
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polydopamine as a bridge to decorate monodisperse gold nanoparticles on Fe3O4 nanoclusters for the catalytic reduction of 4-nitrophenol
Shili Liu,
Aori Qileng,
Junying Huang,
Qiongzhi Gao * and
Yingju Liu*
* and
Yingju Liu*
College of Materials & Energy, South China Agricultural University, Guangzhou 510642, China. E-mail: qiongzhigao@yahoo.com; liuyingju@hotmail.com; Fax: +86-20-85282366; Tel: +86-20-85280319
First published on 25th September 2017
Abstract
Herein, gold nanoparticles (Au NPs) were decorated on magnetic Fe3O4 nanoclusters@polydopamine nanocomposites (Fe3O4@PDA NCs) through a direct and green reduction method. The morphology was investigated via transmission electronic microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS). Results showed that Fe3O4 nanoclusters were successfully coated with a PDA shell layer of 25 nm thickness that could work as a reducing reagent to form Au NPs on the surface of Fe3O4@PDA NCs and prevent the aggregation of Au NPs as well. More interestingly, the concentration of the Au precursor had a great effect on both the size and the dispersion of Au NPs on the surface of Fe3O4@PDA NCs, which directly affected the catalytic activity of Fe3O4@PDA@Au. The catalytic performance of Fe3O4@PDA@Au was determined by reducing 4-nitrophenol to 4-aminophenol in the presence of excessive NaBH4, and the result showed that Fe3O4@PDA@Au prepared from a 130 μM Au precursor exhibited the best catalytic activity with the reaction rate constant of 39.2 s−1 g−1 and a conversion of >99% in ten minutes. After being recycled and reused ten times, the magnetic catalyst still had a conversion of >95%; this suggested that it might have practical applications in the reduction of nitroaromatic compounds.
1. Introduction
4-Nitrophenol is one of the most common organic chemicals used in pesticides, dyes, and explosives. Therefore, possible carcinogenic effects associated with 4-nitrophenol are particularly concerning due to the high toxicity and stability of 4-nitrophenol in water.1 As a typical solution to environmental contamination, an effective strategy for the rapid and efficient removal of nitrophenols from wastewater and contaminated aquifers is highly required.2 In this regard, the catalytic reduction of 4-nitrophenol to 4-aminophenol has gained significant interest since it usually occurs in the aqueous phase without the production of by-products, and the product 4-aminophenol is potentially used in the production of analgesic, antipyretic drugs, anticorrosion lubricants.3,4 Although the reduction of 4-nitrophenol to 4-aminophenol is thermodynamically downhill (Eθ|4-nitrophenol/4-aminophenol = −0.76 V, Eθ|H3BO3/BH4− = −1.33 V versus normal hydrogen electrode), this process still cannot proceed without catalysts due to the kinetic barrier caused by the large potential difference between the donor and the acceptor molecules.5 In the presence of catalysts, the catalytic hydrogenation reaction has the advantages of simplicity, high product quality, and less environmental pollution. To decrease the activation energy, maintain the stability of the catalysts, and prevent them from being reduced by NaBH4, mostly, noble metal materials are used as catalysts in this reaction. Nowadays, the most concerning problems are how to enhance the activity of the catalyst and thus how to decrease the waste of noble metals.Gold nanoparticles (Au NPs) have received significant attention due to their unique physicochemical properties; thus, they have been reported as the most effective noble metals in catalysis, biosensors, optical devices, thermal therapy, and other specific applications.6–9 In catalysis, the initially discovered Au NPs were used to oxidize carbon monoxide by Au NP-supported-transition metal oxide. Recently, several catalytic reactions, such as reduction of alkynols,10 water–gas shift reaction,11 and methanol oxidation,12 can also be easily achieved by Au NPs under mild conditions. However, due to their high active surface atoms and strong van der Waals attraction, Au NPs exhibit a strong tendency to aggregate into larger clusters; this results in a change in their size and shape with deactivation or loss of their catalytic activity.13 To offset this disadvantage, significant efforts, including the use of capping reagents and other supports such as carbon nanotubes,14 silica, and other metal oxides,15,16 have been made to improve the stability of Au NPs. Since the catalytic performance of metal NPs is usually associated with their sizes and crystalline and electronic structures, the support should not block the surface of metal NPs and should maintain the small size and high stability of metal NPs. Additionally, the separation and recycling of catalysts is another important concern. For practical applications, the conventional separation, such as filtration and centrifugation,17 of the catalyst usually involves complex operation and huge loss of products. To overcome these drawbacks, great efforts have been made to introduce magnetic particles into noble metal composites. Although iron oxide nanoclusters (Fe3O4 NCs) have been widely used in biological separation, drug delivery, cancer detection, and therapeutics,18,19 the lack of functional groups on Fe3O4 NCs can still lead to the aggregation of Au NPs on their surface. Therefore, much effort has been focused on the fabrication of water-soluble Fe3O4 NCs with a controllable size, fast magnetic response, and desirable surface property.
Phenol derivatives have been extensively studied in the synthesis of noble metal materials since they are able to serve as both reducing agents and stabilizers.20 As one of the outstanding representatives, polydopamine (PDA) has received significant attention since it can be spontaneously self-polymerized in the presence of oxygen under alkaline conditions.21 Due to the remarkable coating quality of PDA, it has become easy to adhere a uniform and continuous coating layer onto nearly any material, and the layer thickness of PDA can be controlled by changing the concentration of dopamine and the reaction time. Specifically, the PDA film can be used as a reactive platform to template the loading and dispersion of noble metal NPs due to its non-covalent binding with hydroxyl–metal complexes or chelating interactions with metal precursors.19 More importantly, the abundant catechol groups ensure the excellent redox activity of PDA since PDA can reduce noble metal ions such as Au3+ and Ag+.14,22 In addition, the multi-step synthesis strategy is very popular in the preparation of core–shell magnetic polymer-noble composites. For instance, Yao et al. fabricated Fe3O4@polypyrrole-Pd composites,23 where the synthesis strategy involved pre-synthesis of Fe3O4@SiO2 spheres, grafting of PVP on the Fe3O4@SiO2 spheres, deposition of the polypyrrole layer, and reduction of PdCl2 using NaBH4. This fabrication procedure was very complex since some special agents or treatment processes were needed.
In this study, the synthesis of Fe3O4@PDA@Au catalyst was divided into only two steps: self-polymerization of dopamine on Fe3O4 NCs and then the direct reduction of HAuCl4 to form Au NPs on the surface of Fe3O4@PDA NCs, where the PDA layer worked as both the reducing agent and the coupling agent and also effectively prevented agglomeration or leaching of Au NPs. In addition, the Au NPs with different diameters on the core–shell Fe3O4@PDA NCs were controlled by the Au precursor concentrations, and their catalytic properties were investigated in detail.
2. Experimental
2.1 Chemicals and materials
Ferric chloride (FeCl3·6H2O), sodium acetate trihydrate (CH3COONa·3H2O), and ethylenediaminetetraacetic acid disodium salt (C10H14N2Na2O8·2H2O) were bought from Sinopharm Chemical Reagent Co. Ltd. (Beijing, China). 4-Nitrophenol, 4-aminophenol, tris(hydroxymethyl)aminomethane (Tris), gold(III) chloride hydrate (HAuCl4·4H2O, 99.0%), sodium hydroxide (NaOH), and sodium hydroxide (NaBH4) were bought from Aladdin Chemistry Co. Ltd. (Shanghai, China). Dopamine hydrochloride (C8H11NO2HCl) was obtained from Macklin Chemical Reagent Co. Ltd. (Shanghai, China). All other reagents were of analytical grade and used without further purification. Ultrapure water was prepared using a Milli-Q reagent water system (Millipore, Bedford, MA, USA).2.2 Instrumentation
The morphologies of the catalysts were characterized by transmission electron microscopy (TEM, Tecnai 12, FEI, Holland) at the Instrumental Analysis & Research Center in South China Agricultural University. The FTIR spectra and UV-vis spectra were obtained by Fourier transform infrared spectroscopy (FTIR, Nicolet 6700, Thermo, America) and ultraviolet visible spectrophotometry (UV-vis, UV 2550, Shimadzu, Japan), respectively. The surface information of the samples was obtained via X-ray photoelectron spectroscopy (XPS, Thermo-VG Scientific, ESCALAB 250 Xi) using an Al Kα X-ray source at the Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences. The magnetization curves of the catalyst were measured using the Magnetic Property Measurement System (Quantum Design, MPMS XL-703140716) at the Instrumental Analysis & Research Center in Sun Yat-sen University.2.3 Preparation of the Fe3O4@PDA@Au catalyst
Magnetic Fe3O4 NCs were prepared as follows.18 FeCl3·6H2O (0.675 g) was dissolved in 20 mL of ethylene glycol, then 0.2233 g of C10H14N2Na2O8·2H2O and 1.2 g of CH3COONa were added, and magnetic stirring was continued until the solid was dissolved. The mixture was sealed in a 50 mL autoclave and heated at 200 °C for 10 h. The sample was cooled down to room temperature, washed with ethanol and water, separated by an external magnet, and dried in an oven.After this, 11 mg of magnetic Fe3O4 NCs was dispersed in a mixture containing 11 mL of ethanol and 25 mL of ultrapure water. After ultrasonication for 1 h, 12 mL tris solution (pH 8.5, 10 mM Tris–HCl buffer) and 12 mg of dopamine were added. The solution was magnetically stirred for 18 h at room temperature, and then, the product was separated, washed, obtained using an external magnet, and redispersed in 10 mL water for further use.
Finally, 2 mL of the as-prepared Fe3O4@PDA aqueous solution was dispersed in 15 mL of the HAuCl4 solution at 90 °C for 30 min. Different concentrations (30, 80, 130, 160, and 180 μM) of the HAuCl4 solution were used to synthesize Fe3O4@PDA@Au with different Au loadings. After the reaction, Fe3O4@PDA@Au was magnetically separated and washed to remove the free Au NPs. At last, it was dried and dissolved at a concentration of 1 mg mL−1 suspension for the catalytic reduction.
In addition, a small batch of pure Au NPs was prepared as a ref. 24. Herein, 2.0 g of dried Lagerstroemia speciosa leaf powder was dispersed in 50 mL of deionized water at 70 °C for 30 min, centrifuged at 6500 rpm for 10 min, and filtered. Then, the filtrate was used to reduce HAuCl4 at room temperature. The mixture was centrifuged at 8000 rpm for 15 min and washed with ethanol and water. Finally, the Au NPs were dried in vacuum and dissolved at a concentration of 1 mg mL−1 suspension for the catalytic reduction.
2.4 Catalytic study
For the catalytic reduction of 4-nitrophenol, 2 mL of ultrapure water, 0.5 mL of 40 mM 4-nitrophenol, and freshly prepared 0.5 mL of 0.4 mM NaBH4 aqueous solution were continuously mixed in a quartz cuvette. Then, the aqueous suspension of catalysts (1 mg mL−1) was ultrasonically treated. After 0.2 mL of catalyst was added, the color of the solution gradually cleared away as the reaction continued, which was monitored by ultraviolet-visible spectroscopy in the range from 250 nm to 550 nm every 60 s. Then, the catalysts were separated from the mixture using an external magnet, washed with ultrapure water, and reused in the next cycle. The experiment was repeated 10 times to confirm the recycle performance.3. Results and discussion
3.1 Characterization of the Fe3O4@PDA@Au catalyst
TEM was used to characterize the morphology of the samples at each stage. The pristine Fe3O4 NCs exhibit great stability and dispersibility even after stewing for 3 weeks. As shown in Fig. 1A, Fe3O4 NCs are uniformly dispersed with an approximate diameter of 150 nm, which are composed of many interconnected nanoparticles with a size of about 25 nm. Fig. 1B is the TEM image of Fe3O4@PDA, showing that Fe3O4 NCs are decorated with a PDA layer with the average shell diameter of 25 nm. This PDA layer can provide eminent protection against the aggregation of Fe3O4, and complicated modification steps are avoided since the process of synthesis of Fe3O4@PDA NCs contains no reducing agents or thermal treatment for a long time. Then, the PDA layer works as a natural binder to attract HAuCl4 on its surface via non-covalent binding of hydroxyl–metal complexes or chelating interactions. Specifically, the catechol groups of PDA can act as the reducing agent to reduce HAuCl4 and form Au NPs.20 Fig. 1C–G show the TEM image of the as-prepared Fe3O4@PDA@Au with different concentrations of HAuCl4, where the size, shape, and dispersion of Au NPs show strong dependency on the dosage of HAuCl4. At a low dosage of HAuCl4, Au3+ ions may only reach to the surface of the PDA layer where Au3+ ions are reduced to Au0 by the superficial catechol groups and Au nanoparticles are formed (∼8 nm as shown in Fig. 1C). With the increase in the HAuCl4 concentration, a higher amount of monodisperse Au NPs (∼10 nm as shown in Fig. 1D) can be found on the surface of Fe3O4@PDA. The further increase in HAuCl4 concentration can increase the diameters of Au NPs to 20 nm, and some Au NPs can even be embedded in the PDA layer (Fig. 1E). However, if the dosage of HAuCl4 is too high, the size of Au NPs increases to around 40 nm; moreover, the distribution of Au NPs becomes less uniform and obvious agglomeration can be observed (Fig. 1F and G) since Au NPs have a tendency to grow fast in the bulk solution with steric hindrance in the presence of a high amount of HAuCl4. Therefore, 130 μM HAuCl4 was the best concentration to synthesize Fe3O4@PDA@Au. | ||
| Fig. 1 TEM image of (A) pristine Fe3O4 nanoclusters, (B) Fe3O4@PDA NCs, and the as-prepared Fe3O4@PDA@Au with different concentrations of HAuCl4, (C) 30 μM, (D) 80 μM, (E) 130 μM, (F) 160 μM, and (G) 180 μM. | ||
FTIR was further used to characterize the synthesized Fe3O4, Fe3O4@PDA and Fe3O4@PDA@Au composites. As for pristine Fe3O4 (Fig. 2a), the peaks at 584 and 3426 cm−1 can be associated with Fe–O stretching vibration and O–H stretching vibration, respectively, confirming the deposition of Fe3O4.18 The absorption peak at 1400 cm−1 is related to the vibration of COO– groups on the Fe3O4 surface, whereas the broad peak at 1644 cm−1 corresponds to the overlapping of the stretching vibration of carbonyl groups and deformation vibration of O–H groups of Fe3O4. With the PDA coating (Fig. 2b), the characteristic bands at 2960 and 2890 cm−1 belong to the vibrational bands of methylene groups of PDA.25 In addition, the increase in the relative intensity of carbonyl group at 1640 cm−1 and Fe–O group at 584 cm−1 indicates the obvious carbonyl group on the Fe3O4@PDA NCs, whereas the amide N–H shearing, the aromatic ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and the phenolic C–OH stretching vibrations of PDA are demonstrated by the bands at 1509, 1445, and 1301 cm−1, respectively. However, after the modification of Au NPs, the intensities of the corresponding bands, such as 3426, 1640, and 584 cm−1, for Fe3O4@PDA slightly decrease; this suggests that the deposition of Au NPs influences the surface of Fe3O4@PDA NCs.
C, and the phenolic C–OH stretching vibrations of PDA are demonstrated by the bands at 1509, 1445, and 1301 cm−1, respectively. However, after the modification of Au NPs, the intensities of the corresponding bands, such as 3426, 1640, and 584 cm−1, for Fe3O4@PDA slightly decrease; this suggests that the deposition of Au NPs influences the surface of Fe3O4@PDA NCs.
 | ||
| Fig. 2 FTIR spectra of Fe3O4 (a), Fe3O4@PDA NCs (b), and Fe3O4@PDA@Au (c). | ||
The surface chemical information of Fe3O4@PDA and Fe3O4@PDA@Au was also examined by XPS. In Fig. 3A and B, the C, N, and O peaks can be found for both Fe3O4@PDA and Fe3O4@PDA@Au. Moreover, as shown in Fig. 3C, the Au 4f peaks at the binding energies of 83.7 and 87.7 eV can be attributed to Au 4f7/2 and Au 4f5/2, respectively, suggesting the formation of Au NPs on the Fe3O4@PDA NCs. However, Fe cannot be found in both nanocomposites; this indicates that Fe3O4 NCs are confined within the PDA shell structure, consistent with the TEM analyses. Fig. 3D is the C1s core-level spectrum of Fe3O4@PDA NCs, which can be deconvoluted into four peaks located at 284.4, 285.5, 286.4, and 288.5 eV, assigned to the C–C (49.0%), C–N (29.8%), C–O (14.1%), and CO (7.1%) groups, respectively.26 After reaction with HAuCl4, as shown in Fig. 3G, the peaks of C–C (54.1%) and CO (8.6%) are still dominant, but the intensities of the C–N (25.8%), C–O (11.5%) peaks exhibit a slight decline; this suggests that some catechol groups are oxidized. Fig. 3E is the N1s core-level spectrum of Fe3O4@PDA NCs, where the binding energies at 398.9 and 399.9 eV correspond to the N–R and R–NH–R groups; the dominant peak of the N1s core-level spectrum changes from the N–R peak to the R–NH–R peak, as shown in Fig. 3H; this confirms the existence of PDA and further proves the electron transfer.27 As for the O1s core–shell spectrum of Fe3O4@PDA NCs (Fig. 3F), it can be deconvoluted into two peaks at 532.3 eV and 533.2 eV for the C–O and CO group, respectively. After the coating of Au nanoparticles, the percentage of the CO group increased from 47.0% to 51.4% (Fig. 3I), also proving that catechol groups of PDA can release electrons to the corresponding quinone groups, and free Au3+ ions accept these electrons to get reduced to Au0 atoms and achieve exposure of a greater percentage of active sites.20 Therefore, PDA can directly reduce HAuCl4 to synthesize Au NPs on the surface of the Fe3O4@PDA NCs, where no additional reagent is involved and Au NPs are effectively prevented from aggregation via control of the HAuCl4 concentration; this avoids the possible contamination and waste of chemicals.
 | ||
| Fig. 3 XPS survey scan for Fe3O4@PDA NCs (A) and Fe3O4@PDA@Au (B), spectra for Au 4f peaks (C), deconvolution of C1s (D), N1s (E), and O1s (F) peaks of Fe3O4@PDA NCs, and deconvolution of C1s (G), N1s (H), and O1s peaks (I) of Fe3O4@PDA@Au. | ||
3.2 The catalytic ability of the Fe3O4@PDA@Au catalyst on 4-nitrophenol
The catalytic reaction was detected via UV-visible spectroscopy by the change in the absorption peak. As shown in Fig. 4A, the solution of 4-nitrophenol shows a major absorption peak at 317 nm and a slight peak at 400 nm (curve a). After the addition of excess NaBH4 solution, the characteristic absorption peak undergoes a redshift from 317 to 400 nm (curve b), whereas the characteristic absorption peak at 400 nm suggests the formation of a longer conjugative bonds with 4-nitrophenolate ions.13 After the addition of the catalyst, the peak at 400 nm for 4-nitrophenolate ions decreases rapidly, and only a peak at 300 nm in the ultra-violet region can be found (curve c). Correspondingly, as shown in Fig. 4B, the solution immediately changes from light yellow to bright yellow. Without the catalyst, the transformation from 4-nitrophenol to 4-aminophenol is very slow since the maximum absorption peak remains the same. However, after the addition of the catalyst, the solution changes from bright yellow to colorless, as shown in Fig. 4B; this suggests the reduction of 4-nitrophenol to 4-aminophenol, as shown in Fig. 4C. Therefore, the reduction reaction can be summarized as the following equation: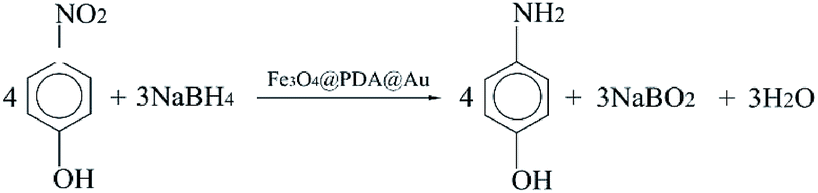
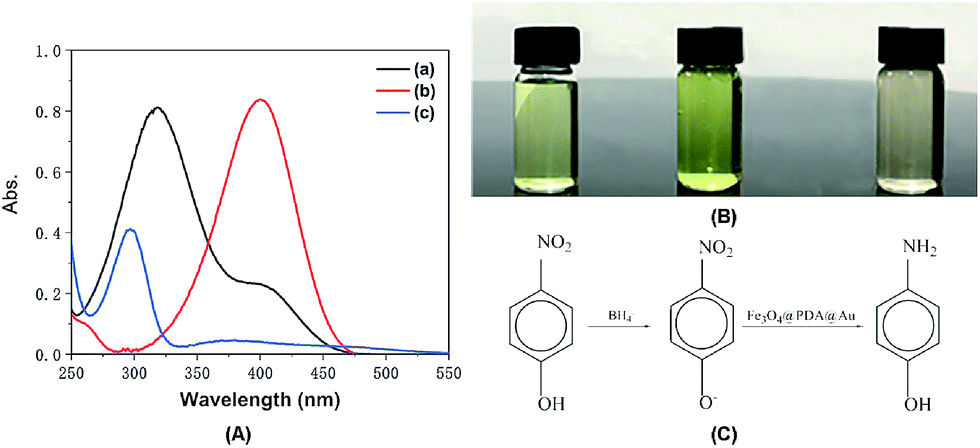 | ||
| Fig. 4 (A) UV-vis absorption spectra of (a) 4-nitrophenol, (b) 4-nitrophenol + NaBH4, and (c) 4-aminophenol. (B) The color change of the corresponding solution, and (C) the transformation of 4-nitrophenol to 4-aminophenol. | ||
To further investigate the mechanism of action of the catalyst, UV-vis spectra were obtained at different times. As shown in Fig. 5A, absorption changes slightly if only Fe3O4@PDA NCs are added. Although Fe3O4 NPs have proven their catalytic ability in the elimination of some substituted phenolic and aniline compounds from solution28 and coordination of the possible ligands to the metal via lone pairs or π-bonds,29 the degradation rate for Fe3O4@PDA NCs is about 25% in the first 10 minutes. The inset of Fig. 5A shows the spectrum of 0.2 mg of pure Au NPs, showing that the catalytic effect is significantly lower even in 30 min. However, if Fe3O4@PDA@Au is added, the intensity of the absorption peak, as shown in Fig. 5B, gradually decreases, and the peak finally disappears; this suggests a much higher catalytic ability of the as-prepared Fe3O4@PDA@Au for the reduction of 4-nitrophenol. Therefore, the modification of Au nanoparticles on Fe3O4@PDA played an important role in this reaction.
 | ||
| Fig. 5 Successive UV-vis absorption spectra for the reduction of 4-nitrophenol in the presence of (A) Fe3O4@PDA NCs (the inset shows the spectrum for pure Au NPs), (B) the as-prepared Fe3O4@PDA@Au obtained from 130 μM HAuCl4, (C) the relationship of ln(Ct/C0) with time, and (D) the relationship of 4-nitrophenol conversion with time. | ||
Since the initial concentration of the NaBH4 solution is set 100 times higher than that of 4-nitrophenol, the reaction rate can be speculated to follow pseudo-first order reaction kinetics, which can be quantitatively described to be a constant from the time-dependent absorption spectra during the reduction process. As shown in Fig. 5C, the linear plot of ln(Ct/C0) versus time is fitted in the following equation:30
| ln(Ct/C0) = ln(At/A0) = −kt |
| k = Km |
In addition, the K value for Fe3O4@PDA@Au obtained from 130 μM HAuCl4 is 39.2 s−1 g−1, which is also greatly higher than those obtained from other concentrations. We have also compared it with the values reported in literature using other different supports, and the abovementioned K value is found to be higher than those of the solid supports such as core–shell SiO2@Au composite particles (1.38 s−1 g−1),35 carbon nanotubes functionalized with cyclotriphosphazene-containing polyphosphazene-loaded Au (5.93 s−1 g−1),36 Au nanoparticles supported on iron oxides (5.25 s−1 g−1),37 polymer-supported Au nanoparticles such as the hexagonal porphyrin-based porous organic polymer (0.25 s−1 g−1),13 and alloy nanoparticles such as silver-gold alloy nanoparticles (34.7 s−1 g−1) synthesized using one-step gamma radiation.38 The possible reason for this high K value is mainly the well-dispersion of Au NPs on Fe3O4@PDA and the effective protection of Au NPs from aggregation by PDA.
Furthermore, as shown in Fig. 5C, it can be found that the absorption intensity of 4-nitrophenol decreases, whereas the absorption intensity of 4-aminophenol increases as the reaction proceeds. Therefore, the conversion of 4-nitrophenol (4-NP) is expressed as
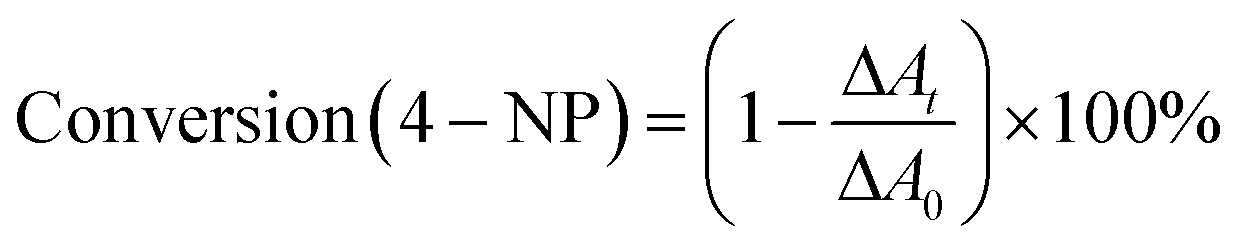
Fe3O4@PDA@Au prepared from 130 μM HAuCl4 shows the best catalytic activity, a conversion of more than 99% in ten minutes, which is more than that obtained using Au NPs at other amounts (30, 80, 160, and 180 μM). Generally, the catalytic performance of Au NPs is affected by their size, site density, stability, and gold–support interactions. Since pure Au NPs will generate different reactant diffusion to the metallic surface and product diffusion away from the particle surface,39 the catalytic abilities of pure Au NPs are very weak (the inset of Fig. 5A). However, for the Fe3O4@PDA@Au catalyst, the electronic activity of Au NPs on the surface of Fe3O4@PDA is much higher than that of pure Au NPs. The activity difference between different Au NPs can be ascribed to the quantity, shape, and more active sites of the suitable dosage of Au NPs, showing that much more catalytic sites are accessible for the reduction of 4-nitrophenol to 4-aminophenol with the perfect amount of gold support. As shown in TEM, with the more uniform Au NPs on the surface, the accessibility to catalytic sites can be significantly enhanced; thus, the catalytic performance can be greatly improved. Hence, the dosage of HAuCl4 plays an important role in controlling the formation of Au NPs and greatly affects the catalytic activity.
Magnetic properties of the prepared Fe3O4@PDA NCs and the Fe3O4@PDA@Au catalyst were determined using the Magnetic Property Measurement System by sweeping the external field between −2 and 2T at room temperature, as shown in Fig. 6A. Both magnetization curves show no remanence and coercivity; this demonstrates the superparamagnetic behavior of the abovementioned catalysts.26,40 Compared to Fe3O4@PDA NCs, the Fe3O4@PDA@Au catalyst shows an obvious decrease in saturation magnetization, which may be attributed to the loading of Au NPs on the surface of the PDA layer and the consequent increase in the thickness of the shell. However, the prepared Fe3O4@PDA@Au catalyst still has a strong magnetization of 33.8 emu g−1, and the catalyst can be easily separated by an external magnet. Therefore, to investigate the reusability of the catalyst, the Fe3O4@PDA@Au catalyst was separated easily and recovered rapidly from the solution within 1 min by a magnet and then redispersed into the new solution for the next cycle. As shown in Fig. 6B, the Fe3O4@PDA@Au catalyst can be successfully reused for ten repeated cycles, and the conversion remains over 95%. Since the time for the complete conversion of different cycles gradually increases, possible deactivation may exist. However, even after 10 cycles, the time for the complete conversion is still only in 50 min, which is acceptable for practical applications. Therefore, the magnetic Fe3O4@PDA@Au catalyst maintains good catalytic performance during the catalytic or separation processes; this suggests that the catalyst possesses excellent recyclability, good stability, and a long life.
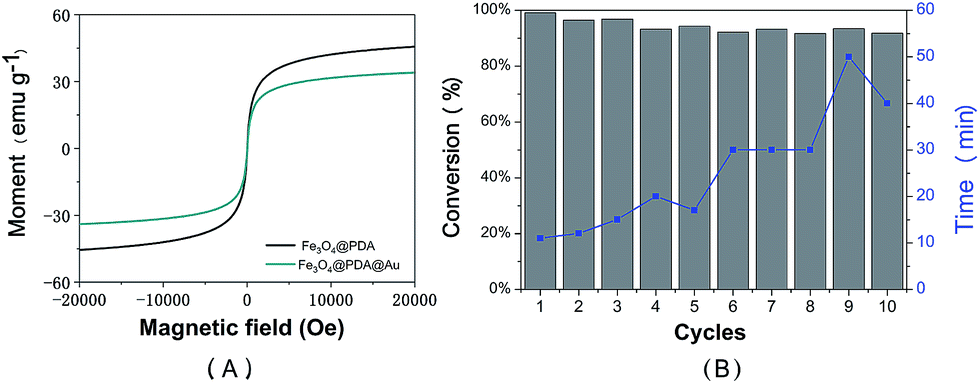 | ||
| Fig. 6 (A) Room-temperature magnetization hysteresis loops of the as-prepared Fe3O4@PDA NCs and Fe3O4@PDA@Au. (B) Recyclability including conversion (black) and time (blue) of the Fe3O4@PDA@Au catalyst in the reduction of 4-nitrophenol. | ||
4. Conclusions
In summary, the well-defined core–shell Fe3O4@PDA@Au catalysts were successfully prepared through a green and facile method, where PDA worked as a reducing agent and coupling agent and effectively prevented agglomeration or leaching of Au NPs without any other reagents or a high thermal treatment. Distinct concentrations of gold precursors were used to decorate core–shell Fe3O4@PDA nanoparticles with different sizes and amounts of Au NPs. Results showed that the magnetic Fe3O4@PDA@Au catalyst prepared from the 130 μM Au precursor exhibited the best catalytic activity with the reaction rate constant of 39.2 s−1 g−1 and a conversion of >99% in ten minutes, which was important for applications under realistic technical conditions. The strategy may provide a facile and versatile guide towards the design of Fe3O4-based nanoparticles for biological, energy, and environmental applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Scientific Foundation of China (21475047), the Science and Technology Planning Project of Guangdong Province (2016B030303010), the Program for the Top Young Innovative Talents of Guangdong Province (2016TQ03N305), and the Foundation for High-level Talents in South China Agricultural University.Notes and references
- N. Sahiner, H. Ozay, O. Ozay and N. Aktas, Appl. Catal., B, 2010, 101, 137–143 CrossRef CAS.
- M. Sivachidambaram, J. Judith Vijaya, K. Kaviyarasu, L. J. Kennedy, H. A. Al-Lohedan and R. J. Ramalingam, RSC Adv., 2017, 7, 38861–38870 RSC.
- C. Wu, X. An, S. Gao and L. Su, RSC Adv., 2015, 5, 71259–71267 RSC.
- A. M. Kalekar, K. K. K. Sharma, M. N. Luwang and G. K. Sharma, RSC Adv., 2016, 6, 11911–11920 RSC.
- M. Liang, R. Su, R. Huang, W. Qi, Y. Yu, L. Wang and Z. He, ACS Appl. Mater. Interfaces, 2014, 6, 4638–4649 CAS.
- C. Fan, L. Zhang, S. Wang, D. Wang, L. Lu and A. Xu, Nanoscale, 2012, 4, 6835–6840 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, Z. Li, L. Au, H. Zhang, M. B. Kimmey, X. Li and Y. Xia, Nano Lett., 2005, 5, 473–477 CrossRef CAS PubMed.
- Y. Wang, K. C. L. Black, H. Luehmann, W. Li, Y. Zhang, X. Cai, D. Wan, S. Liu, M. Li, P. Kim, Z. Li, L. V. Wang, Y. Liu and Y. Xia, ACS Nano, 2013, 7, 2068–2077 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros and J. M. Alonso, Org. Biomol. Chem., 2011, 9, 4405–4416 CAS.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed.
- H. Tang, J. Chen, M. Wang, L. Nie, Y. F. Kuang and S. Z. Yao, Appl. Catal., A, 2004, 275, 43–48 CrossRef CAS.
- Z. Ding, Y. Wang, S. Xi, Y. Li, Z. Li, X. Ren and Z. Gu, Chem.–Eur. J., 2016, 22, 17029–17036 CrossRef CAS PubMed.
- B. Fei, B. Qian, Z. Yang, R. Wang, W. C. Liu, C. L. Mak and J. H. Xin, Carbon, 2008, 46, 1795–1797 CrossRef CAS.
- V. Sudheeshkumar, A. Shivhare and R. W. J. Scott, Catal. Sci. Technol., 2017, 7, 272–280 CAS.
- J. Fang, Y. Zhang, Y. Zhou, C. Zhang, S. Zhao, H. Zhang and X. Sheng, Appl. Surf. Sci., 2017, 392, 36–45 CrossRef CAS.
- J. Zhang, Y. Yuan, K. J. Kilpin, Y. Kou, P. J. Dyson and N. Yan, J. Mol. Catal. A: Chem., 2013, 371, 29–35 CrossRef CAS.
- M. Lin, H. Huang, Z. Liu, Y. Liu, J. Ge and Y. Fang, Langmuir, 2013, 29, 15433–15441 CrossRef CAS PubMed.
- R. Ge, X. Li, M. Lin, D. Wang, S. Li, S. Liu, Q. Tang, Y. Liu, J. Jiang, L. Liu, H. Sun, H. Zhang and B. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 22942–22952 CAS.
- R. Liu, Y. Guo, G. Odusote, F. Qu and R. D. Priestley, ACS Appl. Mater. Interfaces, 2013, 5, 9167–9171 CAS.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353–1359 CrossRef CAS PubMed.
- S. Ma, X. Chen, B. Zhao, H. Dong, Q. Yuan, L. Li, J. Lv, L. Shi and L. Chen, Appl. Catal., A, 2017, 536, 35–44 CrossRef CAS.
- T. Yao, T. Cui, X. Fang, J. Yu, F. Cui and J. Wu, Chem. Eng. J., 2013, 225, 230–236 CrossRef CAS.
- B. C. Choudhary, D. Paul, T. Gupta, S. R. Tetgure, V. J. Garole, A. U. Borse and D. J. Garole, J. Environ. Sci., 2017, 55, 236–246 CrossRef PubMed.
- Y. Tian, Y. Cao, Y. Wang, W. Yang and J. Feng, Adv. Mater., 2013, 25, 2980–2983 CrossRef CAS PubMed.
- T. Zeng, X. Zhang, Y. Ma, H. Niu and Y. Cai, J. Mater. Chem., 2012, 22, 18658–18663 RSC.
- J. Liebscher, R. Mrowczynski, H. A. Scheidt, C. Filip, N. D. Hadade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- S. Zhang, X. Zhao, H. Niu, Y. Shi, Y. Cai and G. Jiang, J. Hazard. Mater., 2009, 167, 560–566 CrossRef CAS PubMed.
- N. Yan, Y. Yuan and P. J. Dyson, Dalton Trans., 2013, 42, 13294–13304 RSC.
- D. Gao, X. Zhang, X. Dai, Y. Qin, A. Duan, Y. Yu, H. Zhuo, H. Zhao, P. Zhang, Y. Jiang, J. Li and Z. Zhao, Nano Res., 2016, 9, 3099–3115 CrossRef CAS.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS.
- Y. Liu, Y. Zheng, B. Du, R. R. Nasaruddin, T. Chen and J. Xie, Ind. Eng. Chem. Res., 2017, 56, 2999–3007 CrossRef CAS.
- N. Goswami, Q. Yao, T. Chen and J. Xie, Coord. Chem. Rev., 2016, 329, 1–15 CrossRef CAS.
- J. Li, R. R. Nasaruddin, Y. Feng, J. Yang, N. Yan and J. Xie, Chem.–Eur. J., 2016, 22, 14816–14820 CrossRef PubMed.
- M. Tang, G. Huang, X. Li, X. Pang and H. Qiu, Mater. Chem. Phys., 2015, 162, 31–40 CrossRef CAS.
- X. Wang, J. Fu, M. Wang, Y. Wang, Z. Chen, J. Zhang, J. Chen and Q. Xu, J. Mater. Sci., 2014, 49, 5056–5065 CrossRef CAS.
- F. Lin and R. Doong, J. Phys. Chem. C, 2011, 115, 6591–6598 CAS.
- K. Hareesh, R. P. Joshi, D. V. Sunitha, V. N. Bhoraskar and S. D. Dhole, Appl. Surf. Sci., 2016, 389, 1050–1055 CrossRef.
- A. Wittstock, B. Neumann, A. Schaefer, K. Dumbuya, C. Kuebel, M. M. Biener, V. Zielasek, H. P. Steinrueck, M. Gottfried, J. Biener, A. Hamza and M. Bäumer, J. Phys. Chem. C, 2009, 14, 5593–5600 Search PubMed.
- D. Wang, H. Duan, J. Lv and C. Lv, J. Mater. Chem. A, 2017, 5, 5088–5097 CAS.
| This journal is © The Royal Society of Chemistry 2017 |