
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling orbital coupling at the CoPc/Bi(111) surface by ab initio calculations and photoemission spectroscopy
Zhaofeng Liangab,
Haoliang Sunab,
Kongchao Shena,
Jinbang Hua,
Bo Songc,
Yunhao Lud,
Zheng Jianga and
Fei Song *a
*a
aShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, 201204, China. E-mail: songfei@sinap.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100100, China
cUniversity of Shanghai for Science and Technology, Shanghai, 200093, China
dCollege of Materials Science and Engineering, Zhejiang University, Hangzhou, 310027, China
First published on 9th November 2017
Abstract
The interfacial electronic structures of cobalt phthalocyanine adsorbed on a semimetal Bi(111) surface have been systematically investigated herein. Our study first indicates that the CoPc molecule is quite sensitive to the adsorption site on the relatively inert Bi(111) surface. Secondly, apparent change of the electronic structures of CoPc has been revealed upon adsorption as compared to that in the gas phase, due to the orbital coupling between the cobalt partial empty state and the surface state from the bismuth substrate and interfacial charge transfer. Interestingly, the local magnetic moment is still retained for the adsorbed CoPc molecule on the diamagnetic Bi(111) surface, which is different to that on other noble metal substrates. Analysis of the charge density difference and the Bader charge provides evident insight on the mechanism of interfacial charge transfer which is chiefly mediated by the central Co atom and the weak vdW dispersion between the π-conjugated macrocyclic ligand and the bismuth substrate, as further confirmed by experimental XPS results. In the end, our report may provide an appealing route towards the fundamental understanding and reliable engineering of interfacial interactions between magnetic and semimetal nanostructures.
1 Introduction
Self-assembly of organic molecules on solid surfaces has been demonstrated as an efficient approach in manufacturing low dimensional nanostructures with appealing applications in nanoelectronic devices, due to its superior capabilities such as well-controlled, tailor-made properties and diverse functionalities.1,2 Among the large variety of organic complexes, the family of phthalocyanines (Pcs) is one of the most frequently investigated π-conjugated organic materials, largely attributed to their high thermal and chemical stability, as well as the propensity to form well-ordered thin films on various surfaces.3–8 Consequently, a broad range of applications have been proposed for Pcs in the field of molecule electronics, for example in light-emitting diodes,9 field effect transistors,10 and photovoltaic cells. Regarding application,11 however, a crucial issue still remains that the device's performance is critically dependent on the interactions between Pc molecules and metal electrodes/substrates, since the physical and chemical properties of the adsorption complex on a certain substrate are governed by the corresponding interface interaction.7,12,13 Cobalt phthalocyanine (CoPc), as a typical 3d transition-metal phthalocyanine (TMPc), has also attracted considerable attention in the past owing to its partial empty Co 3d orbitals and magnetic properties (the molecule structure is shown in Fig. 1a).14,15 Previous studies have showed that the electric structure and magnetic property of CoPc can be flexibly modulated when absorbed on various substrates,5,13,15–17 and the modulation mechanism mostly derives from interfacial orbital hybridization and charge transfer associated with the central Co2+ ion and surface adatoms. For instance, CoPc (as well as other TMPcs) is regarded as a representative prototype for Kondo effect studies.18–20 As reported by Zhao et al., the manipulation of Kondo effect of CoPc molecule can be achieved through the tuning of chemical bonding between CoPc adsorption and the Au(111) substrate.20 Despite considerable investigations have been dedicated to the growth of MPcs on various substrates experimentally and theoretically,4–8,12–34 however, less attention has been paid to the exploration of MPc/semimetal interface in the past. Besides, previous scarce reports mainly concentrate on the morphology investigation of MPc on semimetal surface,3,35 while a systematic exploration of the interface electronic structures is still missing. In fact, bismuth (Bi), as one of the most studied semimetal elements, has exhibited fantastic characters, for example, the Bi(111) or Bi(110) surface with significant spin–orbit coupling have been served as the ideal platform in the exploitation of topological properties.36,37 Hence, it will be interesting to study the CoPc/Bi(111) system as a typical prototype to gain insight in the organic–semimetal framework. In our previous work,38 the adsorption of CoPc on a Bi(111) surface has been investigated mainly by photoemission spectroscopy and it is identified that the interfacial charge transfer occurring from the bismuth substrate to the central Co atom leads to the strong molecule–substrate interaction, even in the case of a relatively invert substrate. Detailed information concerning the adsorption configuration, interfacial electronic structures and the precise molecule–substrate interaction mechanism, however, are still missing, which meantime are of key importance to the fundamental understanding and engineering of the delicate interaction in magnetic/semimetal heterostructures.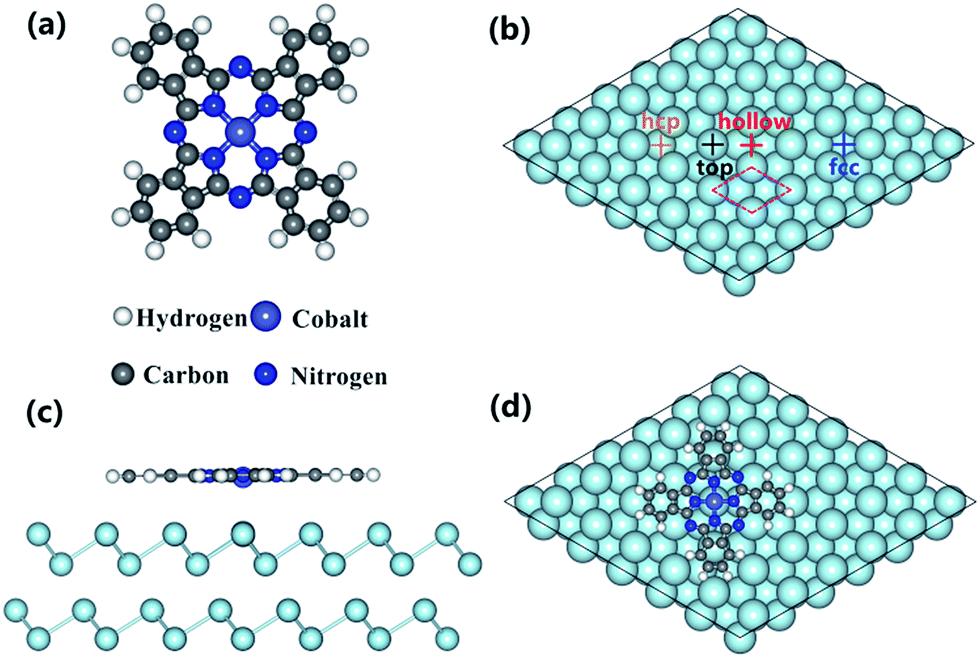 | ||
| Fig. 1 (a) Ball-and-stick model of the CoPc molecule. (b) Schematic drawing of four high-symmetry absorption sites on the Bi(111) surface: top, hcp, fcc and hollow sites. The unit cell of bismuth substrate is indicated by pink dashed line, (c) side view of the top-site adsorption with the lowest absorption energy from DFT calculation, and (d) the top view. | ||
Herein, a comprehensive study is performed for the adsorption of CoPc molecule on a Bi(111) surface, mainly concerning the adsorption configuration, interfacial electronic structures and the mediating mechanism of molecule–substrate interaction by means of ab initio calculations and X-ray photoelectron spectroscopy (XPS). Notably, recent theoretic studies suggest that van der Waals (vdW) interactions may play a key role in the MPc/metal adsorption systems and the calculation results are well coincident with experiment data only if van der Waals interaction effect is considered.15,16,21–23 Indeed, considering the π-conjugated macrocyclic ligand of CoPc as well as the weak interaction when adsorbed on a Bi(111) surface, one can expect that the vdW interactions would be more obvious for the CoPc/Bi(111) system as compared to the case of MPc adsorbed on other relatively active metal surfaces. Consequently, herein, the vdW interaction has been taken into account. Nevertheless, the selection of proper vdW-corrective method is still a crucial issue under debate. Several semi-empirical dispersion-correction schemes have been utilized in MPc/metal adsorption systems, such as DFT-D2 method of Grimme39 applied to the CoPc/Cu(111) interface,15 Tkatchenko–Scheffler van der Waals correction method (DFT-TS)40 applied to the CoPc/Si(111)-B(√3 × √3) system.41 While Bi element is not included in the DFT-D2 function, limited reports in literatures makes it hard to judge whether the DFT-TS method is appropriate for the description of adsorption of CoPc on the Bi(111) surface, a new vdW-corrective approach, called the van der Waals density functional (vdW-DF), can obtain the dispersion interaction from the electron density directly.42 In fact, numerous investigations have shown that vdW-DF method and particularly its modified version vdW-DF2 (ref. 43) are capable of giving excellent descriptions of adsorbate–substrate interaction.44,45 Consequently, vdW-DF2 functional is proposed in this work to make a systematic study of the CoPc/Bi(111) interface.
2 Results and discussion
2.1. Adsorption configurations
Previous studies have shown that MPc molecules interacting with different metal substrates would result in various adsorption geometries and conformations, which can be further characterized by the adsorption site as well as the orientation of macrocyclic ligand on metal surfaces.15,16,23,30 Consequently, it has been devoted first to access the stable configuration for CoPc adsorbed on the Bi(111) surface. In total, four high-symmetry absorption configurations have been constructed by placing the central Co atom of CoPc molecule on the top, hcp, fcc and hollow site, respectively, with respect to the first-layer bismuth atoms from substrate. The adsorption energy (Ead) for the CoPc on the Bi(111) surface complex is calculated by the following definition:| Ead = E[CoPc/Bi(111)] − E[CoPc] − E[Bi(111)] |
From the energy point of view, it can be seen that the top configuration is the most stable adsorption site with the adsorption energy of −2.83 eV, suggesting that the CoPc molecule is favored to be arranged at the top site. Interestingly, it is also notable that the adsorption energy for top site is lower by about 0.44 eV than the rest three configurations, which is apparently different from the CoPc/Ag(111) and CoPc/Au(111) adsorption system, for example, where the adsorption energy for top and hcp configurations is almost same, indicating that the atomic structure/properties of noble metal may hardly influence the adsorption behaviors of MPc molecules. To this point, we propose that, in the weak-interacting CoPc/Bi(111) system, molecules would be more sensitive to the choice of adsorption site at the interface. More precisely, it seems that the interaction between the Co 3d-orbitals and the Bi(111) surface would affect the adsorption configurations in turn. Consequently, we will choose the top site as the most stable configuration to investigate the interfacial electronic properties in the following.
2.2. Interfacial electronic structures
First, spin-polarized projected density of states (PDOS) on the central Co atom and the Pc ligand from CoPc in gas phase and adsorption configuration (top site) are presented, respectively, as well as the PDOS from bismuth substrate in Fig. 2 to intuitively identify the influence of adsorption on interfacial electronic structures. Comparing the PDOS difference of Co atom in gas phase and adsorption form as illustrated in Fig. 2a, one can easily find that the pristine density of states close to the Fermi level from Co atom in gas-phase CoPc have been changed significantly after deposition. One remarkable difference is the disappearance of electronic state with spin-down characteristic at 0.2 eV above the Fermi level, which is attributed to the lowest unoccupied molecule orbitals (LUMO) mostly contributed from the Co atom according to DFT calculations. In fact, analogous behavior has also been reported in the CoPc/Ag(111) adsorption system by Baran et al.,24 which was manifested as a direct charge transfer from substrate surface states to the LUMO of adsorbed CoPc. Since the top configuration of CoPc/Bi(111) has been already confirmed as the preferred adsorption site previously due to its lowest adsorption energy, it can be deduced that the overlap between the unoccupied LUMO orbital of CoPc and surface states from Bi substrate would be maximized when the Co atom is precisely placed above the Bi atom. Such mixing is akin to the charge transfer, leading to modifications in the alignment of orbital levels.46 Interestingly, the transformation of polarization is also found regarding on the Co ion after adsorption, from being pristinely spin-polarized17 to being non-spin-polarized. It should be emphasized that such transformation is not thorough. As observed from PDOS of Co atom in Fig. 2a, the spin-up and spin-down states at −0.7 eV below EF are not completely symmetric, which may imply the incomplete depolarization transformation of the pristine spin-polarized states. Such phenomenon is apparently different from previous reports of CoPc/Au(111),20 CoPc/Cu(111)15 and CoPc/Ag(111) systems,24 where the PDOS of Co ion have turned into non-polarized completely after adsorption.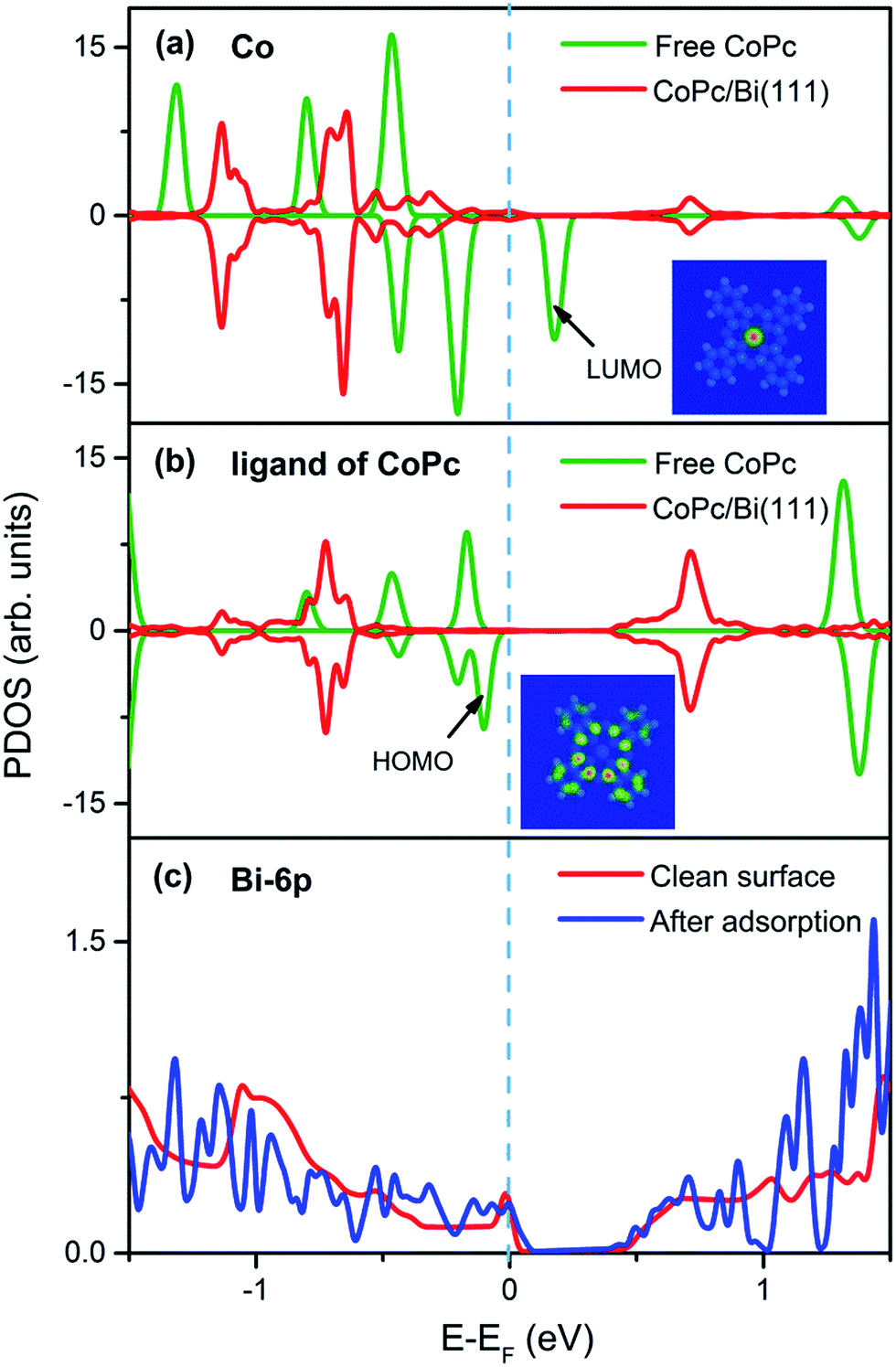 | ||
| Fig. 2 Calculated spin-polarized PDOS of (a) the central Co atom and (b) the Pc ligand from CoPc in the form of gas phase and adsorption configuration (top site), respectively. (c) PDOS of the bismuth atom from a clean surface and that after the deposition of CoPc organics. The Fermi level is indicated by blue dashed line as a guide to eye. The electron cloud distribution of the LUMO and HOMO from the gas-phase CoPc is present in the inset in (a) and (b), respectively. | ||
Regarding on the PDOS of Pc ligand as displayed in Fig. 2b, the peak at around −0.1 eV below the Fermi level can be easily identified as the highest occupied molecule orbital (HOMO) contributed by the π-orbital of macrocyclic ligand. Notably, the state has been shift by about 0.6 eV further away from the Fermi level after adsorption, while the rest states around Fermi level have also been shift in a similar way. Considering the difference of Fermi level between the CoPc molecule and the Bi(111) substrate, such shift is can be explained as the Fermi level pining and consequently, changes of PDOS of Pc ligand in Fig. 2b are not so obvious except the varying of intensity, suggesting that the electronic structures of Pc ligand are less affected by the adsorption.
Moreover, changes of the PDOS induced by adsorption have also been distinguished from the substrate Bi atom, as depicted in Fig. 2c. A sharp surface state from the clean bismuth substrate can be easily identified around the Fermi level, which is attributed to the Bi 6p orbitals and it gets quenched after adsorption with more features showing up below the Fermi level.47 The attenuation of bismuth surface state and the appearance of rich structures below EF in the PDOS of Bi 6p orbitals after adsorption can therefore be proposed as a result of interfacial charge transfer and molecule–substrate interaction, whereas Bi atoms are supposed to donate electrons/charge to the adsorbed CoPc molecule. As a result, it can be drawn that electronic structures at the CoPc/Bi(111) interface strongly depend on coupling between the unoccupied LUMO of CoPc and the bismuth 6p orbital.
Furthermore, the projected density of states of Co atom from CoPc/Bi(111) interface on corresponding Co-3d orbitals are also displayed in Fig. 3, to unveil the molecule–substrate interaction and interfacial electronic structures in detail. Schematic sketch of the cobalt 3d orbital splitting to the D4h tetragonal distortion in the CoPc molecule is also shown inset for easy illustration, and are classified by the magnetic quantum number m. Since the dx2−y2 orbital is located far away above the Fermi level, its ability to attract electrons from substrate Bi atoms and the corresponding contribution towards the interfacial charge transfer can be ignored in principle.17 Similarly, previous STM experiments for 3d-metal Pc molecules also demonstrated that dx2−y2 orbitals couldn't be directly imaged due to its faintest coupling with metal tip states.20,24,31 Therefore, one can expect that, only the dz2 orbital, which dominates the PDOS in the vicinity of the Fermi level can contribute to the interfacial charge transfer and the molecule–substrate interaction. In fact, as discussed previously, the LUMO of CoPc is mainly contributed by the central Co atom as judged from its electron cloud distribution. Therefore, the LOMO contains a major Co dz2 contribution, whereas the HOMO is dxz/dyz dominated.
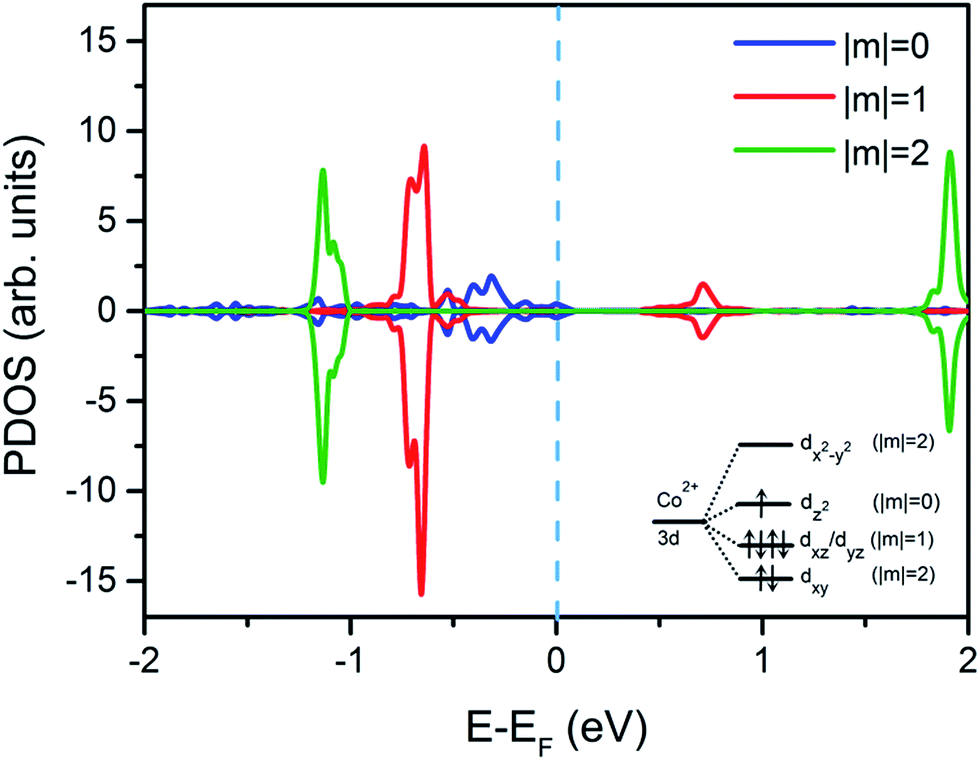 | ||
| Fig. 3 Calculated projected density of states of the Co 3d orbitals from the CoPc molecule adsorbed on a Bi(111) surface. Blue, red, and green lines represent its dz2 (|m| = 0), dxz/dyz (|m| = 1) and dxy/dx2−y2 (|m| = 2) orbitals, respectively, where m is the magnetic quantum number. The Fermi level is marked by blue dashed line as a guide to eye. | ||
Nevertheless, as seen in Fig. 3, behaviors of these orbitals are differential. The dz2 orbital becomes non-polarized while the almost-degenerate orbitals dxz and dyz are still polarized, to some extent. A corollary to such abnormal polarization properties, which are obviously different from the observation of CoPc adsorbed on other metallic surfaces,15,20,24 may be ascribed to the weak interfacial interaction. At this stage, it can be concluded that the dz2 orbital has the strongest coupling with the bismuth surface state along z-direction, since it is the most proximate frontier orbital around the Fermi level. Consequently, the partially empty dz2 orbital preferentially accepts charge/electron transferred from the Bi 6p orbital, resulting in the original density of Co 3dz2 states quenched. Given rise to the asymmetric density of states for dxz/dyz orbitals below the 3dz2 orbital as shown in Fig. 3, local magnetic moments is still retained in the adsorbed CoPc molecule. Different to the adsorption on other noble metals with paramagnetic surfaces such as Ag or Cu, the magnetization of the Co atom in CoPc on Bi(111) is not completely destroyed by the influence of the bismuth diamagnetic surface,15,16 mostly due to the fact that the substrate 4s/5s electrons from Cu or Ag surface can magnetically screen the spin of Co atom of CoPc, while the bismuth 6p electrons cannot.15
2.3. Interfacial charge transfer
In general, charge transfer is an important aspect of interfacial electric structures, which provides a beneficial approach to reflect the molecule–substrate interaction. To understand the role of Co atom in the interfacial interaction and the mechanism of charge transfer between CoPc molecule and Bi(111) surface, the charge density difference is then calculated using the following definition:| Δρ = ρ[Bi(111)/CoPc] − ρ[Bi(111)] − ρ[CoPc] |
 | ||
| Fig. 4 (a) Visualization of charge density difference upon adsorption. Color scale range is 5 × 10−3e/a03 with a0 the Bohr radius. (b) A schematic drawing of the charge transfer for the top-site configuration of the CoPc/Bi(111) system. Black atoms represent the most peripheral carbon atoms (CB) in the benzene ring of CoPc, while red (blue) arrow and positive (negative) value represent the reception (lose) of electron for sectional atoms (CB, Co and its underneath Bi atom), respectively. | ||
In addition, Bader charge analysis has also been performed for the free CoPc molecule and adsorbed CoPc on the Bi(111) surface utilizing a program developed by Henkelman, Arnaldsson, and Jónsson.49 As seen in Fig. 4b, the Co atom obtained 0.21 |e| net charge in total, while the underneath Bi atom donate about 0.14 |e| and the neighboring Bi atoms also contribute certain electron donation, suggesting the reception of electron to Co atom is mainly contributed by the donation of electron from the underneath and neighboring Bi atoms. Moreover, CB atom only received about 0.01 |e|, which can be almost neglected compared with that for the Co atom.
Nevertheless, the good accordance between qualitative description of charge-density difference and quantitative Bader charge analysis makes it convinced to draw such a conclusion that, the mechanism of charge transfer at interface is chiefly mediated by the central metal Co atom, segmentally, combined with the vdW dispersion interaction between π-conjugated macrocyclic ligand and Bi substrate.
2.4. X-ray photoelectron spectroscopy
So far, theoretical electronic structures of the CoPc/Bi(111) system through DFT calculation has been acquired. In order to verify the our calculation results, the Co 2p core-level photoemission spectra was also measured to get deeper insight of the electronic structures for CoPc/Bi(111) system. As shown in Fig. 5, the evolution of the Co 2p3/2 core-level at the CoPc/Bi(111) interface is depicted as a function of annealing temperature. For the convenient comparison, the Co 2p3/2 XP spectrum for a thick CoPc film adsorbed on the Bi(111) surface was also recorded and present at the bottom of Fig. 5. According to previous reports, the major component located at 780.6 eV should be regarded as the characteristic of the Co(II) oxidation state,50,51 which arises from the original CoPc in gas phase.52 Interestingly, another intense peak is visibly recognized at the binding energy of 778.5 eV, which shall be attributed to the cobalt metal state (Co(0)).50,51 The emergence of Co metal state upon adsorption on Bi(111) herein can only be assigned to the interfacial charge transfer and the related molecule–substrate interaction, which make the original partial-empty Co 3d state (for example, the dz2 orbital) filled and consequently, a new chemical state located at lower binding energy is resolved.34 Moreover, an additional component at 782.6 eV is also resolved for both thin and thick CoPc film, which might be assigned to the shake-up peak associated with Co(II) oxidation state.51 At the initial annealing stage up to 373 K, no visible change of the spectrum is observed compared to that at RT, indicating that the Co 2p3/2 spectrum annealed to 373 K was still mainly dominated by the Co(II) component. With the annealing temperature continually rises to 573 K, however, the Co(II) component undergo significant attenuation in intensity as well as its shake-up satellite. On the contrary, the Co(0) component gradually becomes predominant. It can also be now drawn that interfacial charge transfer and the molecule–substrate interaction gets greatly enhanced by thermal annealing, which enables more free Bi adatoms available from substrate and therefore promote the charge transfer to cobalt atoms. As generally reported, thermal annealing easily induce interfacial charge rearrangement and the self-assembly of organic molecules on metal substrates.32,46 In a word, the change of chemical state for the central cobalt ion from Co(II) to Co(0) depicted in the XPS spectra is a pronounced tendency that, the central Co atom accepted electrons donated from the Bi substrate, owing to its partially empty 3d orbital. Similar phenomenon has also been reported for the analogous systems of TMPcs on Ag and Au surfaces.33,50 Besides, the vanishing of shake-up satellite at 573 K is again another evident proof of the above-mentioned conclusion. In the case of multilayer coverage of CoPc at RT, the Co(0) component is fairly feeble, owing to the fact that the Bi surface primarily interacted with the Co atom only from the first-layer CoPc, and hardly have interaction with upper CoPc layers, where the cobalt ion is still maintained as in gas-phase with the pristine Co(II) state. In general, the XPS measurement delivers same message with the previous DFT calculations.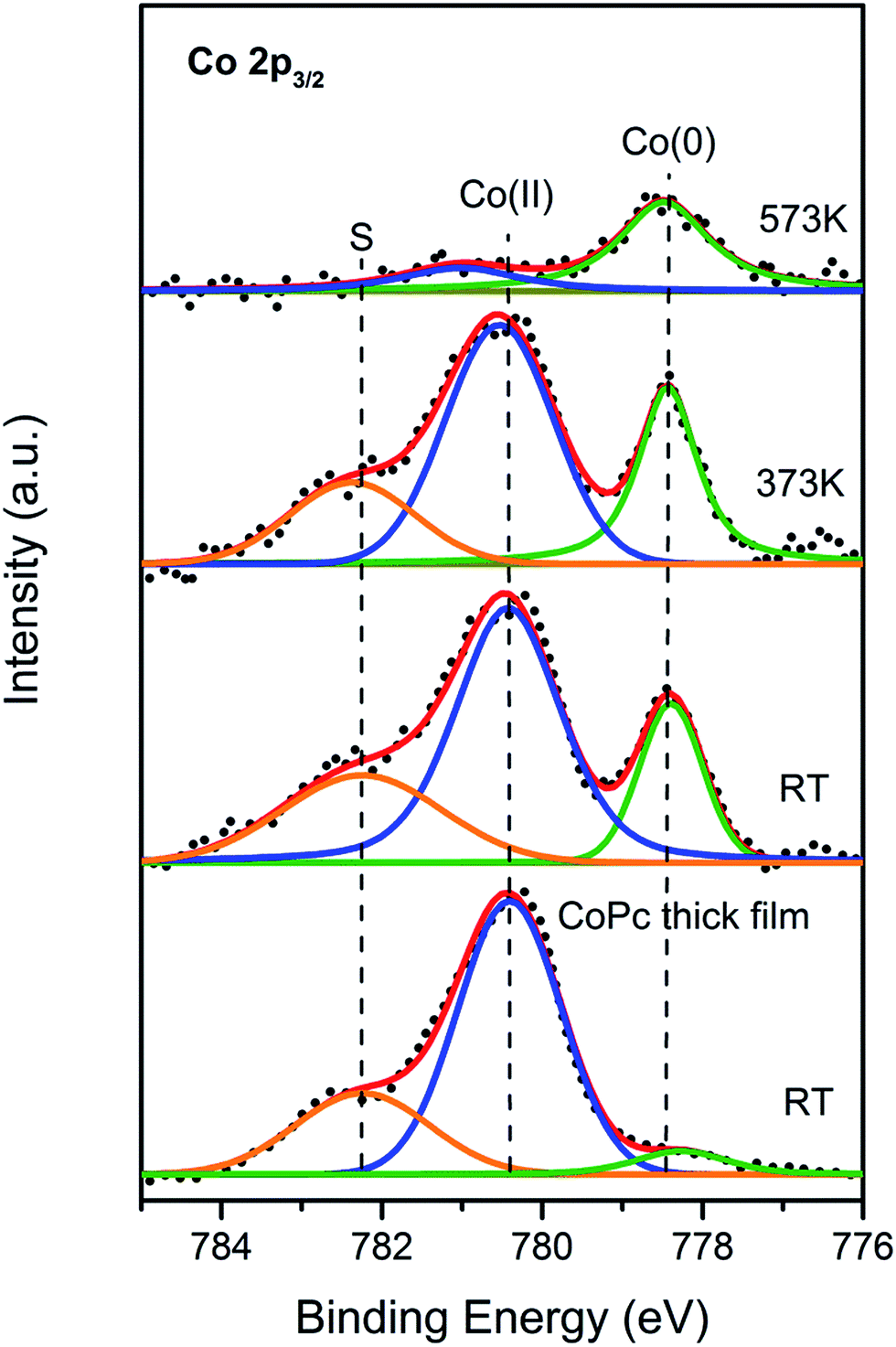 | ||
| Fig. 5 XP spectrum of Co 2p3/2 for thin CoPc film deposited on Bi(111) surface kept at RT followed by thermal annealing. Black dots: raw data; blue line: fitted Co(II) oxidation state; green line: fitted component with Co(0) state; orange line: the shake-up component associated to the Co(II) state. As a reference, a thick CoPc film was also prepared and recorded as shown in the bottom. | ||
3 Materials and methods
3.1. DFT calculations
Ab initio calculations were performed based on density functional theory (DFT)53,54 by employing the VASP55,56 code with vdW-DF2 functional. The projector augmented wave (PAW) approach was used.57 By referring previous work on analogic systems,17,23,37 the kinetic energy cutoff for the plane-wave basis was set to 400 eV. The convergence criterion for electronic self-consistent relaxation is set to 10−5 eV. A hexagonal slab was used to simulate the unit cell of the adsorption model, with a dimension of 22.74 Å × 22.74 Å × 20.96 Å including four layers of Bi(111) and 15 Å vacuum. As revealed from previous STM investigations of CoPc grown on Bi(111) surface,35 the first layer is flat-lying absorbed on substrate surface. Thus, the isolated CoPc molecule was placed in the x-y plane with initial molecule–substrate distance of 3.5 Å. Moreover, four high-symmetry absorption configurations were considered by placing central Co atom at top, hcp, fcc and hollow sites as shown in Fig. 1b. During geometry optimization, the top layer of the substrate and the molecule were fully relaxed with a force convergence criterion of 0.05 eV Å−1. Only a single gamma point was used in all calculations for CoPc/Bi(111) because of the large number of atoms in the slab. In addition, the free MPc molecule was optimized within the same framework and its total energy was calculated with the same energy cutoff and convergence criteria as MPc/Bi(111).3.2. Photoemission spectroscopy experiments
Photoelectron spectroscopy experiments were done under ultra-high vacuum with the base pressure better than 2 × 10−10 mbar at room temperature (RT). XPS measurements were carried out in the lab using a monochromatic Al-Kα radiation (hν = 1486.6 eV), while the photon energy was calibrated by comparing the binding energy of Ta 4f peak and the metal Fermi level (EF) from the sample holder. The Bi(111) substrate (Mateck) was cleaned prior to organic film deposition by cycles of Ar+ sputtering at 800 eV and post annealing to about 400 K. The CoPc molecule (Sigma-Aldrich) was thermally evaporated onto the Bi(111) surface from a temperature controlled evaporation cell after overnight outgassing, while the film thickness was estimated from a quartz microbalance calibrated by the intensity attenuation of XPS peak from Bi substrate.4 Conclusion
We have presented a comprehensive study of the CoPc molecule adsorbed on a semimetal Bi(111) surface by means of ab initio method and photoemission spectroscopy. DFT calculations manifest that CoPc is quite sensitive to the adsorption site on the relatively inert Bi(111) surface and favourites to adsorb on the top site due to the maximized orbital coupling between the partial empty Co 3d state from CoPc and the surface state (Bi 6p) from substrate. A comparison of PDOS for CoPc in gas-phase and the adsorption manner demonstrates that the change of interfacial electronic structures is mostly dominated by the central Co atom, particularly by its LUMO (namely the dz2 state) while the Pc ligand seems to be less affected by the adsorption. Interestingly, our study reveals an incomplete transformation process of depolarization in CoPc after adsorption on the diamagnetic Bi(111) surface mainly contributed by the central Co atom. In the end, the schematic profile of charge density difference and quantitative Bader charge analysis points out that the mechanism of interfacial charge transfer is chiefly mediated by the Co atom in CoPc combining with the weak vdW dispersion between the π-conjugated macrocyclic ligand and substrate, as further confirmed by XPS study of the Co 2p3/2 core level. With a detailed investigation and convincing interpretation of the magnetic-molecule/semimetal complex, our report may be beneficial to the dedicated exploit of semimetal–organic devices based on such heterostructures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by National Key Research and Development Program of China (SQ2016YFJC030064), National Natural Science Foundation of China (91545101), The Hundred Talents Program of Chinese Academy of Sciences, and Shanghai Pujiang Program.Notes and references
- L. Bartels, Nat. Chem., 2010, 2, 87–95 CrossRef CAS PubMed.
- A. Kühnle, Curr. Opin. Colloid Interface Sci., 2009, 14, 157–168 CrossRef.
- T.-T. Zhang, C.-J. Wang, K. Sun, H.-K. Yuan and J.-Z. Wang, Appl. Surf. Sci., 2014, 317, 1047–1051 CrossRef CAS.
- M. Casarin, M. Di Marino, D. Forrer, M. Sambi, F. Sedona, E. Tondello, A. Vittadini, V. Barone and M. Pavone, J. Phys. Chem. C, 2010, 114, 2144–2153 CAS.
- S. R. Wagner, B. Huang, C. Park, J. Feng, M. Yoon and P. Zhang, Phys. Rev. Lett., 2015, 115, 096101 CrossRef PubMed.
- P. Järvinen, S. K. Hämäläinen, M. Ijäs, A. Harju and P. Liljeroth, J. Phys. Chem. C, 2014, 118, 13320–13325 Search PubMed.
- K. Xiao, W. Deng, J. K. Keum, M. Yoon, I. V. Vlassiouk, K. W. Clark, A. P. Li, I. I. Kravchenko, G. Gu, E. A. Payzant, B. G. Sumpter, S. C. Smith, J. F. Browning and D. B. Geohegan, J. Am. Chem. Soc., 2013, 135, 3680–3687 CrossRef CAS PubMed.
- T. Bathon, P. Sessi, K. A. Kokh, O. E. Tereshchenko and M. Bode, Nano Lett., 2015, 15, 2442–2447 CrossRef CAS PubMed.
- P.-C. Kao, S.-Y. Chu, Z.-X. You, S. J. Liou and C.-A. Chuang, Thin Solid Films, 2006, 498, 249–253 CrossRef CAS.
- R. W. I. de Boer, A. F. Stassen, M. F. Craciun, C. L. Mulder, A. Molinari, S. Rogge and A. F. Morpurgo, Appl. Phys. Lett., 2005, 86, 262109 CrossRef.
- M. Schwarze, W. Tress, B. Beyer, F. Gao, R. Scholz, C. Poelking, K. Ortstein, A. A. Gunther, D. Kasemann, D. Andrienko and K. Leo, Science, 2016, 352, 1446–1449 CrossRef CAS PubMed.
- C. Stadler, S. Hansen, I. Kröger, C. Kumpf and E. Umbach, Nat. Phys., 2009, 5, 153–158 CrossRef CAS.
- E. Annese, J. Fujii, I. Vobornik, G. Panaccione and G. Rossi, Phys. Rev. B, 2011, 84, 174443 CrossRef.
- E. Salomon, P. Amsalem, N. Marom, M. Vondracek, L. Kronik, N. Koch and T. Angot, Phys. Rev. B, 2013, 87, 075407 CrossRef.
- X. Chen and M. Alouani, Phys. Rev. B, 2010, 82, 094443 CrossRef.
- J. D. Baran and J. A. Larsson, J. Phys. Chem. C, 2013, 117, 23887–23898 CAS.
- Y. Y. Zhang, S. X. Du and H. J. Gao, Phys. Rev. B, 2011, 84, 125446 CrossRef.
- Y. Wang, X. Zheng and J. Yang, J. Chem. Phys., 2016, 145, 154301 CrossRef PubMed.
- K. J. Franke, G. Schulze and J. I. Pascual, Science, 2011, 332, 940–944 CrossRef CAS PubMed.
- A. D. Zhao, Q. X. Li, L. Chen, H. J. Xiang, W. H. Wang, S. Pan, B. Wang, X. D. Xiao, J. L. Yang, J. G. Hou and Q. S. Zhu, Science, 2005, 309, 1542–1544 CrossRef CAS PubMed.
- R. Cuadrado, J. I. Cerda, Y. Wang, G. Xin, R. Berndt and H. Tang, J. Chem. Phys., 2010, 133, 154701 CrossRef PubMed.
- S. Li, J. Hao, F. Li, Z. Niu, Z. Hu and L. Zhang, J. Phys. Chem. C, 2014, 118, 27843–27849 CAS.
- A. Mugarza, R. Robles, C. Krull, R. Korytár, N. Lorente and P. Gambardella, Phys. Rev. B, 2012, 85, 155437 CrossRef.
- J. D. Baran, J. A. Larsson, R. A. J. Woolley, Y. Cong, P. J. Moriarty, A. A. Cafolla, K. Schulte and V. R. Dhanak, Phys. Rev. B, 2010, 81, 075413 CrossRef.
- U. G. Perera, H. J. Kulik, V. Iancu, L. G. Dias da Silva, S. E. Ulloa, N. Marzari and S. W. Hla, Phys. Rev. Lett., 2010, 105, 106601 CrossRef CAS PubMed.
- Z. P. Hu, B. Li, A. D. Zhao, J. L. Yang and J. G. Hou, J. Phys. Chem. C, 2008, 112, 13650–13655 CAS.
- A. Scarfato, S.-H. Chang, S. Kuck, J. Brede, G. Hoffmann and R. Wiesendanger, Surf. Sci., 2008, 602, 677–683 CrossRef CAS.
- S. R. Wagner and P. Zhang, J. Phys. Chem. C, 2014, 118, 2194–2201 CAS.
- F. Song, J. W. Wells, K. Handrup, Z. S. Li, S. N. Bao, K. Schulte, M. Ahola-Tuomi, L. C. Mayor, J. C. Swarbrick, E. W. Perkins, L. Gammelgaard and P. Hofmann, Nat. Nanotechnol., 2009, 4, 373–376 CrossRef CAS PubMed.
- Y. L. Huang, E. Wruss, D. A. Egger, S. Kera, N. Ueno, W. A. Saidi, T. Bucko, A. T. Wee and E. Zojer, Molecules, 2014, 19, 2969–2992 CrossRef PubMed.
- A. Mugarza, C. Krull, R. Robles, S. Stepanow, G. Ceballos and P. Gambardella, Nat. Commun., 2011, 2, 490 CrossRef PubMed.
- G. Antczak, W. Kaminski, A. Sabik, C. Zaum and K. Morgenstern, J. Am. Chem. Soc., 2015, 137, 14920–14929 CrossRef CAS PubMed.
- L. Giovanelli, P. Amsalem, T. Angot, L. Petaccia, S. Gorovikov, L. Porte, A. Goldoni and J. M. Themlin, Phys. Rev. B, 2010, 82, 125431 CrossRef.
- K. Shen, B. Narsu, G. Ji, H. Sun, J. Hu, Z. Liang, X. Gao, H. Li, Z. Li, B. Song, Z. Jiang, H. Huang, J. W. Wells and F. Song, RSC Adv., 2017, 7, 13827–13835 RSC.
- M.-L. Tao, Y.-B. Tu, K. Sun, Y. Zhang, X. Zhang, Z.-B. Li, S.-J. Hao, H.-F. Xiao, J. Ye and J.-Z. Wang, J. Phys. D: Appl. Phys., 2016, 49, 015307 CrossRef.
- Z. F. Wang, L. Chen and F. Liu, Nano Lett., 2014, 14, 2879–2883 CrossRef CAS PubMed; F. Song, J. W. Wells, Z. Jiang, M. Saxegaard and E. Wahlström, ACS Appl. Mater. Interfaces, 2015, 7, 8525–8532 Search PubMed.
- Y. Lu, W. Xu, M. Zeng, G. Yao, L. Shen, M. Yang, Z. Luo, F. Pan, K. Wu, T. Das, P. He, J. Jiang, J. Martin, Y. P. Feng, H. Lin and X. S. Wang, Nano Lett., 2015, 15, 80–87 CrossRef CAS PubMed.
- H. Sun, Z. Liang, K. Shen, J. Hu, G. Ji, Z. Li, H. Li, Z. Zhu, J. Li, X. Gao, H. Han, Z. Jiang and F. Song, Surf. Sci., 2017, 661, 34–41 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- R. G. A. Veiga, R. H. Miwa and A. B. McLean, Phys. Rev. B, 2016, 93, 115401 CrossRef.
- M. Dion, H. Rydberg, E. Schroder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- K. Lee, É. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B, 2010, 82, 081101 CrossRef.
- K. Berland, V. R. Cooper, K. Lee, E. Schroder, T. Thonhauser, P. Hyldgaard and B. I. Lundqvist, Rep. Prog. Phys., 2015, 78, 066501 CrossRef PubMed.
- W. Liu, V. G. Ruiz, G.-X. Zhang, B. Santra, X. Ren, M. Scheffler and A. Tkatchenko, New J. Phys., 2013, 15, 053046 CrossRef.
- T. C. Tseng, C. Urban, Y. Wang, R. Otero, S. L. Tait, M. Alcami, D. Ecija, M. Trelka, J. M. Gallego, N. Lin, M. Konuma, U. Starke, A. Nefedov, A. Langner, C. Woll, M. A. Herranz, F. Martin, N. Martin, K. Kern and R. Miranda, Nat. Chem., 2010, 2, 374–379 CrossRef CAS PubMed.
- M. Bianchi, F. Song, S. Cooil, Å. F. Monsen, E. Wahlström, J. A. Miwa, E. D. L. Rienks, D. A. Evans, A. Strozecka, J. I. Pascual, M. Leandersson, T. Balasubramanian, P. Hofmann and J. W. Wells, Phys. Rev. B, 2015, 91, 165307 CrossRef.
- B. Chilukuri, U. Mazur and K. W. Hipps, Phys. Chem. Chem. Phys., 2014, 16, 14096–14107 RSC.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- F. Petraki, H. Peisert, I. Biswas and T. Chasse, J. Phys. Chem. C, 2010, 114, 17638–17643 CAS.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H. P. Steinruck, J. Phys. Chem. C, 2007, 111, 3090–3098 CAS.
- J. Uihlein, M. Polek, M. Glaser, H. Adler, R. Ovsyannikov, M. Bauer, M. Ivanovic, A. B. Preobrajenski, A. V. Generalov, T. Chassé and H. Peisert, J. Phys. Chem. C, 2015, 119, 15240–15247 CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–561 CrossRef CAS.
- H. M. Petrilli, P. E. Blochl, P. Blaha and K. Schwarz, Phys. Rev. B, 1998, 57, 14690–14697 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |