
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualization of filler network in silicone rubber with confocal laser-scanning microscopy
Min Liu,
Ming Kang *,
Yongren Mou,
Kexu Chen and
Rong Sun
*,
Yongren Mou,
Kexu Chen and
Rong Sun
College of Materials Science and Engineering, Southwest University of Science and Technology, Mianyang 621010, China. E-mail: mkang311@126.com; Tel: +86 13547133979
First published on 21st November 2017
Abstract
Most of the present studies of filler network structure basically stay in some phenomenological descriptions and classical electron microscopes are restricted by conventional 2-D imaging. This paper completed the visualization of filler networks in silica-reinforced silicone rubber (MVQ). Using a special network visualization technique based on fluorescent markers embedded in silica and Confocal Laser-Scanning Microscopy with an optical lateral resolution of about 200 nm, insights into the precise 3-D morphology and detailed micro-structure of filler networks were gained. A kind of sensitized-silica phosphor was prepared, with the excitation wavelength of 405 nm, which is consistent with the laser wavelength of Confocal Laser-Scanning Microscopy. Combining Image J software and tensile measurements, shapes, network structures, size distributions of aggregates in the silicone rubber matrix with silica loading from 10 to 60 phr were directly observed and the loading threshold for the formation of filler network in this experiment was obtained.
1. Introduction
• Rubber is an important polymeric material because of its unique viscoelastic properties. However, the reinforcement of this soft matrix is essential to realize the required properties for practical applications. Therefore, fillers are extensively used in the rubber industry, not only to reinforce the rubber matrix but also to improve the processing performance of rubber and, in some cases, to reduce the price of the final material.1 As confirmed, filler particles are not evenly distributed in the rubber matrix, but are distributed according to the different methods of preparation to form the so-called filler network structures, which have been demonstrated to play a key role in enhancing toughness and strength of rubber materials.2–4 Strain-induced deformation, destruction, and reconstruction of filler networks were directly observed under cyclic loading for the first time by synchrotron radiation X-ray nano-computed tomography technique and the reinforcing and toughening effects of filler networks were quantitatively assigned to three mechanisms, namely elastic deformation, destruction, and friction of filler network,5 so the understanding of filler network structure is of the utmost importance for the performance of rubber.• However, due to the complexity of the micro-structure of filled-rubber and the restriction of research methods and technology, it is very difficult to observe the formation of the filler network structure and its contribution to the mechanical behavior of rubber directly. Although the synchrotron radiation X-ray nano-computed tomography technique has a great advantage in the study of filler network structure, this technology is based on large-scale scientific devices with limited resources, which limits its widely use in filler network research. Thus, the present studies of filler network structure basically stay in some phenomenological description and lack relatively ideal technology for detecting the filler network structure. On the one hand, a number of theories and models about Payne effect have been proposed to interpret the importance of the filler aggregates and networks, such as the Kraus model, the links–nodes–blobs (LNB) model,6,7 the chain slippage models, the bound rubber/entanglement network model,8 the localized glassy layer model9 and so on. The result that the Payne effect of CB gel (CBG) is frequency-independent and does not exhibit hysteresis during loading–unloading cycle indicates that the CBG network rather than the entanglement network dominates the instant recovery of the highly filled compounds,10 supporting the deduction of Satoh et al. about the quick healing behavior of filler network.11
• On the other hand, in general for the purpose of detailed microscopic characterization and fine-scale identification, other conventional microscopic techniques or additional high-resolution imaging tools such as scanning electron microscopy (SEM), environmental scanning electron microscope (ESEM), transmission electron microscopy (TEM), or atomic force microscopy (AFM) have been applied to observe the filler network structure in rubber. Atitaya Tohsan used TEM and AFM to observe the morphological features of peroxide cross-linked in situ silica-filled nanocomposites.12 It can be seen that spherical aggregates with an average size of 500 nm were detected and a filler network structure was also generated. Yong Lin13 and Jun Yang14 also used TEM to describe filler network structures generated from silica/reduced graphene oxide and silica, respectively. Jingyi Wang used SEM to observe the fracture surfaces of NR vulcanizates with different fillers.15 It can be seen that pristine filler agglomerated highly with large-size agglomerates in NR matrix. However, the agglomerates dealt with Si69 were smaller and dispersed well in NR matrix. However, SEM can only detect the morphology and distribution of filler in the rubber surface but can not observe the spatial distribution of filler inside the rubber. For AFM, although three-dimensional images of fillers in the surface can be obtained through the different value of the force between micro-probe and rubber matrix, micro-probe and fillers, it requires extreme level of roughness on the surface of the sample for the reason that its maximum scanning height is 2 μm. Besides, the soft nature of the silicone rubber also has a great influence on the accuracy of scan probe. For TEM, as a result of weak electronic penetration ability, the sample is required to be thin enough, which results in that only partially spatial distribution information of filler inside rubber can be obtained. Besides, because of the co-existence of rubber phase and the various kinds of filler aggregation, a pure filler phase that is covered by rubber matrix could not be achieved by SEM, TEM or AFM, which results in that it is difficult to define a so-called “percolation threshold” for forming model filler network in the rubber matrix.
Here, fluorescent markers and Confocal Laser-Scanning Microscopy (CLSM) with optical lateral resolution about 200 nm are employed to study the filler network structure of silica in a large volume of silicone rubber matrix. A kind of sensitized, spherical, and mono-disperse silica phosphor (SiO2@SiO2:Eu(DBM)3phen) was prepared by the sol–gel method, implementing the fluorescent markers of silica. What's more, the particle size and luminescence properties of this kind of filler are controllable, by adjusting the reaction conditions, therefore it is feasible to prepare the filler that is suitable for fluorescent markers. For CLSM, it is a general technique to draw a series of sliced images of a specimen, which can be reconstructed as 3-D images. The non-invasive and non-destructive laser-raster technique is applied to characterize fluorescent silica fillers and the in depth examinations along Z-axis in XY, XZ, or YZ scanning modes at increments of 150 nm are capable to reconstruct 3-D internal structure of them.16,17 It demonstrated that CLSM is more advantageous in (1) detecting the micro-structure of fluorescent silica fillers at micron to sub-micron scale resolution, (2) obtaining pure filler phase in the composites, (3) reconstructing 3-D morphology of fluorescent silica fillers, (4) observing the morphology of silica filler network structure and the aggregates size through fluorescence distribution, (5) in situ observation of samples without serious damages.18–23
2. Experimental
2.1 Materials
Tetraethyl orthosilicate (TEOS), acetone, Cetyl-Trimethyl Ammonium Bromide (CTAB), 1,10-phenanthroline (phen) and dicumyl peroxide (DCP) were obtained from Chengdu Kelong Chemical Reagent Industry (China). Eu(NO3)3·6H2O (99.99%) and dibenzoyl methane (DBM) were purchased from Aladdin Reagent Co. The silicone rubber (type: 110-2, Mn = 6.5 × 106 g mol−1, 0.23% mol−1 vinyl content, MVQ) used in the study was purchased from Zhejiang Xin'an Chemical Group Co., Ltd. (China). The other materials are all commercially available.2.2 Preparation of silica phosphors (SiO2@SiO2:Eu(DBM)3phen)
The rare earth (RE) complexes Eu(DBM)3phen were synthesized as report.24 The mono-disperse SiO2@SiO2:Eu(DBM)3phen phosphors were synthesized in a seeded growth way, which our group had prepared successfully as following steps.25 The formation process for the SiO2@SiO2:Eu(DBM)3phen phosphors was shown in Fig. 1. In the first step, a certain amount of TEOS was rapidly dropped into a mixture that consisted of anhydrous ethanol, distilled water, and ammonia, with the volume ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1, stirring for 30 min. The size of seeds were controllable by adjusting the ratio of ammonia. In the second step, anhydrous ethanol, distilled water and ammonia with the same volume ratio were added rapidly into the above mixture, stirring for 15 min at room temperature. Then CTAB and Eu(DBM)3phen acetone solution (the molar composition of Eu(DBM)3phen:TEOS(secondary addition) was 5:100) were added slowly(using a micro-injector) into the mixture in the sonicator. Finally, a certain amount of TEOS was added slowly again and reacted for 5 h. The samples were separated by centrifugation at 8000 rpm for 5 min, washed by acetone and anhydrous ethanol, and then dried at 60 °C for 24 h in air. In order to remove residuary CTAB, the as-dried precursors were calcined at 350 °C for 3 h in nitrogen atmosphere. The heating rate was 20 °C min−1.
:5:1, stirring for 30 min. The size of seeds were controllable by adjusting the ratio of ammonia. In the second step, anhydrous ethanol, distilled water and ammonia with the same volume ratio were added rapidly into the above mixture, stirring for 15 min at room temperature. Then CTAB and Eu(DBM)3phen acetone solution (the molar composition of Eu(DBM)3phen:TEOS(secondary addition) was 5:100) were added slowly(using a micro-injector) into the mixture in the sonicator. Finally, a certain amount of TEOS was added slowly again and reacted for 5 h. The samples were separated by centrifugation at 8000 rpm for 5 min, washed by acetone and anhydrous ethanol, and then dried at 60 °C for 24 h in air. In order to remove residuary CTAB, the as-dried precursors were calcined at 350 °C for 3 h in nitrogen atmosphere. The heating rate was 20 °C min−1.
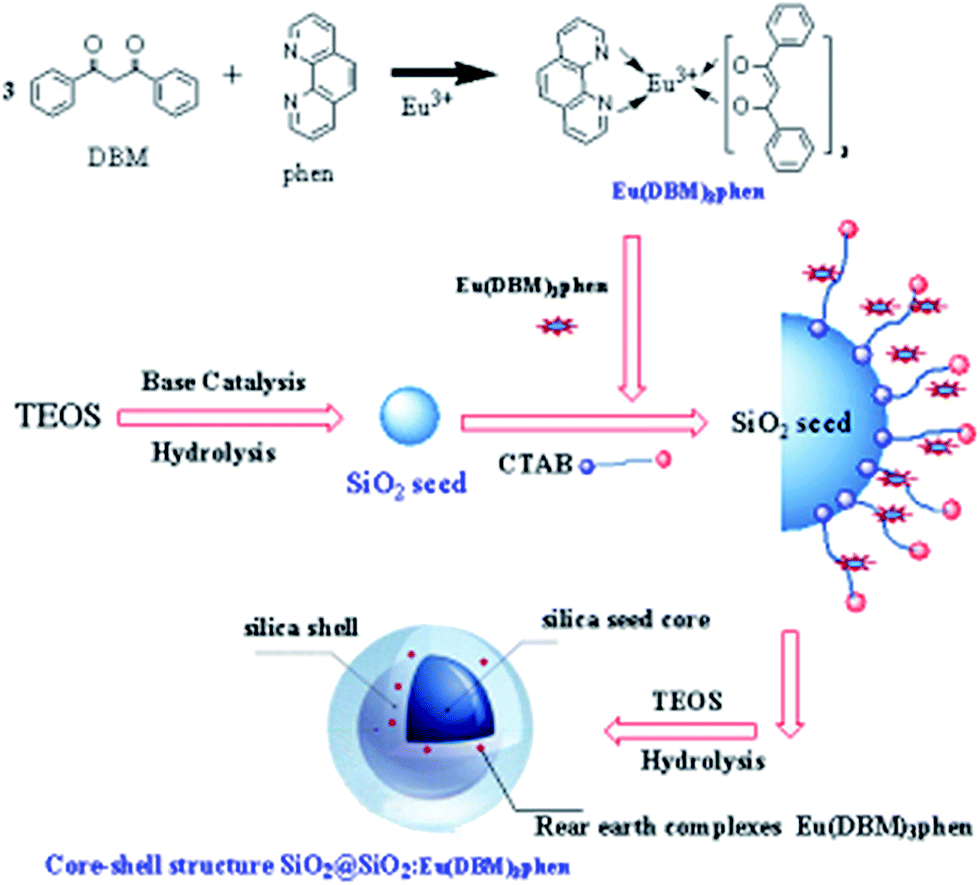 | ||
| Fig. 1 The formation mechanism of SiO2@SiO2:Eu(DBM)3phen phosphors. | ||
2.3 Preparation of silica phosphors filled MVQ
The compounds were prepared using an internal mixer at 105 °C and 90 rpm for 30 min (weight content of silica phosphors were 10 to 60 phr). As the structure control agent, a small amount of hydroxyl silicone oil was added in the process of mixing. After 1 week of adsorption at room temperature, the cross-linking agent DCP was added into the compounds at room temperature with 60 rpm for 15 min (weight content of DCP was 2.5 phr) and then were cured to a film by a compression molding machine at 160 °C and 10 MPa for 10 min to obtain the vulcanizates. The formulations and cure conditions for preparation of the rubber sample were shown in Table 1. The films were then cut into rectangular-shaped specimens with thickness of 50 μm, for CLSM imaging.The samples silica phosphors filled MVQ coded as MVQx, in which x denotes the silica phosphors loading in phr (parts per hundred rubber) of the original compounds.
2.4 Visualization of the filler network structure
Because of the limitation of focal depth of CLSM imaging system, samples were cut to thin films with thickness of 50 μm for CLSM imaging, operated with an Ar laser (405 nm). The principle of the CLSM was shown in Fig. 2: focusing of laser light on a narrow point inside the specimen, detection of only the in-focus light through a confocal pinhole and the light from out-of-focus structures was faded out.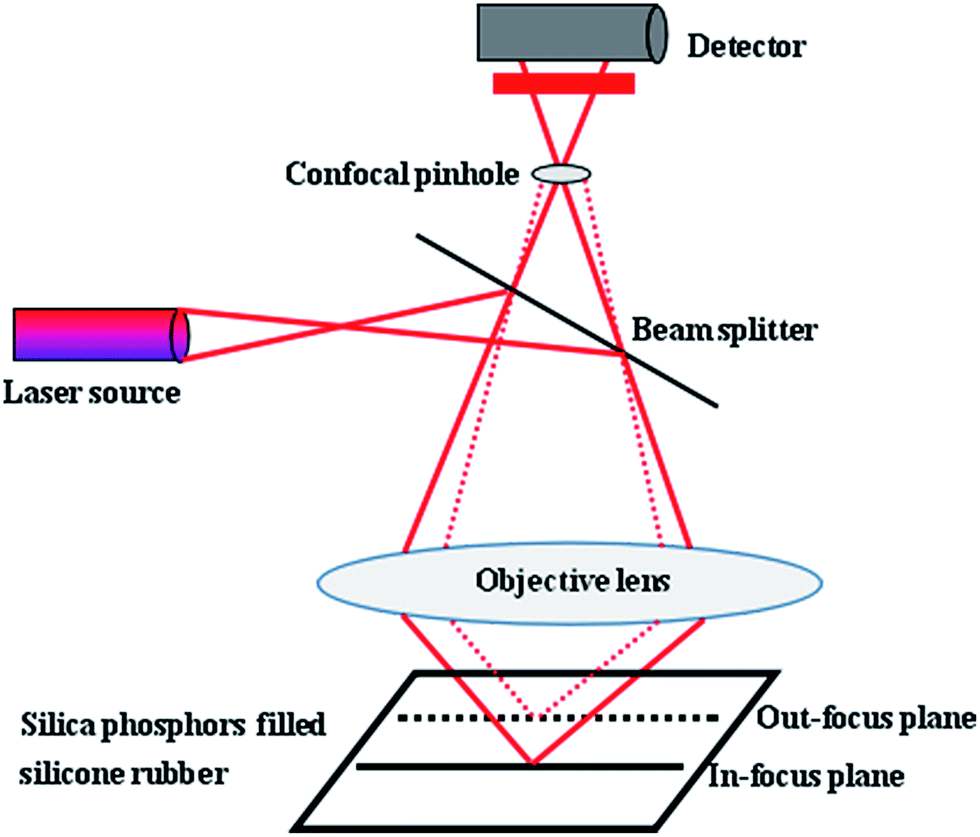 | ||
| Fig. 2 Principle of the CLSM. | ||
The resulting optical section contains only information from one focus plane. To ensure the fluorescence of the silica phosphors filled silicone rubber and to enable a high-quality analysis of the filler network structures, during image acquisition in XY, XZ, YZ, and XYZ modes, line average was 2 and images were captured as 2-D images in digital image resolution of up to 1024 × 1024 pixel. The confocal images were captured in jpg format. The 200 consecutive confocal images, obtained at increments of up to 0.15 μm, were stacked as optical sections in z-direction. The CLSM settings for the evaluation of the experiments were presented in Table 2. Finally, Image J software was used to complete 3-D reconstruction.
| Parameter [unit] | Laser wavelength [nm] | Magnification [—] | Line average [—] | Channel 1 type [nm] | Channel 2 type [nm] | Image size [μm2] | Pixel size [nm2] | Z-Step size [μm] | Nr of steps [—] | Format [—] |
|---|---|---|---|---|---|---|---|---|---|---|
| 405 | 20× | 2 | PMT (417–517) | PMT (588–800) | 581.25 × 581.25 | 568.18 × 568.18 | 0.15 | 200 | 1024 × 1024 |
2.5 Characterization
The surface morphology of the silica phosphors were analyzed by a field emission scanning electron microscope (FESEM) (TESCAN MAIA3 SEM) operating at 20 kV after sputtering treatment. Thermal gravimetric analysis and differential scanning calorimetry (TGA-DSC) data were recorded with a thermal analysis instrument (TGA/SDTA851e, METTLER TOLEDO, Switzerland) at a heating rate of 20 °C min−1 in an air flow of 100 mL min−1. The photo-luminescence excitation (PLE) and emission (PL) spectra of silica phosphors were measured at room temperature by a luminescence spectrophotometer (Model F-4500, Hitachi) with a 150 W xenon lamp as the excitation source. Mixed rubber was prepared by a laboratory size internal mixer (RC400P, HAAKE Co., Ltd, Germany). Cross-linked silicone rubber sheets were prepared in a platen press (P300E, DR COLLIN Co., Ltd, Germany). Confocal images and 3-D reconstructions were produced using confocal laser-scanning microscopy (Leica TCS Sp8) and Image J software, respectively. The tensile tests were performed at room temperature on RSA-G2 (TA Instruments) at a stretching rate of 5 mm min−1.3. Results and discussion
3.1 Characterization of silica phosphor and fluorescence properties of MVQ10-60
Fig. 3 showed the FESEM images and TGA curves of SiO2@SiO2:Eu(DBM)3phen phosphors. As shown in Fig. 3a, the obtained silica phosphors were spherical and mono-disperse that most diameters of the phosphors were 275 nm (as confirmed by size distribution histogram in Fig. 3b), ensuring a consistent size with commercial sol–gel silica.26 In order to demonstrate the thermal stability of SiO2@SiO2:Eu(DBM)3phen phosphors at vulcanizing temperature (160 °C), the thermogravimetric analysis (TGA) of pure SiO2, pure Eu(DBM)3phen and SiO2@SiO2:Eu(DBM)3phen particles were adopted, as shown in Fig. 3c. It showed that for Eu(DBM)3phen particle, it started to decompose at about 360 °C. For SiO2@SiO2:Eu(DBM)3phen phosphors, a small weight loss from 30 °C to 150 °C was observed, which was due to the loss of the adsorbed water molecules and a large weight loss from 250 °C to 600 °C attributed to the condensation among silanol groups (the same as pure SiO2) was observed and the decomposition of Eu(DBM)3phen, so its mass loss rate was 1.4% more than pure SiO2. Therefore, it could be concluded that whether pure Eu(DBM)3phen particles or SiO2@SiO2:Eu(DBM)3phen phosphors, they won't decompose under the curing temperature, which ensures the final fluorescent properties of SiO2@SiO2:Eu(DBM)3phen phosphors filled vulcanized rubber.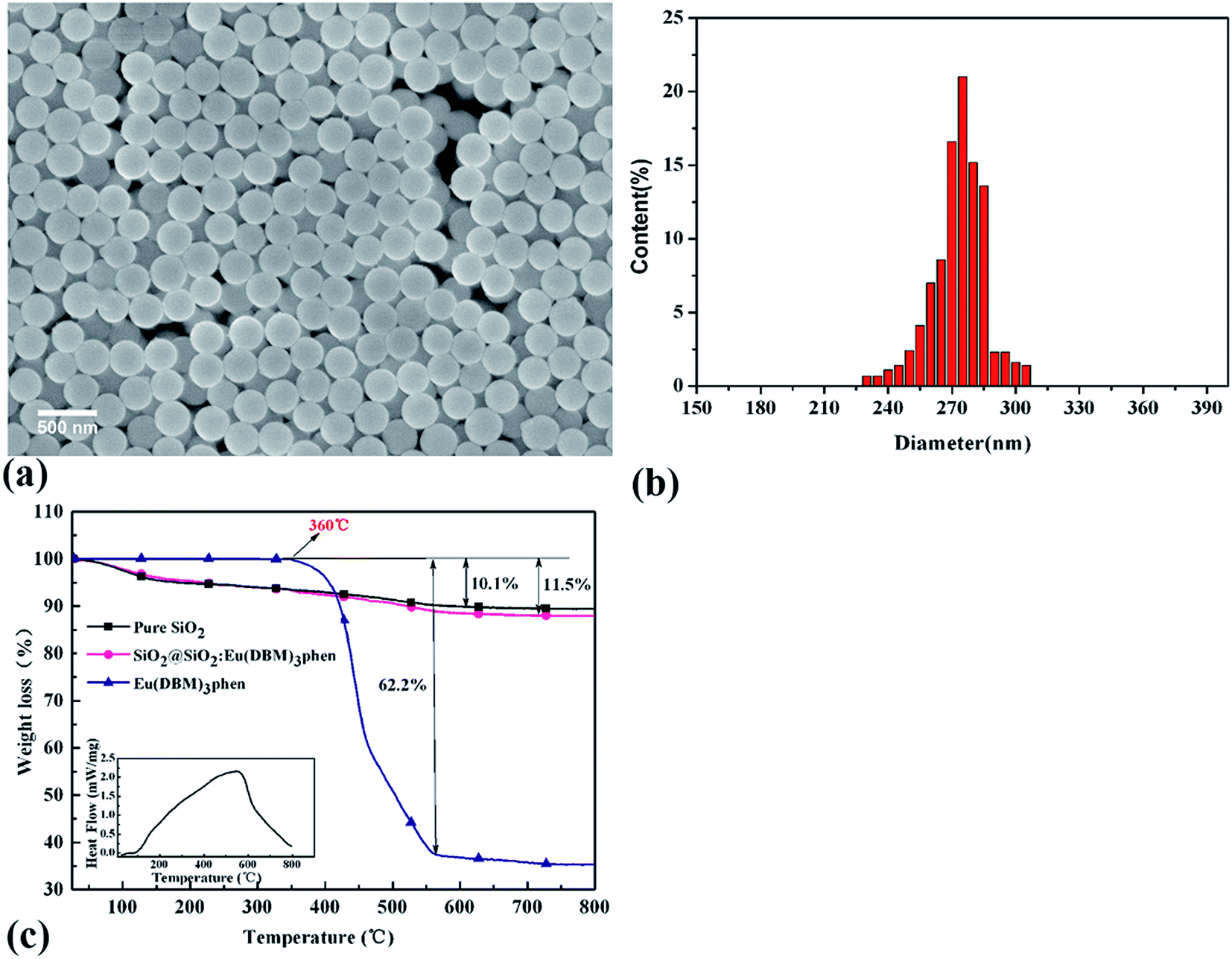 | ||
| Fig. 3 (a) FESEM images of SiO2@SiO2:Eu(DBM)3phen phosphors, (b) histograms of size distribution of SiO2@SiO2:Eu(DBM)3phen phosphors, and (c) TGA curves of pure SiO2, pure Eu(DBM)3phen and SiO2@SiO2:Eu(DBM)3phen phosphors. | ||
Since the wavelength of four independent solid-state lasers in CLSM we used are 405 nm, 488 nm, 552 nm and 638 nm, the control of peak position of emission spectra is necessary. The excitation and emission spectra of SiO2@SiO2:Eu(DBM)3phen phosphors were shown in Fig. 4a and b. The excitation spectrum was obtained by monitoring the emission of the 5D0–7F2 transition of the Eu3+ ions at 610 nm (Fig. 4a). The excitation spectrum of the SiO2@SiO2:Eu(DBM)3phen phosphors was composed of a broad band centered at 405 nm, which was consistent with the laser wavelength of CLSM. Upon excitation at 405 nm, the strongest red emission peaking at 610 nm arose (Fig. 4b), which was consistent with the testing results of CLSM. Fig. 4a1 and b1 present the excitation and emission spectra of MVQ10-60. They all showed excellent fluorescence performance, and no shift of characteristic peak position when compared with silica phosphors, indicating that the fluorescent marker of filler network has been completed.
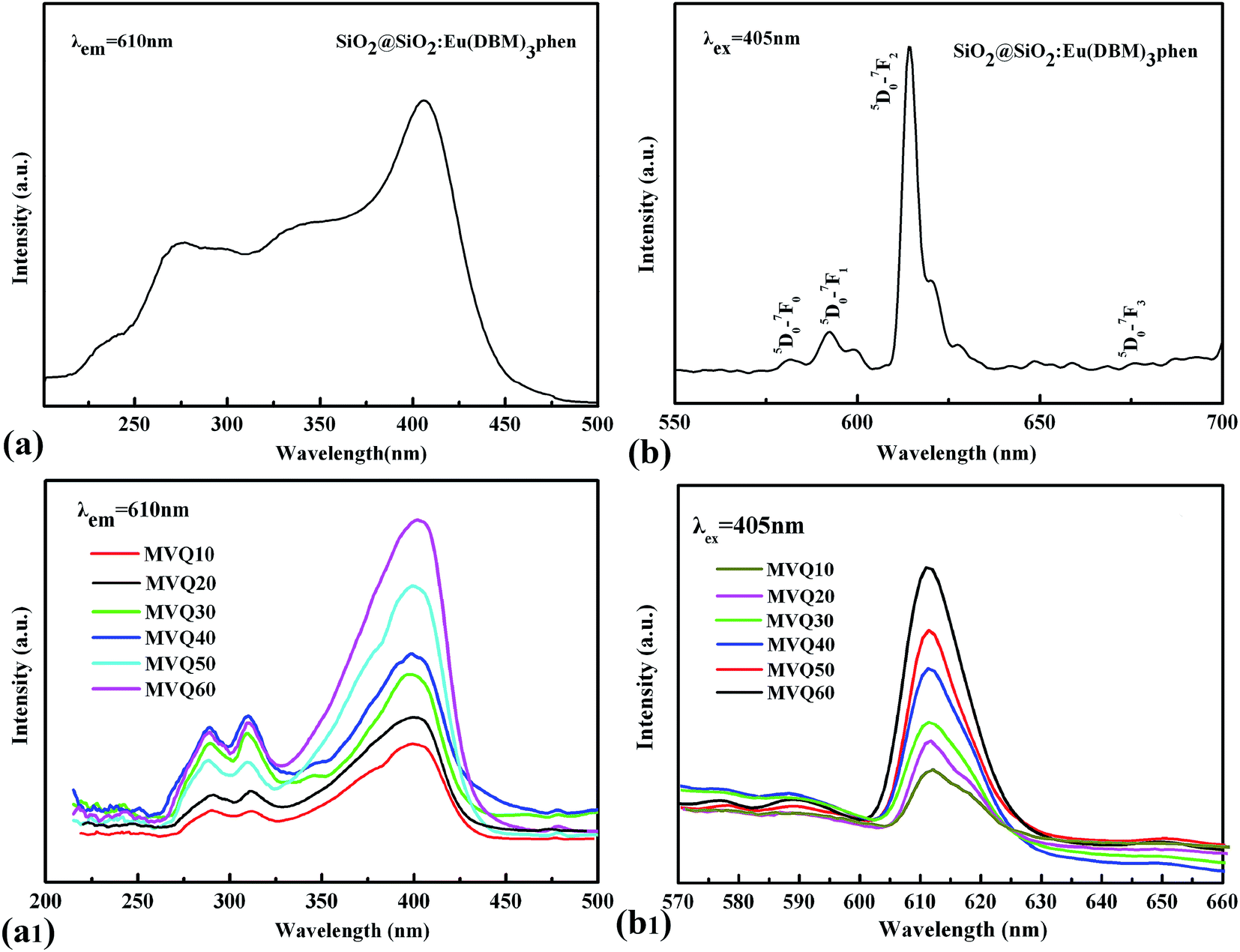 | ||
| Fig. 4 The excitation spectra and emission spectra of (a), (b) silica phosphors, and (a1), (b1) MVQ10-60. | ||
3.2 Filler network as observed by CLSM
As the filler network of MVQ60 was relatively typical, CLSM observation of MVQ60 was carried out as shown in Fig. 5, which were obtained by channel 1. In the 2-D CLSM image, red and dark phases were attributed to silica phosphors and rubber phases, respectively. It was observed that the silica phosphors primary particles appeared to be unevenly dispersed in rubber matrix and mutually fused to form their higher ordered structures, i.e., the aggregates, because of the formation of hydrogen bonds between the abundant silanol groups on its surface.15 Additionally, it should be pointed out that the silica phosphors phases, which were covered by silicone rubber, were so fuzzy, so the observation of filler network structure and the statistics of aggregate size have not been completed yet. Therefore, 3-D reconstruction with the consecutive 2-D con-focal images was adopted to describe the integrated morphology of filler network of silica phosphors.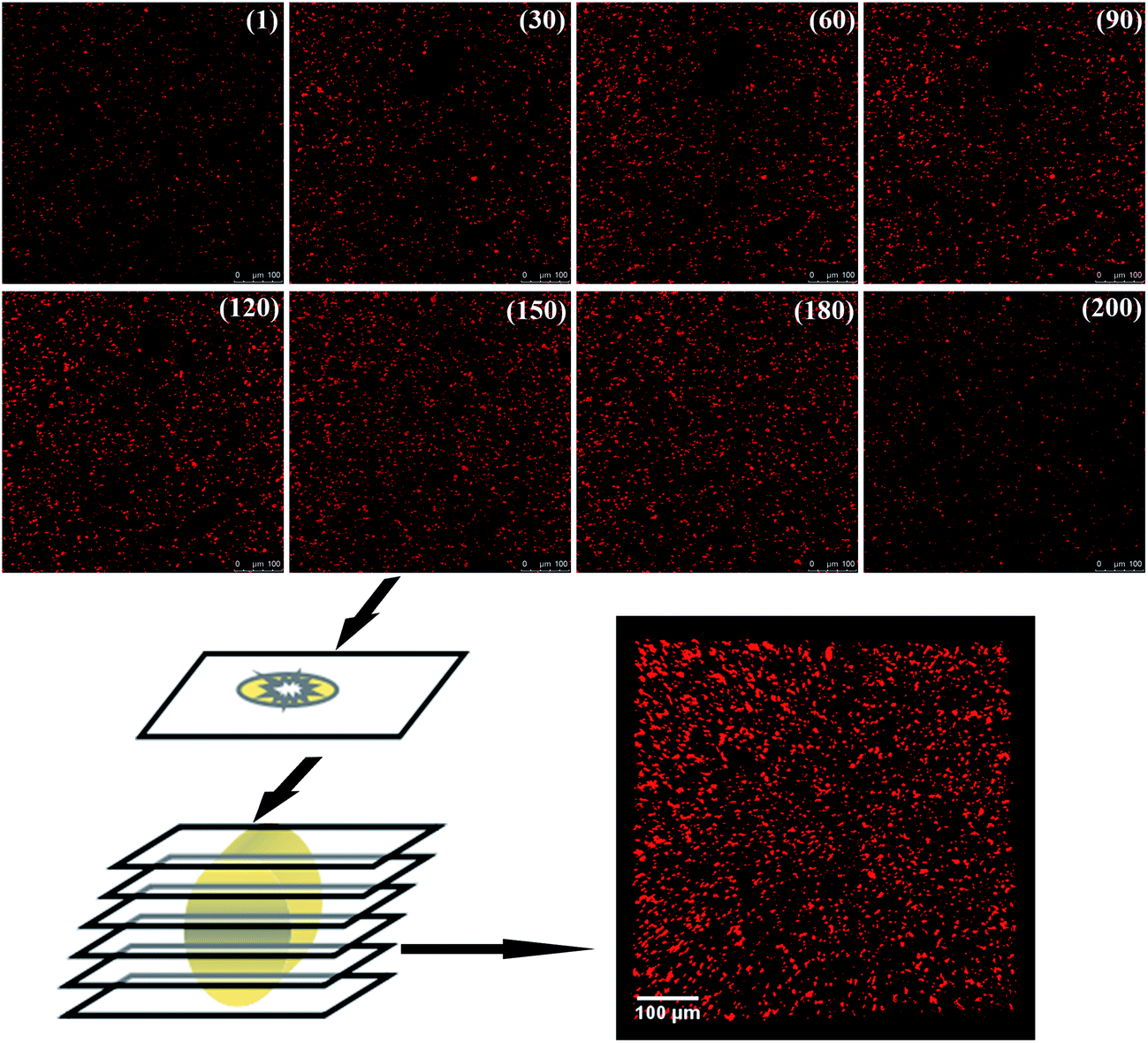 | ||
| Fig. 5 The process of 3-D reconstruction of silica phosphors network obtained from MVQ60. | ||
Image J software was used to realize the establishment of 3-D reconstruction. Briefly, 200 consecutive confocal images were imported through image sequence program and 3-D images were obtained from 3D plugins, from which the structure characteristics of the sample could be observed easily at any angle. Fig. 5 showed the process of 3-D reconstruction of silica phosphors network obtained from MVQ60 and a representative front view 3-D image of silica phosphors dispersing was shown at the bottom right corner of Fig. 5, where the rubber matrix was made transparent. As it showed, the aggregates were dispersed throughout and could be found with various morphologies, such as rod-like, semilunar, elliptical, and spherical and with size of a few micrometers.5 In order to describe the filler network structure more intuitively, the silica phosphors networks were constructed along the backbone of their aggregates from reconstruction imaging Briefly, 3-D images of silica phosphors networks were converted to binary imaging and skeletoned using Image J software, and a full scale of filler network associating with the aggregates dispersing were obtained (as shown in Fig. 6).
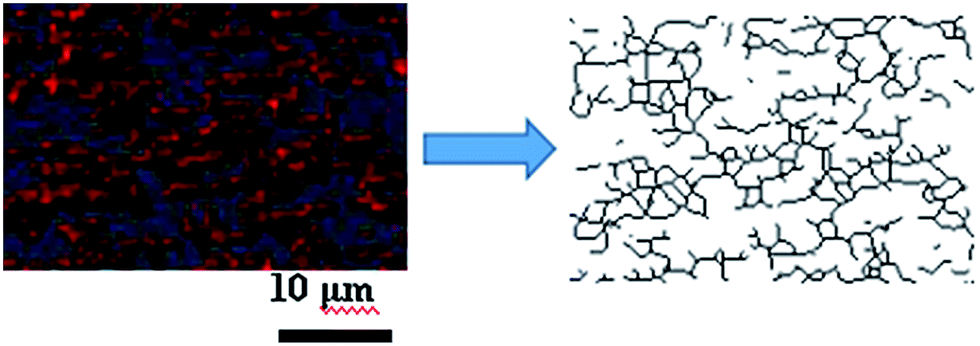 | ||
| Fig. 6 Step of filler network construction. A small zone of the filler network was constructed along the backbone of silica phosphors aggregates by Image J software. | ||
3.3 Filler network structure evolution under different filler content loading
Fig. 7 presented the filler network structure of MVQ10-60 as observed by SEM and CLSM and the skeletonization of filler networks.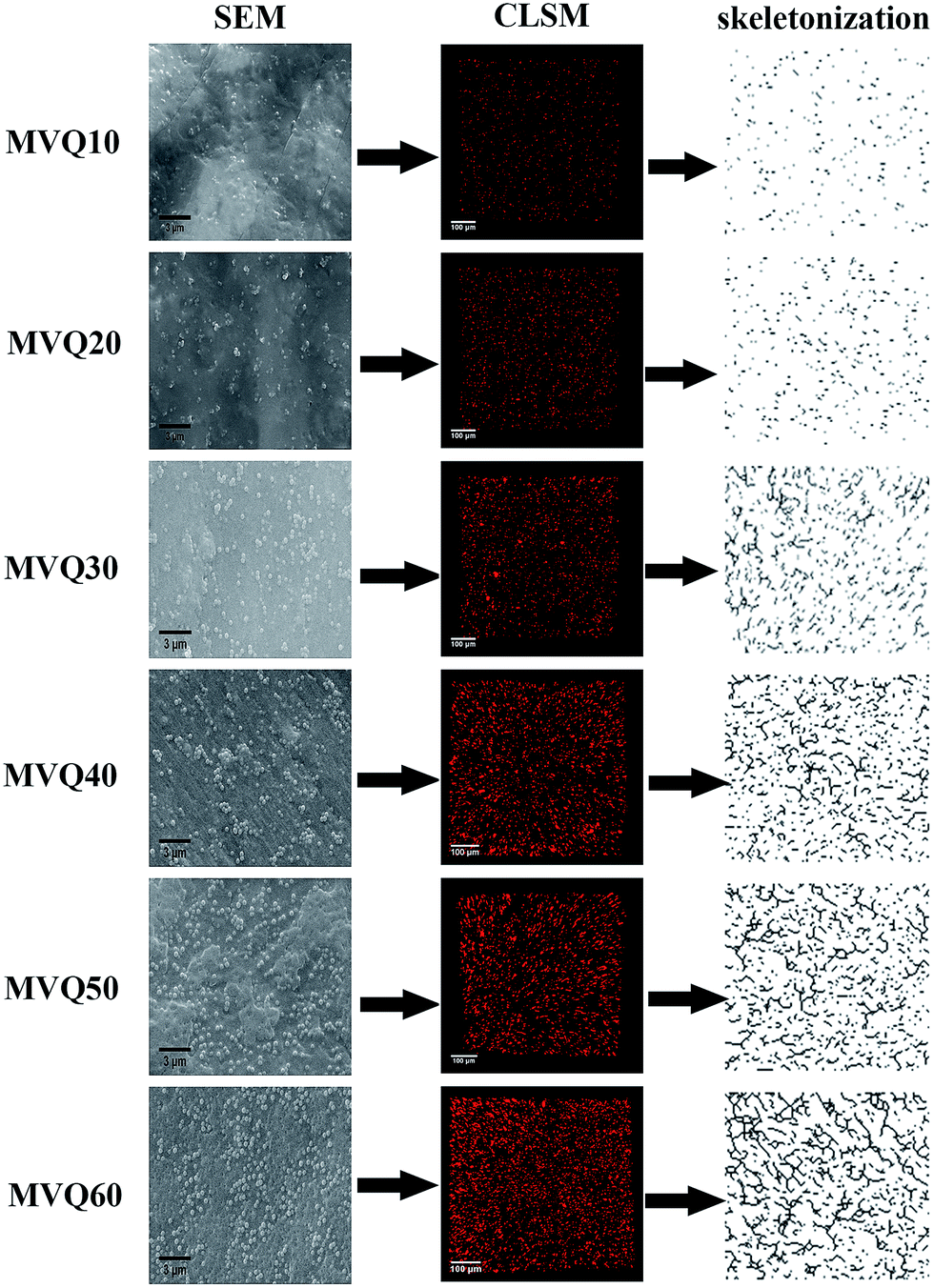 | ||
| Fig. 7 SEM images, CLSM images and skeletonization of silica phosphors networks obtained from MVQ10-60. | ||
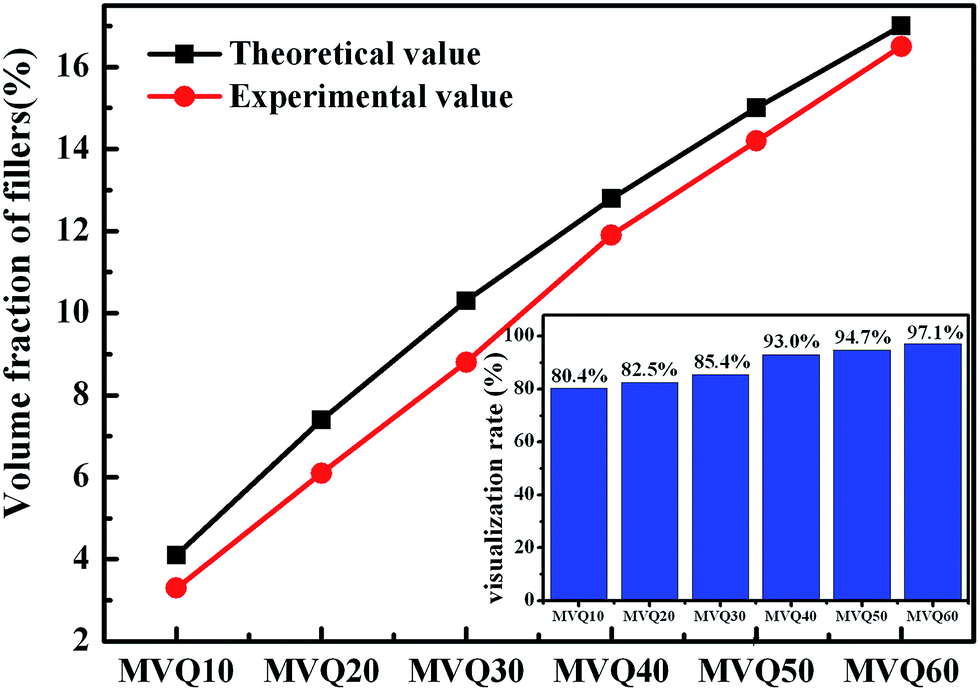
As shown in SEM images, the white and spherical parts showed the silica phosphors, which fused into aggregates of a few micrometers and there were no distinct large-scale agglomerates. Because of the limitations on SEM testing principle, it could only observe the distribution in the surface of sample, therefore the 3-D structure of filler network still could not be observed clearly. It could be seen from CLSM images and skeletonization results that filler networks were generated at the loading of filler over 30 phr and some different characteristics were reasonably detected. Notably, the filler network of sample MVQ60 seemed to be larger and clearer than that of other samples. Adding more silica phosphors resulted in a denser and more connected network and the size of their aggregates became larger. It could be explained by the reason that hydrogen bonds between the abundant silanol groups on its surface were significantly increased with the increase of the silica content, so it can be reasonably concluded that the silica phosphors with a high content tended to undergo severe aggregation. In order to verify the feasibility of this method, the experimental value, theoretical value of filler volume fraction and the related visualization rate were calculated and shown in Fig. 8. The theoretical value of filler volume fraction (υf) were calculated by the following eqn (1):
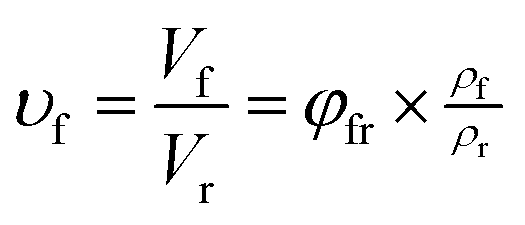 | (1) |
 | ||
| Fig. 8 Volume fraction of fillers obtained from MVQ10-60. | ||
The theoretical value of MVQ10-60 were 4.1%, 7.4%, 10.3%, 12.8%, 15.0% and 17.0%, while the experimental value were 3.3%, 6.1%, 8.8%, 11.9%, 14.2% and 16.5%. The related visualization rate were 80.5%, 82.4%, 85.4%, 93.0%, 94.7% and 97.1% (the ratio of the experimental value with the theoretical value). The deviation of the volume fraction could be explained by the reason that a small number of mono-disperse silica in the rubber matrix with diameters less than 200 nm could not be observed by CLSM. However, based on the fact that visualization rate attained 90% above with filler content over 40 phr, it was feasible to conduct the thorough research to the filler network structure through this new method, including the statistics of aggregates size, the calculation of the network connectivity, the strain-induced deformation, destruction, and reconstruction. With the increase of filler content, the visualization rate increased rapidly, which would be explained later. So, it provided a new method to detect the structural evolution under loading and a new direction for revealing the reinforcement mechanism of filler network.
Moreover, based on CLSM imaging, the size distributions of silica phosphors aggregates in the silicone rubber matrix could be counted directly. The reconstruction imaging was analyzed by Image J software with the assumption that aggregates were spherical, and then the diameter distributions of aggregates were obtained. Fig. 9a–f were the size distribution of silica phosphors aggregates obtained from MVQ10-60. For all samples, the sizes of aggregates were mainly in the range of 2.5–7 μm and occupied about half of the total counted results, while large ones with sizes more than 7 μm and small ones with sizes less than 2.5 μm took a minor fraction. Thus, the contents of aggregates with sizes smaller than 2.5 μm and larger than 7 μm (denoted as PS and PL) were selected to evaluate the aggregation degree of aggregates. Fig. 9g depicted PS and PL at different content of silica phosphors. Upon increasing the content of fillers, PS showed a monotonic decrease. For MVQ10, PS was about 64%, which reached to 8% at content of 60 phr (MVQ60). Meanwhile, PL increased from 1% at content of 10 phr to 39% at content of 60 phr. The synchronous changing trends of PS and PL revealed silica phosphors with a high content tended to undergo severe aggregation,27 which was the reason why CLSM was more applicable to high filler content rubber.
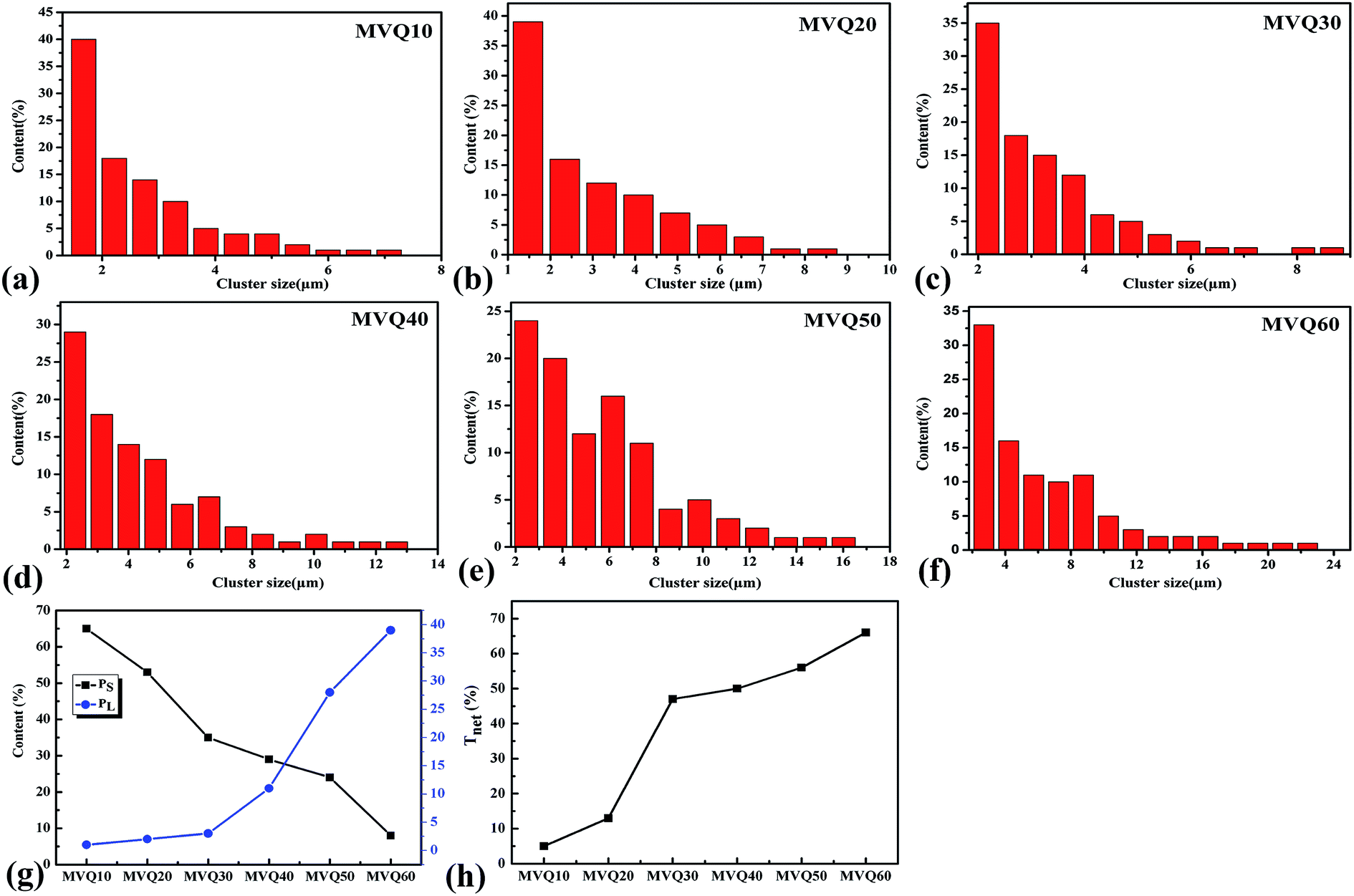 | ||
| Fig. 9 The size distribution of silica phosphors aggregates obtained from (a) MVQ10, (b) MVQ20, (c) MVQ30, (d) MVQ40, (e) MVQ50, (f) MVQ60, (g) contents of aggregates with sizes smaller than 2.5 μm (PS) and larger than 7 μm (PL), and (h) network connectivity obtained from MVQ10-60. | ||
The network connectivity (Tnet) is defined as an indicator to explore the network structures of the samples with various silica phosphors amounts. With the aid of imaging analysis from CLSM and the numerical statistical analysis from Image J software, the effective inter-particle distance of adjacent silica phosphors aggregates (dc) and the percentage of the filler network connectivity Tnet could be calculated by the following equations:28
| dc = (0.86υ−1/3f − 1)dave | (2) |
| Tnet = N(dl)/N(di) | (3) |
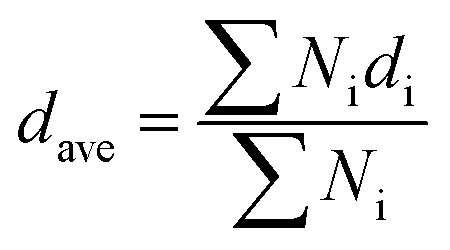 , where di is the diameter of the silica phosphors aggregates and Ni is the number of the silica phosphors aggregates. According to this arrangement, we can consider that silica phosphors aggregates have sizes larger than the effective inter-particle distance will keep the network connected.27 Tnet was calculated and plotted vs. the silica phosphor content in Fig. 9h. Originally, Tnet was 5% for the low silica phosphor amounts of 10 phr and it was 13% for MVQ20. Increasing the silica phosphor content to 30 phr caused a sharp increase in Tnet to 47%. In this case, we believed that the filler network had been formed at filler content over 30 phr. In fact, for the low silica phosphor amounts, the large effective inter-distance, small size of the aggregates and weak interaction of adjacent silica phosphor aggregates made them inadequate for forming a complete filler network. Upon increasing the silica phosphor content, the filler network connectivity increased.
, where di is the diameter of the silica phosphors aggregates and Ni is the number of the silica phosphors aggregates. According to this arrangement, we can consider that silica phosphors aggregates have sizes larger than the effective inter-particle distance will keep the network connected.27 Tnet was calculated and plotted vs. the silica phosphor content in Fig. 9h. Originally, Tnet was 5% for the low silica phosphor amounts of 10 phr and it was 13% for MVQ20. Increasing the silica phosphor content to 30 phr caused a sharp increase in Tnet to 47%. In this case, we believed that the filler network had been formed at filler content over 30 phr. In fact, for the low silica phosphor amounts, the large effective inter-distance, small size of the aggregates and weak interaction of adjacent silica phosphor aggregates made them inadequate for forming a complete filler network. Upon increasing the silica phosphor content, the filler network connectivity increased.
3.4 Tensile and mechanical fracture measurements
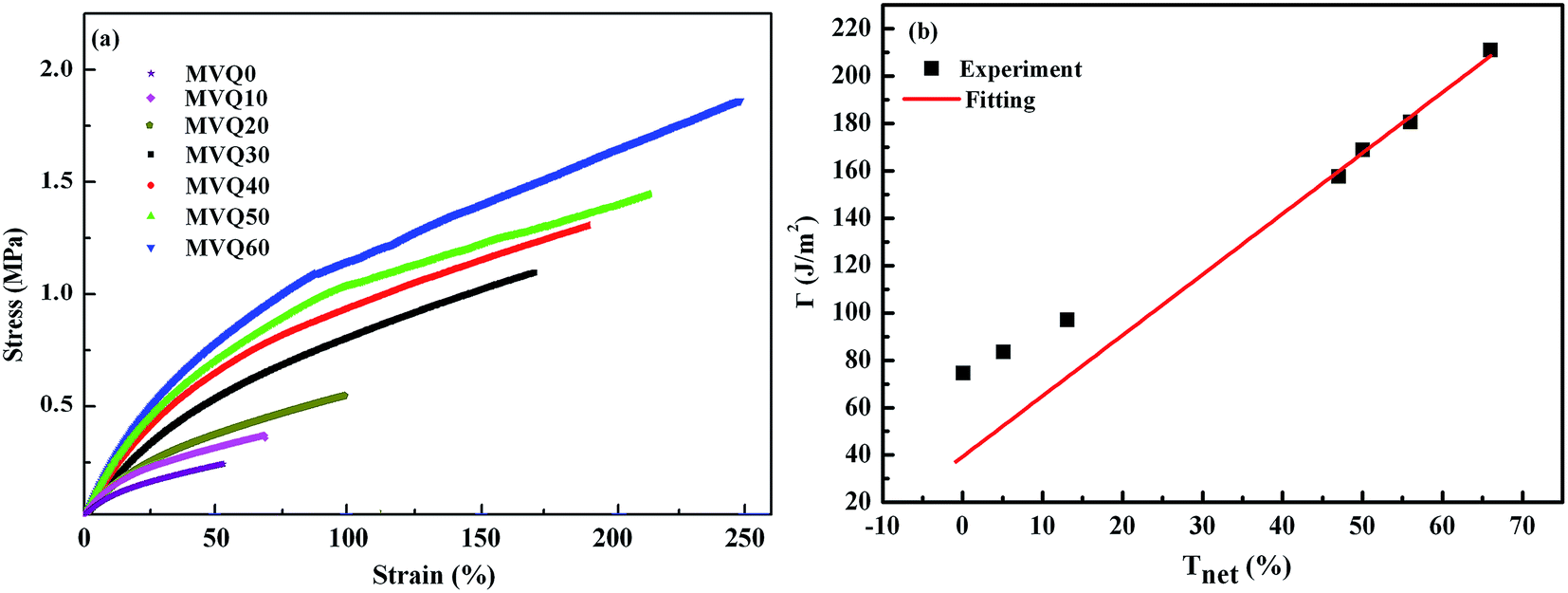
After presenting the network connectivity results on silica filler network, we compared the mechanical properties of filled silicone rubbers with different silica content loading. Fig. 10a showed the stress–strain curves of MVQ0-60 samples during tensile deformation. For a quantitative comparison, we calculated the fracture energy Γ of filled silicon rubbers, which is the work required to generate a crack on unit area of sample and expressed as eqn (4).29
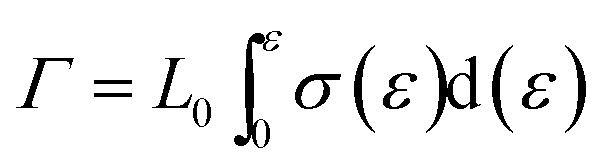 | (4) |
 | ||
| Fig. 10 (a) Engineering stress–strain curves obtained from MVQ10-60, (b) fracture energy as a function of network connectivity. The solid line shows a linear fitting of experimental data. | ||
Based on eqn (4), the fracture energy Γ of MVQ0-60 samples were calculated to be from 72.37 to 216.92 J m−2, indicating continuous reinforcement and toughening effects. Fig. 10b showed the fracture energy as a function of network connectivity and the solid line showed a linear fitting of experimental data. At the loading of silica below 30 phr, filler network was not formed and the reinforcing performance was mainly affected by the rubber cross-linked chains, resulting in deviation from the linear relationship in Fig. 10b. At the loading of filler over 30 phr (the network connectivity over 37%), the excessive stress resulted in fracture of bundles or phase morphology of filler network, so the fracture energy was consistent with the network connectivity and the correlation coefficient R2 was 0.997. It also demonstrated that the loading threshold for the formation of filler network in this experiment was 30 phr, over which the filler network was crucial for rubber reinforcing.
4. Conclusions
Spherical silica phosphors(sol–gel silica), prepared by in situ synthesis, with primary particle sizes around 250–300 nm and 2.5–7 μm of the aggregates in the silicone rubber matrix were took as an example to complete the visualization of filler network structure by fluorescent markers embedded in silica and CLSM with optical lateral resolution about 200 nm, and the corresponding 3-D structure, skeletonization and size distributions of aggregates under different filler content loading were obtained by Image J software. The visualization rate of the silica phosphors aggregates indicated that CLSM was feasible to conduct the thorough research to the filler network structure, since the visualization rate attained 90% above with filler content over 40 phr. Through the analysis of network connectivity and fracture energy, the result that the loading threshold for the formation of filler network was 30 phr in this experiment could be obtained. The current study also demonstrates that CLSM has considerable advantages when compared with SEM and synchrotron radiation X-ray nano-computed tomography technique. It has both: intuition and efficiency in examining 3-D lateral and vertical morphology and micro-structure of filler networks, which helps to explore the strain-induced deformation, destruction, and reconstruction of filler network more easily and efficiently.Conflicts of interest
There are no conflicts to declare.References
- J. L. Valentín, I. Morabarrantes, J. Carreterogonzález, M. A. Lópezmanchado, P. Sotta, D. R. Long and K. Saalwächter, Macromolecules, 2010, 43, 334–346 CrossRef.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- A. S. Hoffman, P. S. Stayton, T. Shimoboji, G. Chen, Z. Ding and A. Chilkoti, Macromol. Symp., 2015, 118, 553–562 CrossRef.
- S. S. Sarkawi, W. K. Dierkes and J. W. M. Noordermeer, Eur. Polym. J., 2014, 54, 118–127 CrossRef CAS.
- L. Chen, W. Zhou, J. Lu, J. Li, W. Zhang, N. Huang, L. Wu and L. Li, Macromolecules, 2015, 48, 7923–7928 CrossRef CAS.
- C. R. Lin and Y. D. Lee, Macromol. Theory Simul., 1996, 5, 1075–1104 CrossRef CAS.
- A. Lion, Rubber Chem. Technol., 1999, 72, 410–429 CrossRef CAS.
- J. M. Funt, Rubber Chem. Technol., 1988, 61, 842–865 CrossRef CAS.
- Y. Fukahori, Rubber Chem. Technol., 2003, 76, 548–566 CrossRef CAS.
- S. Gan, Z. L. Wu, H. Xu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 1454–1463 CrossRef CAS.
- Y. Satoh, S. Fujii, S. Kawahara, Y. Isono and S. Kagami, e-Journal of Soft Materials, 2007, 3, 29–40 CrossRef.
- A. Tohsan, R. Kishi and Y. Ikeda, Colloid Polym. Sci., 2015, 293, 2083–2093 CAS.
- Y. Lin, S. Liu, J. Peng and L. Liu, Composites, Part A, 2016, 86, 19–30 CrossRef CAS.
- J. Yang and C.-R. Han, J. Phys. Chem., 2013, 117, 20236–20243 CAS.
- J. Wang, H. Jia, L. Ding and X. Xiong, Polym. Adv. Technol., 2015, 26, 1168–1175 CrossRef CAS.
- A. J. Rocker, A. R. E. Weiss, J. S. Lam, T. J. Van Raay and C. M. Khursigara, J. Microbiol. Methods, 2015, 117, 85–94 CrossRef PubMed.
- K. Chatzimeletiou, E. E. Morrison, Y. Panagiotidis, P. Vanderzwalmen, N. Prapas and Y. Prapas, Hum. Reprod., 2012, 27, 106–113 CrossRef PubMed.
- Y. Zou, M. Kaestner and E. Reithmeier, Optic Laser Eng., 2015, 74, 40–46 CrossRef.
- J. Taendl, S. Nambu, A. Orthacker, G. Kothleitner, J. Inoue, T. Koseki and C. Poletti, Mater. Charact., 2015, 108, 137–144 CrossRef CAS.
- T. Kimura, M. Shintate and N. Miyamoto, Chem. Commun., 2015, 51, 1230–1233 RSC.
- J. Kus, Int. J. Coal Geol., 2015, 137, 1–18 CrossRef CAS.
- L. Deillon, J. Zollinger, D. Daloz, M. Založnik and H. Combeau, Mater. Charact., 2014, 97, 125–131 CrossRef CAS.
- F. Sondej, A. Bück and E. Tsotsas, Powder Technol., 2016, 287, 330–340 CrossRef CAS.
- L. R. Melby, N. J. Rose, E. Abramson and J. C. Caris, J. Am. Chem. Soc., 1964, 86, 5117–5125 CrossRef CAS.
- Y. Mou, M. Kang, F. Wang, M. Liu, K. Chen and R. Sun, J. Sol-Gel Sci. Technol., 2017, 83, 447–456 CrossRef CAS.
- H. Zhou, L. Song, A. Lu, T. Jiang, F. Yu and X. Wang, RSC Adv., 2016, 6, 15155–15166 RSC.
- L. Chen, L. Song, J. Li, P. Chen, N. Huang and L. Li, Macromol. Mater. Eng., 2016, 301, 1390–1401 CrossRef CAS.
- S. Wolff, M. J. Wang and E. H. Tan, Rubber Chem. Technol., 1993, 66, 163–177 CrossRef CAS.
- L. Song, Z. Wang, X. Tang, L. Chen, P. Chen, Q. Yuan and L. Li, Macromolecules, 2017, 50, 7249–7257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |