7. In this control reaction, compound 4a was obtained as the major product, along with a trace amount of a minor product. This minor product was isolated and further characterized by spectroscopic analysis to be the imine 8 (Scheme 3). In the proton NMR spectrum, a singlet peak at 2.75 ppm was clearly observed and assigned to be the methyl absorption of 8. The detection of formation of the intermediate imine 8 in this pseudo three-component reaction provides solid evidence that the mechanism involves the first condensation of aminocoumarin with acetophenone to give the imine 8.
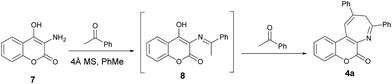 |
| | Scheme 3 Formation of the intermediate 8 and its conversion to the product 4a. | |
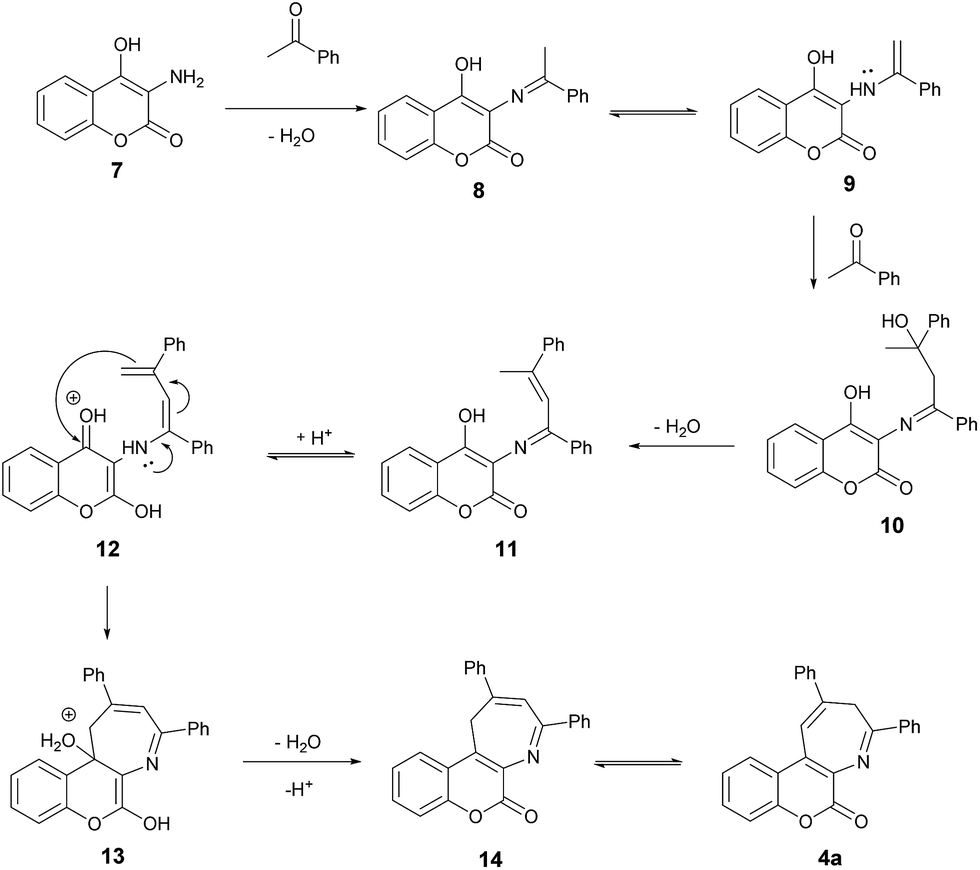
Scheme 4 depicts the proposed mechanism for this pseudo three-component synthesis of coumarin-annulated azepines in details. It presumably starts with dehydration of 3-amino-4-hydroxycoumarin (7) with acetophenone to give the imine 8 which is in equilibrium with the enamine 9. The enamine 9 further reacts with the second molecule of acetophenone to form the tertiary alcohol 10. The subsequent dehydration to remove the second water molecule affords the imine 11 which again is in equilibrium with the enamine 12. The intramolecular addition of enamine to the 4-position of the coumarin ring of 12 yields the cyclized 13. The elimination of the third water molecule along with deprotonation of 13 furnishes the 16. Final acid-catalyzed isomerization of 14 affords the product azepine 4a. This methodology provides an easy and quick access to the coumarin[3,4-b]azepines.
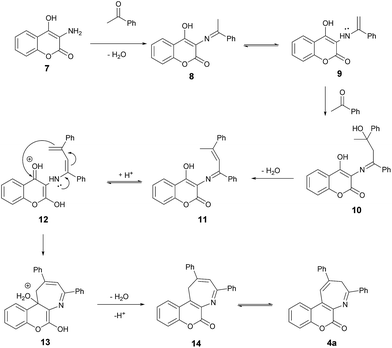 |
| | Scheme 4 Proposed mechanism for the formation of 4a. | |
Conclusions
In summary, we have demonstrated that coumarin-annulated azepines can be efficiently constructed via p-TsOH-catalyzed pseudo three-component coupling of 3-amino-4-hydroxycoumarin with two equiv. of acetophenone. The scope of the reaction is illustrated by preparation of 15 coumarin[3,4-b]azepine analogs with moderate to good yields. The biological evaluation of the prepared coumarin-annulated azepines is currently underway.
Experimental
Instrumentation
Melting points were determined on a Mel-Temp melting point apparatus in open capillaries and are uncorrected. Infrared (IR) spectra were recorded using 1725XFT-IR spectrophotometer. High resolution mass spectra (HRMS) were obtained on a Thermo Fisher Scientific Finnigan MAT95XL spectrometer using magnetic sector analyzer1 H NMR (400 MHz) and 13C NMR (100, or 150 MHz) spectra were recorded on a Varian VXR300 or Bruker 400/600 spectrometer. Chemical shifts were reported in parts per million on the δscale relative to an internal standard (tetramethylsilane, or appropriate solvent peaks) with coupling constants given in hertz. 1H NMR multiplicity data are denoted by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60G-254 plates (25 mm) and developed with the solvents mentioned. Visualization was accomplished by using portable UV light, ninhydrin spray and iodine chamber. Flash chromatography was performed in columns of various diameters with Merck silica gel (230–400 mesh ASTM 9385 kieselgel 60H) by elution with the solvent systems. Solvents, unless otherwise specified, were reagent grade and distilled once prior to use. All new compounds exhibited satisfactory spectroscopic and analytical data.
Synthesis of 4-hydroxy-3-nitrocoumarin (6)
To a stirred suspension of 4-hydroxycoumarin (5, 4.0 g, 24.67 mmol, 1.0 equiv.) in AcOH (50 mL) was added sodium nitrite (17 mg, 0.25 mmol, 0.01 equiv.) in one portion and HNO3 (3.5 mL) drop wise. The resulting mixture was stirred at room temperature for 5 min and followed by heating at 70 °C for 30 min in an oil bath. As the brown solution attained to room temperature, the pure compound crystallized out from the solution. These crystals were filtered, washed with hexanes (6 × 50 mL), and dried in vacuo to afford 3-nitro-4-hydroxycoumarin (6) as off-yellow shiny crystals. 4.6 g; yield 90%; Rf = 0.15 (10% MeOH/DCM); mp 176–177 °C (lit.16 177 °C); 1H NMR (CDCl3, 300 MHz) δ 9.41 (bs, 1H), 7.88 (dd, J = 7.8, 1.5 Hz, 1H), 7.52 (td, J = 8.1, 1.8 Hz, 1H), 7.24–7.16 (m, 2H).
Synthesis of 3-amino-4-hydroxycoumarin (7)
To a suspension of 4-hydroxy-3-nitrocoumarin (6, 4.0 g, 22.58 mmol) in ethanol (200 mL) was added 10% Pd/C (50 mg) at room temperature. The resulting solution was stirred under H2 atmosphere for 6 h at that temperature. After the completion of the reaction, the suspension was filtered through the celite pad, washed extensively with MeOH (3 × 30 mL), acetone (3 × 30 mL), and concentrated to afford an off-white solid. 2.82 g; yield 82%; Rf = 0.16 (10% MeOH/DCM); mp 216 °C (lit.17 222–224 °C); 1H NMR (CDCl3, 300 MHz) δ 8.19 (bs, 2H), 7.84 (dd, J = 8.0, 1.6 Hz, 1H), 7.47 (td, J = 7.6, 1.6 Hz, 1H), 7.25–7.21 (m, 2H).
General procedure for the preparation of coumarin-fused azepines (4)
To a 100 mL round bottom flask was charged with 3-amino-4-hydroxycoumarin (1.0 equiv.), acetophenone derivative (2.1 equiv.), p-TsOH (0.2 equiv.), and 4 Å molecular sieves in anhydrous toluene (25 mL). The resulting mixture was refluxed for about 8–10 h. The dark solution was allowed to attain to the room temperature and the solvent was evaporated in vacuo. The residue was redissolved in DCM (50 mL), filtered, and washed with copious amounts of DCM. The resulting solvent was concentrated in vacuo and the product was subjected to flash column chromatography to afford the coumarin-fused azepine derivative.
4a. Off-yellow solid; yield 80%; Rf = 0.42 (30% EtOAc/hexanes); mp 220–221 °C; 1H NMR (CDCl3, 400 MHz) δ 7.89 (dd, J = 8.0, 1.6 Hz, 2H), 7.83 (dd, J = 7.8, 1.2 Hz, 1H), 7.62–7.60 (m, 2H), 7.50–7.40 (m, 5H), 7.38 (s, 1H), 7.39–7.35 (m, 1H), 7.33–7.29 (m, 3H), 5.01 (bs, 1H), 2.18 (bs, 1H); 13C NMR (CDCl3, 150 MHz) δ 159.9, 152.0, 150.2, 138.4, 137.7, 136.0, 132.4, 131.1, 131.0, 129.8, 129.4, 129.1, 128.7, 128.5, 127.9, 124.4, 124.2, 119.7, 119.2, 117.2, 36.9; IR νmax (ATR) 3427, 3062, 1721, 1602, 1446, 1280, 1186, 1067, 765, 690 cm−1; HRMS (EI) calcd for C25H17NO2 [M+] 363.1259 found 363.1252.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the Ministry of Science and Technology of the Republic of China, Taiwan, for financially supporting this research under Contract No. MOST 105-2113-M-029-001.
Notes and references
- N. A. Nedolya and B. A. Trofimov, Chem. Heterocycl. Compd., 2013, 49, 152 CrossRef CAS.
- I. Khan, Anupama and B. Singh, Sci. Rev. Chem. Commun., 2015, 5, 13 CAS.
- J. A. Balfour and K. L. Goa, Drugs, 1991, 42, 511 CrossRef CAS PubMed.
- J. Weinstock, D. L. Ladd, J. W. Wilson, C. K. Brush, N. C. F. Yim, G. Gallagher Jr, M. E. McCarthy, J. Silvestri and H. M. Sarau, J. Med. Chem., 1986, 29, 2315 CrossRef CAS PubMed.
- B. M. Smith, J. M. Smith, J. H. Tsai, J. A. Schultz, C. A. Gilson, S. A. Estrada, R. R. Chen, D. M. Park, E. B. Prieto, C. S. Gallardo, D. Sengupta, P. I. Dosa, J. A. Covel, A. Ren, R. R. Webb, N. R. A. Beeley, M. Martin, M. Morgan, S. Espitia, H. R. Saldana, C. Bjenning, K. T. Whelan, A. J. Grottick, F. Menzaghi and W. J. Thomsen, J. Med. Chem., 2008, 51, 305 CrossRef CAS PubMed.
- J. W. Coe, P. R. Brooks, M. G. Vetelino, M. C. Wirtz, E. P. Arnold, J. Huang, S. B. Sands, T. I. Davis, L. Lebel, C. B. Fox, A. Shrikhande, J. H. Heym, E. Schaeffer, H. Rollema, Y. Lu, R. S. Mansbach, L. K. Chambers, C. C. Rovetti, D. W. Schulz, F. D. Tingley and B. T. O'Neill, J. Med. Chem., 2005, 48, 3474 CrossRef CAS PubMed.
-
(a) C. A. James, A. L. Coelho, M. Gevaert, P. Forgione and V. Snieckus, J. Org. Chem., 2009, 74, 4094 CrossRef CAS PubMed;
(b) N. Thasana, R. Worayuthakarn, P. Kradanrat, E. Hohn, L. R. Young and S. Ruchirawat, J. Org. Chem., 2007, 72, 9379 CrossRef CAS PubMed;
(c) I. Kostova, Curr. HIV Res., 2006, 4, 347 CrossRef CAS PubMed;
(d) T. L. Yao, D. W. Yue and R. C. Larock, J. Org. Chem., 2005, 70, 9985 CrossRef CAS PubMed;
(e) F. Borges, F. Roleira, N. Milhazes and L. Santana, Curr. Med. Chem., 2005, 12, 887 CrossRef CAS PubMed;
(f) D. L. Yu, M. Suzuki and L. Xie, Med. Res. Rev., 2003, 23, 322 CrossRef CAS PubMed.
-
(a) B. N. Goguen, A. Aemissegger and B. Imperiali, J. Am. Chem. Soc., 2011, 133, 11038 CrossRef CAS PubMed;
(b) Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610 CrossRef CAS PubMed;
(c) L. Chen, T. S. Hu and Z. Yao, Eur. J. Org. Chem., 2008, 6175 CrossRef CAS;
(d) K. Komatsu, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13447 CrossRef CAS PubMed;
(e) M. T. Lee, C. K. Yen, W. P. Yang, H. H. Chen, C. H. Liao, C. H. Tsai and C. H. Chen, Org. Lett., 2004, 6, 1241 CrossRef CAS PubMed;
(f) K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC;
(g) E. Quanten, P. Adriaens, F. C. Deschryver, R. Roelandts and H. Degreef, Photochem. Photobiol., 1986, 43, 485 CrossRef CAS PubMed.
- J. J. Chen, K. T. Li and D. Y. Yang, Org. Lett., 2011, 13, 1658 CrossRef CAS PubMed.
- K. T. Li, Y. B. Lin and D. Y. Yang, Org. Lett., 2012, 14, 1190 CrossRef CAS PubMed.
- C. H. Lin and D. Y. Yang, Org. Lett., 2013, 15, 2802 CrossRef CAS PubMed.
-
(a) S. Ranatunga, C.-H. A. Tang, C. W. Kang, C. L. Kriss, B. J. Kloppenburg, C.-C. A. Hu and J. R. Del Valle, J. Med. Chem., 2014, 57, 4289 CrossRef CAS PubMed;
(b) Y. Kuroki, R. Akao, T. Inazumi and M. Noguehi, Tetrahedron, 1994, 50, 1063 CrossRef CAS;
(c) M. A. Waly, M. M. Mashaly, M. N. Khodeir and A. Omar, Boll. Chim. Farm., 1994, 113, 698 Search PubMed;
(d) K. R. Prasad and M. Darbarwar, Synth. Commun., 1990, 20, 1379 CrossRef CAS.
- K. Ramig, S. Alli, M. Cheng, R. Leung, R. Razi, M. Washington and L. V. Kudzma, Synlett, 2007, 18, 2868 CrossRef.
- C. G. Neochoritis, C. A. Tsoleridis, J. Stephanidou-Stephanatou, C. A. Kontogiorgis and D. J. Hadjipavlou-Litina, J. Med. Chem., 2010, 53, 8409 CrossRef CAS PubMed.
- N. Kausar, P. Mukherjee and A. R. Das, RSC Adv., 2016, 6, 88904 RSC.
-
(a) K. B. Manjappa, W. F. Jhang, S. Y. Huang and D. Y. Yang, Org. Lett., 2014, 16, 5690 CrossRef CAS PubMed;
(b) K. B. Manjappa, J. R. Syu and D. Y. Yang, Org. Lett., 2016, 18, 332 CrossRef CAS PubMed;
(c) K. B. Manjappa, Y. T. Peng, W. F. Jhang and D. Y. Yang, Tetrahedron, 2016, 72, 853 CrossRef CAS.
- N. C. Ganguly, M. Datta, P. De and R. Chakravarty, Synth. Commun., 2003, 33, 647 CrossRef CAS.
- I. Brady, D. Leane, H. P. Hughes, R. J. Forster and T. E. Keyes, Dalton Trans., 2004, 21, 334 RSC.
- Crystallographic data (excluding structure factors) for 4a, 4j, and 4n have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC-1568069, 1568070, and 1568068, respectively.†.
-
(a) K. Ramig, E. M. Greer, D. J. Szalda, S. Karimi, A. Ko, L. Boulos, J. Gu, N. Dvorkin, H. Bhramdat and G. Subramaniam, J. Org. Chem., 2013, 78, 8028 CrossRef CAS PubMed;
(b) K. Ramig, Tetrahedron, 2013, 69, 10783 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure for the synthesis of 4a–4o and CIF files. CCDC 1568068–1568070. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09289j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a