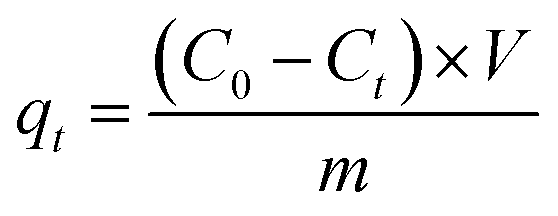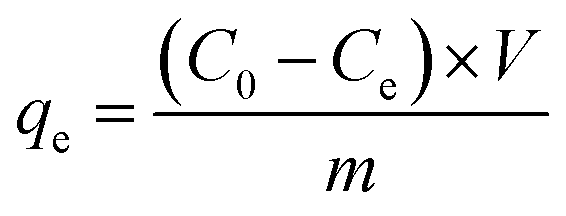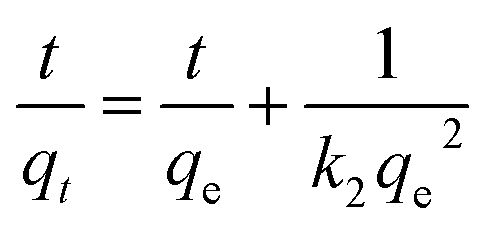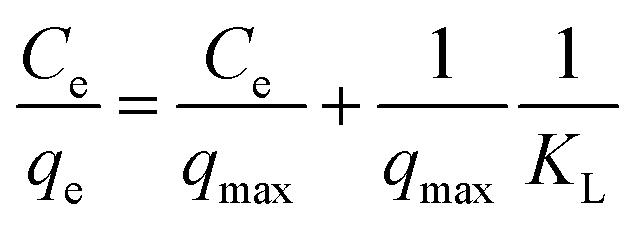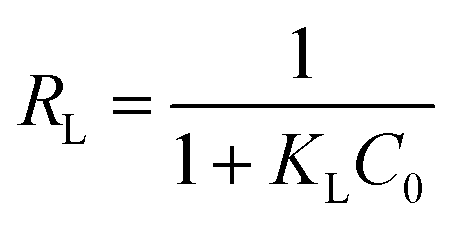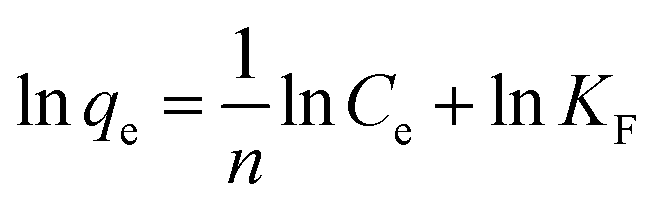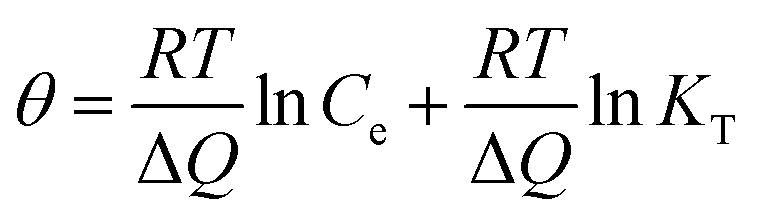DOI:
10.1039/C7RA10009D
(Paper)
RSC Adv., 2017,
7, 53362-53372
Solar light assisted green synthesis of photoreduced graphene oxide for the high-efficiency adsorption of anionic dyes
Received
8th September 2017
, Accepted 13th November 2017
First published on 21st November 2017
Abstract
Graphene oxide (GO) with unique physical and chemical properties, such as high specific surface area, chemical stability and environmental friendliness, has been considered as an excellent adsorbent to remove organic dyes from polluted water. However, because of the electrostatic repulsion between the GO and anionic dyes, the adsorption capacity for anionic dyes is undesirable. Here photoreduced GO (PRGO) was prepared by solar light irradiation and used to investigate the adsorption capacity for anionic dyes. The obtained GO and PRGO were characterized by UV-visible absorption, Raman spectroscopy, atomic force microscopy (AFM), scanning electron microscopy (SEM), Fourier transform infrared (FTIR) and X-ray photoelectron spectroscopy (XPS). The adsorption kinetics and isotherms were evaluated through adsorption experiments of the anionic orange II (OII) dye adsorbed on GO or PRGO. The results revealed that the adsorption was an exothermic process and followed pseudo-second order kinetics. Four more typical anionic dyes, methyl orange (MO), Ponceau S (PS), Azo Rubine (AR), and Trypan Blue (TB), were also used to compare the adsorption capacities between GO and PRGO. It was demonstrated that the adsorption capacity of anionic dyes was significantly enhanced by PRGO, and the maximum enhancement was up to 8 fold. In conclusion, PRGO reduction by solar light irradiation in a green, convenient and cost effective manner, offers promise for the high-efficiency removal of anionic dyes in water treatment.
1. Introduction
Today, the shortage of fresh and clean water has become one of the most important challenges around the world; the serious water pollution arising from the rapid development of industry endangers human health.1,2 As an important source of water pollution, organic dyes mainly originate from the printing, textile, leather tanning and paper-making industries.3 Most of the dyes with heterocyclic or aromatic structures have been identified as toxic, mutagenic and even carcinogenic.4 Even more crucial, they are easily transported within the aqueous environments but hardly biodegradable. Various methods have been developed for the removal of organic dyes from wastewater, including membrane separation,5 ion exchange,6 chemical precipitation7 and so on. Of all the methods proposed, adsorption is the most commonly used because of its versatility, wide applicability and economic feasibility.8 With the development of nanomaterials, especially the discovery of graphene with unique chemical and physical properties,9,10 such as the large specific surface, chemical stability and environmental friendliness, graphene and its derivatives have emerged as a key materials for the removal of dyes.11–13 Among them, graphene oxide (GO) presents outstanding adsorption capacity for cationic dyes, arising from the π–π interaction and electrostatic interaction between dyes and GO surface.14,15 However, the adsorption capacity of anionic dyes by GO is extremely low, due to the strong repulsion interaction between anionic dyes and electronegative GO.
In view of the electronegative nature of GO arising from the oxygen-containing functional groups on GO basal plane,16 the reduction processes have been designed to improve the adsorption capacity of anionic dyes, by removing the functional groups and restoring the sp2 π-conjugated network to enhance the π–π interaction between the anionic dyes and reduced GO (RGO). In 2011, Ramesha and coworkers reduced GO using hydrazine hydrate and investigated the adsorption property of RGO on anionic dye of orange G (OG).17 The results showed that the RGO could act as a good adsorbent for anionic dyes, and the maximum adsorption capacity of OG was up to 5.98 mg g−1. Later, Sun group compared the adsorption capacities of GO and RGO on the anionic dye of acridine orange (MO), by in situ reducing GO with sodium hydrosulfite.18 They found that the maximum adsorption capacity of MO was improved by 2.35 folds after chemical reduction. Recently, Xiao et al. reported that the adsorption capacity of GO on anionic indigo carmine (IC) dye was enhanced by 3.63 folds, after reduction by L-cysteine.19 Hence, the RGO reduced by chemical reductants shows a promising alternative for the removal of anionic dyes from pollution water. While, the secondary pollution and energy waste during the chemical reduction still limits its further applications.
Here, we present an environmentally friendly and cost-free method to prepare photoreduced GO (PRGO) dispersion and investigate the adsorption capacities of GO and PRGO on anionic dyes. In the experiment, PRGO was synthesized by exposing GO dispersion to solar light irradiation in the sealed glasses with different duration time.20,21 Compared with other routes, this method is simple and easy for mass production, and it is not subject to experimental conditions. More importantly, this method avoids secondary pollution for the environment. The reduction degree was characterized by UV-visible absorption, Raman spectra, atomic force microscope (AFM), scanning electron microscope (SEM), Fourier transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS), respectively. The adsorption kinetics and isotherm of GO and PRGO on anionic Orange II (OII) dye have been measured and analyzed to explore the adsorption mechanism. The adsorption of OII dye on the PRGO materials was rapidly increased in the first 5 min and almost reaches the equilibrium in less than half an hour. Compared with GO, the adsorption rate was enhanced by 33 folds. The maximum adsorption capacity of OII was improved by 4 folds after solar reduction. Adsorption behavior on other four typical anionic dyes further evidence that the PRGO reduction by solar light irradiation is a green, convenient and cost-effective method without secondary pollution for the high-efficiency adsorption of anionic dyes.
2. Experimental section
2.1 Materials
The GO powder (the diameter of 0.5–5 μm and thickness of 1–3 nm) was supplied by Nanjing XFNANO Materials Tech Co. Ltd. Orange II (OII), Methyl Orange (MO), Ponceau S (PS), Azo Rubine (AR), Trypan Blue (TB), and Ethylene Glycol (EG) were purchased from J&K Scientific Ltd. All purchased dyes were used directly without any further purification. 0.22 μm Millipore syringe filters were obtained from Tianjin Navigator Lab Instrument Co., Ltd. An Elix® Advantage system (Millipore Q, USA) was used to produce deionized water.
2.2 Preparation of PRGO
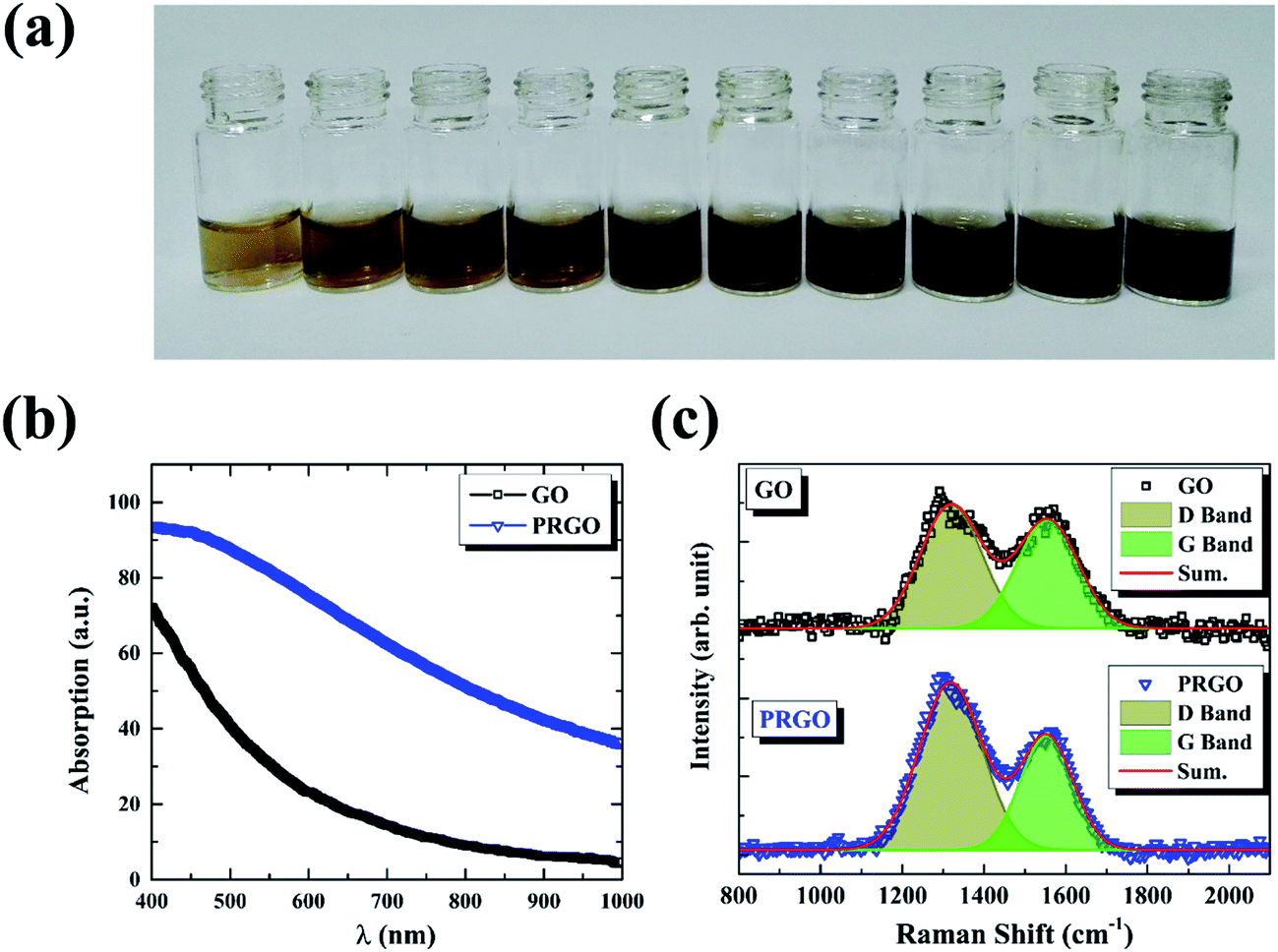
The GO powder was dissolved in deionized water (2 mg mL−1) by ultrasonic dispersion to form a yellow-brown GO suspension, as shown in Fig. 1a. The as-prepared dispersion was stored in sealed transparent glass bottles. The reduction of GO was carried out by exposing 0.67 mg mL−1 dispersion to solar light with the duration time from 8 h to 72 h. In order to maintain the same reduction condition, the experiments were performed in the same weather temperature (30 °C) and atmospheric environment (cloudless). Initial pH value of the dispersion was measured to be 5.0 ± 0.3 and no significant change was observed after photoreduction.
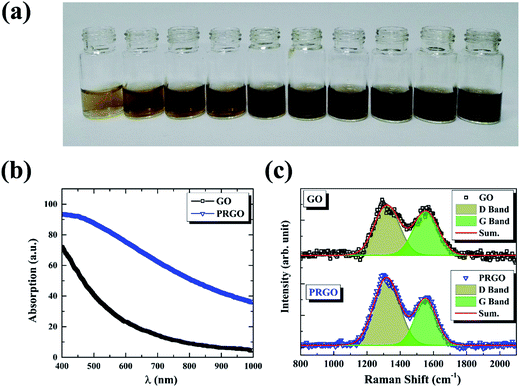 |
| | Fig. 1 (a) Photographic images of GO (left first) and PRGO with irradiation duration time of 8, 16, 24, 32, 40, 48, 56, 64, and 72 h, respectively. (b) and (c) are absorption and Raman spectra of GO and PRGO with irradiation duration time of 72 h, respectively. | |
2.3 Sample characterization
GO and PRGO were characterized by a UV-visible absorption spectra on a Maya2000Pro Vis-NIR spectrometer (Ocean Optics), which was also used to measure the concentration of anionic dyes in the adsorption experiments. Raman spectra was carried out on a home-built scanning confocal system equipped with a 532 nm laser source and a long focus monochromator (Horbia Jobin Yvon, 1250M), which can be found in our previous works.22–24 The morphology of nanosheets was observed using SEM (SU8010, Hitachi, Japan). AFM (5000N, JEOL, Japan) was used to measure the height of GO and PRGO, respectively. FTIR used to characterize the chemical compositions of GO and PRGO was recorded on the Si substrate with a commercial Bruker FTIR spectrometer (Thermo Scientific Nicolet iS50). XPS analysis was conducted on an AXIS ULTRA DLD spectrometer (Thermo Kratos British) with Al Kα irradiation as the exciting source (300 W).
2.4 Dye adsorption experiments
The adsorption capacities of GO and PRGO with different irradiation duration time were investigated by five anionic dyes, including OII, MO, PS, AR, and TB, whose concentrations were determined using UV-Vis spectrophotometer (Ocean Optics May2000Pro) based on their maximum absorbance wavelengths at 510, 462, 520, 515, and 598 nm, respectively. Calibration plots of dye concentrations were established based on Lambert–Beer's Law. The adsorption experiments were performed by mixing 2 mg GO or PRGO with a series of dye solutions with different initial concentrations at room temperature (22 °C) in the sealed bottles. After shaking at 80 rpm in a shaker, the suspensions were withdrawn and filtered with 0.22 μm Millipore syringe filters, and the residual concentrations of dyes were calculated according to UV-Vis absorption spectra. In the adsorption kinetics study, the dye aqueous with the volume of 3 mL and initial concentrations C0 of 20 mg L−1 were mixed with 2 mg GO or PRGO. After shaking, the concentration of residual dye solution Ct were measured respectively at 5, 10, 15, 20, 40, 90, 180, 330, 450, 570, 750, and 1440 min (24 h) from the beginning. The adsorption capacities qt for dyes adsorbed onto GO or PRGO were determined according the following equation:| |
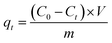 | (1) |
where V is the volume of the solution (L) and m is the mass of GO or PRGO (g), respectively. In the equilibrium isotherm experiments, 3 mL dyes solutions with initial concentrations C0 of 6.7, 13, 20, 26.7, 33, 40, 46.7, 53, 60, and 67 mg L−1 were mixed with 2 mg GO or PRGO at room temperature. The amount of dyes adsorption at equilibrium qe (mg g−1) can be obtained by the equation:| |
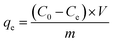 | (2) |
where Ce is the concentrations of residual dyes solutions after shaking for 24 h.
2.5 Desorption and recycling test
The adsorption stability of PRGO was investigated by recycling the material through dyes adsorption–desorption experiments. In a typical experiment, 3 mL dyes with concentration of 60 mg L−1 was mixed with 2 mg PRGO and shaking for 24 h at room temperature. The adsorbents were subsequently removed from the solutions by filtration and washed with EG and deionized water. The residual dyes were measured to calculate the adsorption capacities. The EG washed GO or PRGO was then dissolved with deionized water again and used in the second cycle adsorption experiment. This process was repeated in four cycles for OII dye.
3. Results and discussion
3.1 Photoreduction and characterization
The reduction of GO was carried out by exposing its suspension hold in sealed transparent glass bottles to solar light. Fig. 1a presents the photograph of GO suspensions before and after exposition with duration time from 8 to 72 h. Clearly, the color of the suspensions changed from yellow-brown to deep black gradually as the increasing of exposition time. Conventional absorption spectroscopy was performed on the initial (black line) and the last (72 h, blue line) suspensions, as shown in Fig. 1b. The PRGO can be significantly distinguished from the GO spectrum by an increasing of absorption in the range of 400–1000 nm,25 in accordance with their color change. The increased absorption corresponds to the removal of oxygen-containing functional groups and the restoration of the sp2 π-conjugated network in the PRGO nanosheets. Fig. 1c displays the Raman spectra from suspensions before (black line) and after (blue line) solar irradiation with 72 h, respectively. The two prominent peaks can be determined in both materials, which are corresponding to D and G band, respectively. The G band is usually represented the first order scattering of the E2g phonon of sp2 carbon, while the D band is associated with structural defects and disorders.26 The D and G bands are fitting by Gauss function with the center values of 1320, 1555 cm−1 for GO, and 1318 and 1553 cm−1 for PRGO, respectively. A slight redshift can be explained by a graphitization of the sample, as reported by the previous literature.25,27 Furthermore, the intensity ratio of D to G band (ID/IG) can be used to denote the disorder degree and averaged crystal size of the sp2 network.28,29 The increased ID/IG ratio from 1.17 for GO to 1.76 for PRGO indicates the decreased sizes but the increased numbers of sp2 frameworks.30
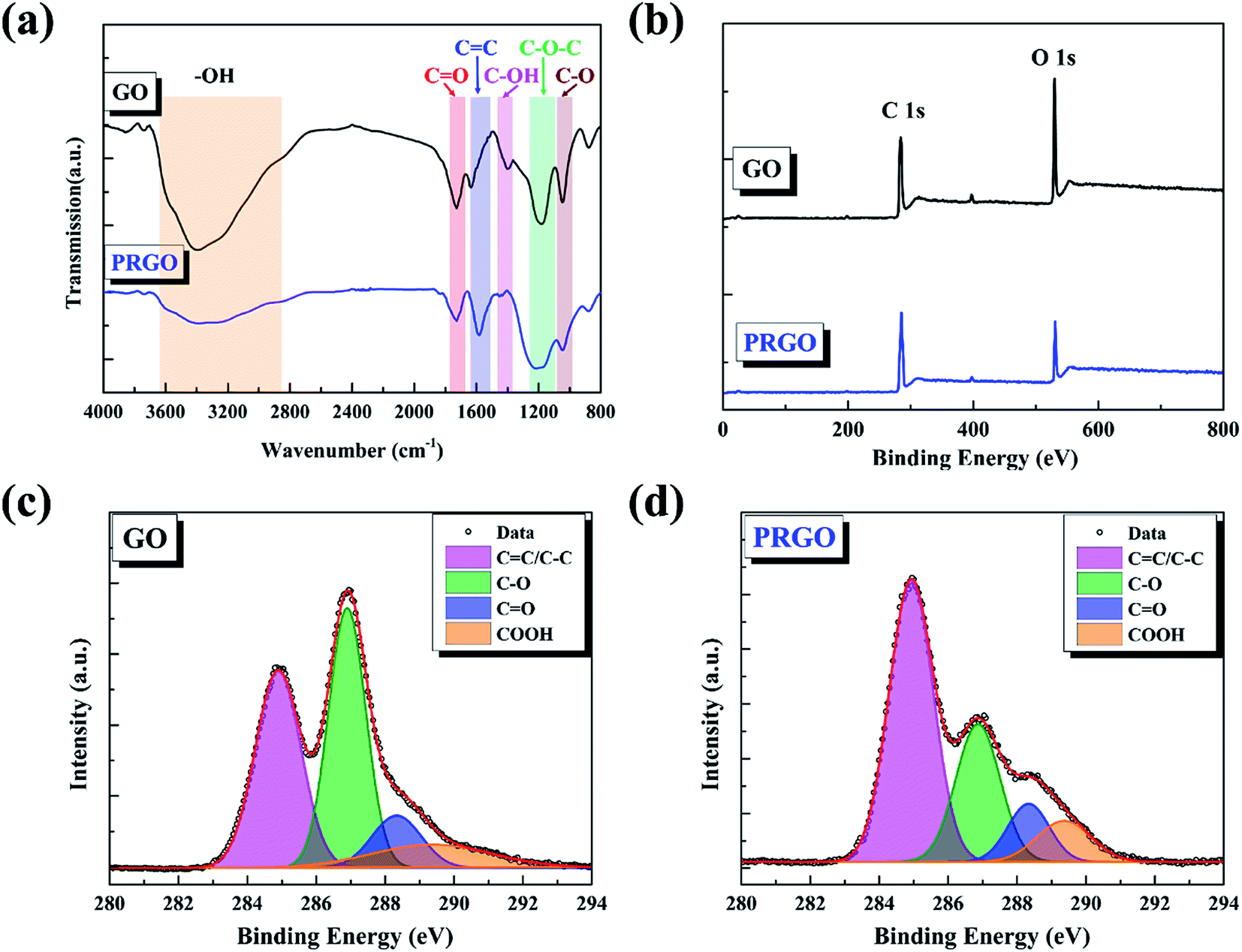
The chemical compositions of GO before and after solar light irradiation were investigated using FTIR measurements. As shown in Fig. 2a, the band between 3600 and 2800 cm−1 is attributed to the O–H stretching vibration arising from hydroxyl groups, the bands at 1730, 1620, and 1045 cm−1 can be described as the stretching modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, and C–O groups, and the peaks located at 1410 and 1195 cm−1 are assigned to the bending vibration of C–OH groups as well as C–O–C asymmetric stretching vibration of epoxy group, respectively.31,32 The decrease or even disappear of hydroxyl and epoxy groups, as well as the increases of carboxyl strongly indicates the reduction during light irradiation. The position of CC peak shifts from 1620 cm−1 to 1590 cm−1, possibly because of the restoration of sp2 structures. Here, we also used XPS to quantify the degree of reduction. The C/O ratio can be calculated from the atomic percentages obtained from the survey XPS spectra,29 as shown in Fig. 2b. The ratio of GO sample before and after solar light irradiation increases from 2.54 to 6.19, revealing the removal of oxygen-containing functional groups from GO basal plane. Fig. 2c and d present the C 1s deconvolution spectra of GO before and after irradiation, respectively. The spectra can be curve-fitted into four peak components with binding energy at about 284.9, 286.9, 288.4, and 289.4 eV, corresponding to the CC/C–C, C–O (hydroxyl and epoxy), CO (carbonyl), and OC–O (carboxyl) species, respectively.33 The increase of CC peak and the decrease of C–O peak hint that the conjugation sp2 structures were restored and the epoxy and alkoxy groups on the GO surface were removed, respectively. Nevertheless, the carbonyl and carboxyl at the sheet edges have little change, which is similar to the reduction by chemical agents.34
O, CC, and C–O groups, and the peaks located at 1410 and 1195 cm−1 are assigned to the bending vibration of C–OH groups as well as C–O–C asymmetric stretching vibration of epoxy group, respectively.31,32 The decrease or even disappear of hydroxyl and epoxy groups, as well as the increases of carboxyl strongly indicates the reduction during light irradiation. The position of CC peak shifts from 1620 cm−1 to 1590 cm−1, possibly because of the restoration of sp2 structures. Here, we also used XPS to quantify the degree of reduction. The C/O ratio can be calculated from the atomic percentages obtained from the survey XPS spectra,29 as shown in Fig. 2b. The ratio of GO sample before and after solar light irradiation increases from 2.54 to 6.19, revealing the removal of oxygen-containing functional groups from GO basal plane. Fig. 2c and d present the C 1s deconvolution spectra of GO before and after irradiation, respectively. The spectra can be curve-fitted into four peak components with binding energy at about 284.9, 286.9, 288.4, and 289.4 eV, corresponding to the CC/C–C, C–O (hydroxyl and epoxy), CO (carbonyl), and OC–O (carboxyl) species, respectively.33 The increase of CC peak and the decrease of C–O peak hint that the conjugation sp2 structures were restored and the epoxy and alkoxy groups on the GO surface were removed, respectively. Nevertheless, the carbonyl and carboxyl at the sheet edges have little change, which is similar to the reduction by chemical agents.34
 |
| | Fig. 2 (a) FTIR spectra of GO and PRGO with irradiation duration of 72 h, respectively. (b)–(d) are XPS survey spectra and curve fit of C 1s spectra of GO and PRGO with irradiation duration of 72 h, respectively. | |
The surface morphology of GO before and after solar irradiation were analyzed by AFM and SEM. From AFM images (Fig. 3a and b), it can be found that GO nanosheets are exfoliated into monolayer with the lateral size in the region of 0.5–5 μm and the height of ∼1.3 nm. After solar irradiation, the nanosheets are still monolayer with the height of ∼0.7 nm, indicating the effective removal of functional groups on the basal plane of GO. Fig. 3c and d present the SEM images of GO and PRGO, respectively. As reported in the previous works,18 GO exhibits ordered structures, while PRGO shows exfoliated nanosheets and folding nature. Because of abundant hydrophilic functional groups on the basal plane of GO (such as –OH), many interlayered hydrogen bonds will be formed during filtration and evaporation progresses, and lead to the formation of ordered GO nanosheets. After solar irradiation, the PRGO becomes less hydrophilic due to the loss of functional groups. The decreased interlayer interaction results in less compact and more crumpled, as well as the high surface area for effective adsorption.
 |
| | Fig. 3 (a) and (b) are AFM images of GO and PROG, respectively. (c) and (d) are SEM images of GO and PROG, respectively. | |
The reduction of GO by solar light irradiation can be explained by photochemical effect. The solar light has a broad spectral distribution from ultraviolet to infrared (∼4.27–1.55 eV). As proved by Ida and Antonietti,35,36 the ultraviolet irradiation is very effective in reducing GO through light-generated electron–hole pairs, which can be immediately formed during irradiation. Then the generated electron–hole pairs move to the functional groups on the GO basal plane, and result in the remove of hydroxyl groups (–OH). While a certain number of carbonyl (CO) and carboxyl (OC–O) groups still remain, as evidenced by FTIR and XPS spectra shown in Fig. 2. The non-uniform reduction results on functional groups can be attributed to their quite different bonding energies. Theoretical calculations have shown that the binding energy of hydroxyl group adsorbed onto graphene plane is estimated to be 1.5 eV, while that of C–C bond in carbonyl and carboxyl exceeds 5.8 eV.37,38 As a consequent, only the hydroxyl group was dissociated efficiently. Furthermore, on the contrast to the solar reduction by a convex lens,20 which resulted in the rapid heating and temperature increasing, the reduction under this experiment is mild without significant temperature change. Thus, photothermal effect can be ignore.
3.2 Adsorption kinetics
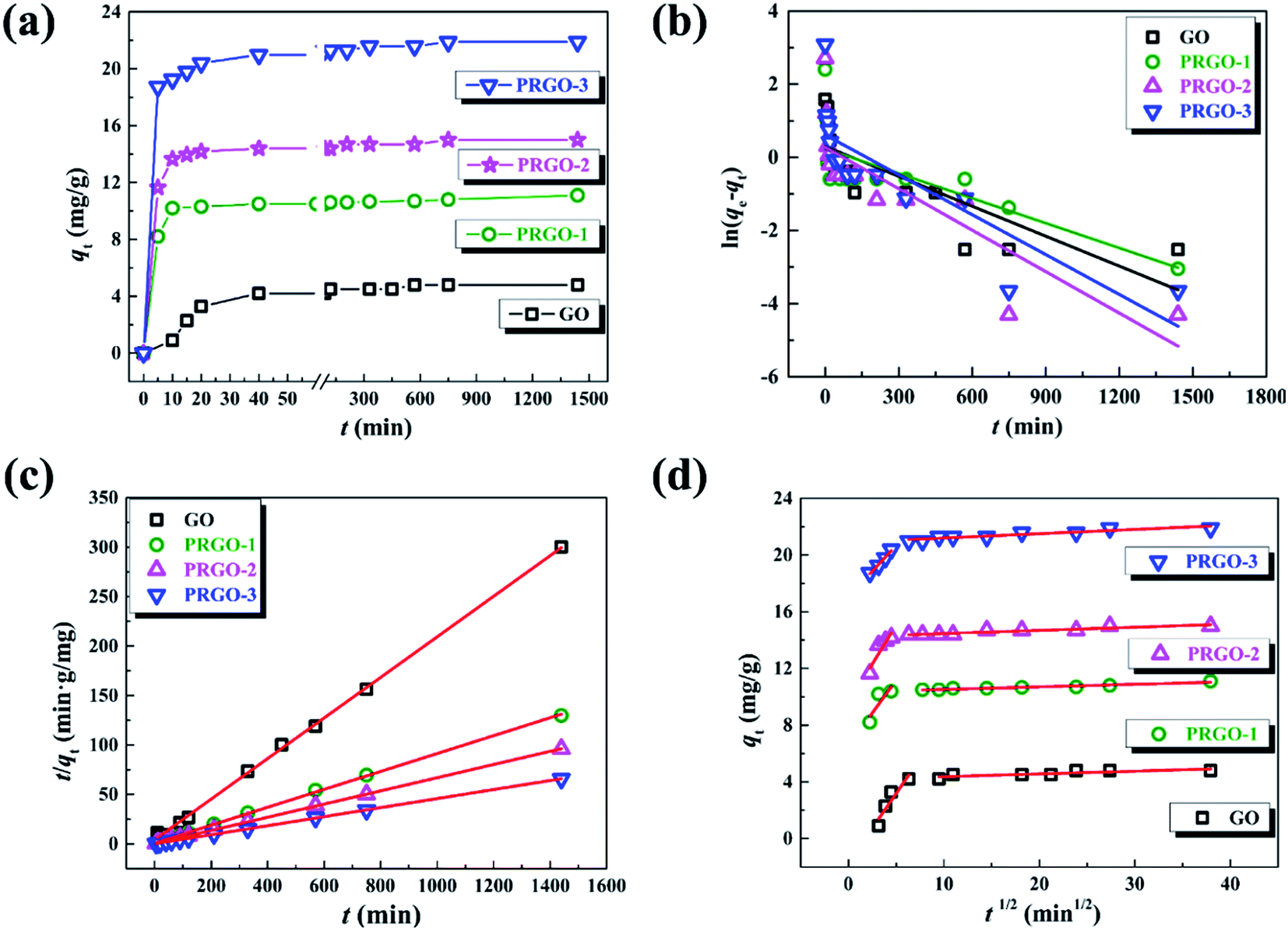
In order to understand the mechanism of the adsorption process and evaluate the adsorption capacity, the adsorption kinetics experiments were performed by measuring the time-dependent dye adsorption behaviors. Given the similar adsorption kinetics for the anionic dyes used in our experiments, here we only display the experimental results of OII dye. Fig. 4a presents the adsorption capacity of OII varying as the equilibrium time in the range of 5 min to 24 h for GO and PRGO, respectively. As expected, the adsorption capacity of OII on PRGO are much higher than that of GO materials. Here, PRGO-1, PRGO-2, and PRGO-3 denote the GO exposing to the solar light with the duration time of 24, 48, and 72 h, respectively. Furthermore, the adsorption of OII dye on all PRGO materials is rapidly increased in the first 5 min and almost reaches the equilibrium in less than half an hour. However, the adsorption on GO is relatively slow and reaches the equilibrium at about 10 h. The high adsorption capacity as well as fast adsorption rate can be attributed to the availability of a large number of adsorption sites on PRGO surface, resulting from the restoration of small but abundant sp2 π-conjugated structures. These sites make the aromatic dyes contacting immediately with the surface of PRGO with the aid of strong π–π interaction, while the GO surface with many functional groups slows down the adsorption rate due to the steric effect and less sp2 structures.
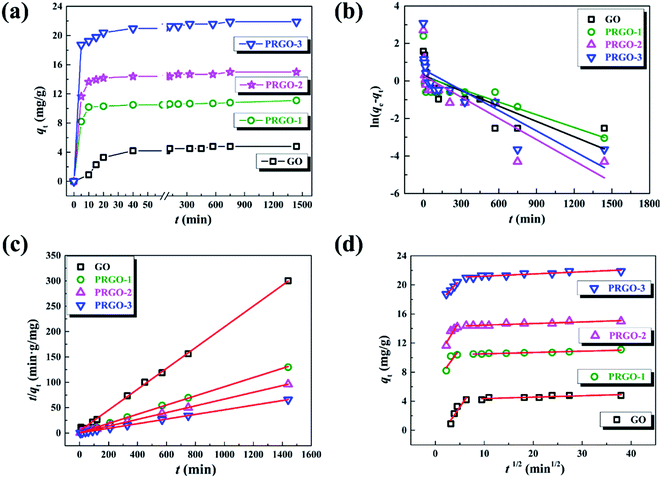 |
| | Fig. 4 (a) Adsorption kinetics of OII on GO and PRGO-1, 2, 3 with irradiation duration of 24, 48, and 72 h, respectively. (b)–(d) are the plots of pseudo-first order model, pseudo-second order and intraparticle diffusion models for the OII adsorption on GO and PRGO, respectively. | |
To get insight into the adsorption processes and determine the kinetic parameters, the experimental data are fitted by three different kinetic models: (1) the pseudo-first order model, (2) pseudo-second order model and (3) intraparticle diffusion model, respectively.
The pseudo-first order model describes the adsorption that the change of adsorption rate over time is directly proportional to the difference between equilibrium and current adsorption capacity.39 Its linear form can be expressed as the following:
| | |
ln(qe − qt) = ln(qe) − k1t
| (3) |
where
qt and
qe are the adsorption capacities in mg g
−1 at equilibrium and at time
t in min, and
k1 is the rate constant of adsorption in min
−1, respectively. The
qe and
k1 can be determined from the slopes and intercepts of ln(
qe −
qt) against the
t plots, as shown in
Fig. 4b. The resulted kinetic parameters for GO and PRGO with different reduction degree are presented in
Table 1. The results show that the linear correlation coefficients (
R2) are among 0.537 to 0.686, indicating the pseudo-first order model cannot be used to describe the adsorption process.
Table 1 Parameters of pseudo-first order and pseudo-second order kinetic models
| |
Parameters |
GO |
PRGO-1 |
PRGO-2 |
PRGO-3 |
| Pseudo-first order |
qe (mg g−1) |
1.35 |
1.64 |
1.35 |
1.91 |
| k1 (min−1) |
3.0 × 10−3 |
4.0 × 10−3 |
4.0 × 10−3 |
4.0 × 10−3 |
| R2 |
0.613 |
0.537 |
0.686 |
0.683 |
| Pseudo-second order |
qe (mg g−1) |
4.88 |
11.1 |
15.0 |
21.9 |
| V0 (mg (g−1 min−1)) |
0.237 |
1.45 |
3.98 |
7.87 |
| k2 (g (mg−1 min−1)) |
1.0 × 10−2 |
1.2 × 10−2 |
1.8 × 10−2 |
1.6 × 10−2 |
| R2 |
0.999 |
0.999 |
0.999 |
0.999 |
On the other hand, the pseudo-second order model is generally used to depict the chemisorption process with a rate-limiting step,39 which can be represented as equation:
| |
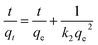 | (4) |
similarly,
qe is the adsorption capacity at equilibrium and
qt is the adsorption amount of dyes at time
t, respectively. The
k2 parameter in g (mg
−1 min
−1) denotes the rate constant of the pseudo-second order kinetic model. Specifically, the value
V0 represents the adsorption rate at initial time,
34 where
All the parameters can be determined by linearly fitting the plots of t/qt against time t, and the results are presented in Table 1. The large linear correlation coefficients, R2 > 0.999, suggest that the OII absorbed on GO and PRGO follows the pseudo-second order kinetic model. More importantly, the equilibrium adsorption capacity of PRGO-3 with solar light exposing duration of 72 h is 4.5-fold higher than that of GO suspensions. And the initial adsorption rate of PRGO-3 is even enhanced by 33 folds, in accord with the experimental results shown in Fig. 4a.
The intraparticle diffusion is also used to reveal the possible diffusion mechanism, which describes the successive processes of dyes diffusion through the boundary layer, intraparticle diffusion and adsorption on the surfaces of GO and PRGO.40 The equation could be expressed as the following form:
where
kpi is the intraparticle diffusion rate constant of stage
i in mg (g
−1 min
−1/2), and
Ci in mg g
−1 is the intercept of stage
i, which gives the information about the thickness of the boundary layer. That is the larger the
Ci, the greater the boundary-layer effect.
40 Fig. 4d presents the plots of
qt vs. t1/2 and their fitting lines, the calculated parameters have been presented in
Table 2. Even though the correlation coefficients are pretty low, two distinguished stages still can be determined. The first sharp stage can be attributed to external diffusion that is the diffusion of dyes through the solution to the bare or restored sp
2 π-conjugated sites on the surface of monolayer nanosheets. The second stage is intra-particle diffusion, which describes the diffusion of dyes to the small sp
2 structures surrounding by oxygen-containing functional groups. As presented in
Table 2, the diffusion rates of
kp1 for GO and PRGO are much higher that
kp2, while the
C1 are slightly lower than
C2. This phenomenon indicates that in the beginning a large amount of adsorbed sites are availability on the GO and PRGO surface, resulting in the high adsorption rate. This results are also coincide with the rapidly increased adsorption in the first 5 min, as shown in
Fig. 4a. In the second step, the adsorption rate was controlled by the rate of the dyes transported from the bare-sp
2 sites to the sp
2 sites surrounding by oxygen-containing functional groups. The steric effect of functional groups and electrostatic repulsion between functional groups and anionic dyes results in the extremely slow adsorption rates. Furthermore, either in the first stage or in the second stage, the
Ci values increases with the increasing of duration time under solar light, which can be attributed to the increasing of sp
2 π-conjugated structures during photoreduction process.
Table 2 Parameters of interparticle diffusion kinetic model
| |
Parameters |
GO |
PRGO-1 |
PRGO-2 |
PRGO-3 |
| First stage |
kp1 (mg (g−1 min−1/2)) |
9.8 × 10−1 |
9.3 × 10−1 |
1.1 × 100 |
1.0 × 10−1 |
| C1 (mg g−1) |
1.70 |
6.54 |
9.53 |
17.0 |
| R2 |
0.817 |
0.471 |
0.774 |
0.973 |
| Second stage |
kp2 (mg (g−1 min−1/2)) |
1.9 × 10−2 |
1.8 × 10−2 |
2.2 × 10−2 |
3.0 × 10−2 |
| C2 (mg g−1) |
4.17 |
10.3 |
14.2 |
20.9 |
| R2 |
0.636 |
0.930 |
0.848 |
0.852 |
3.3 Adsorption isotherm
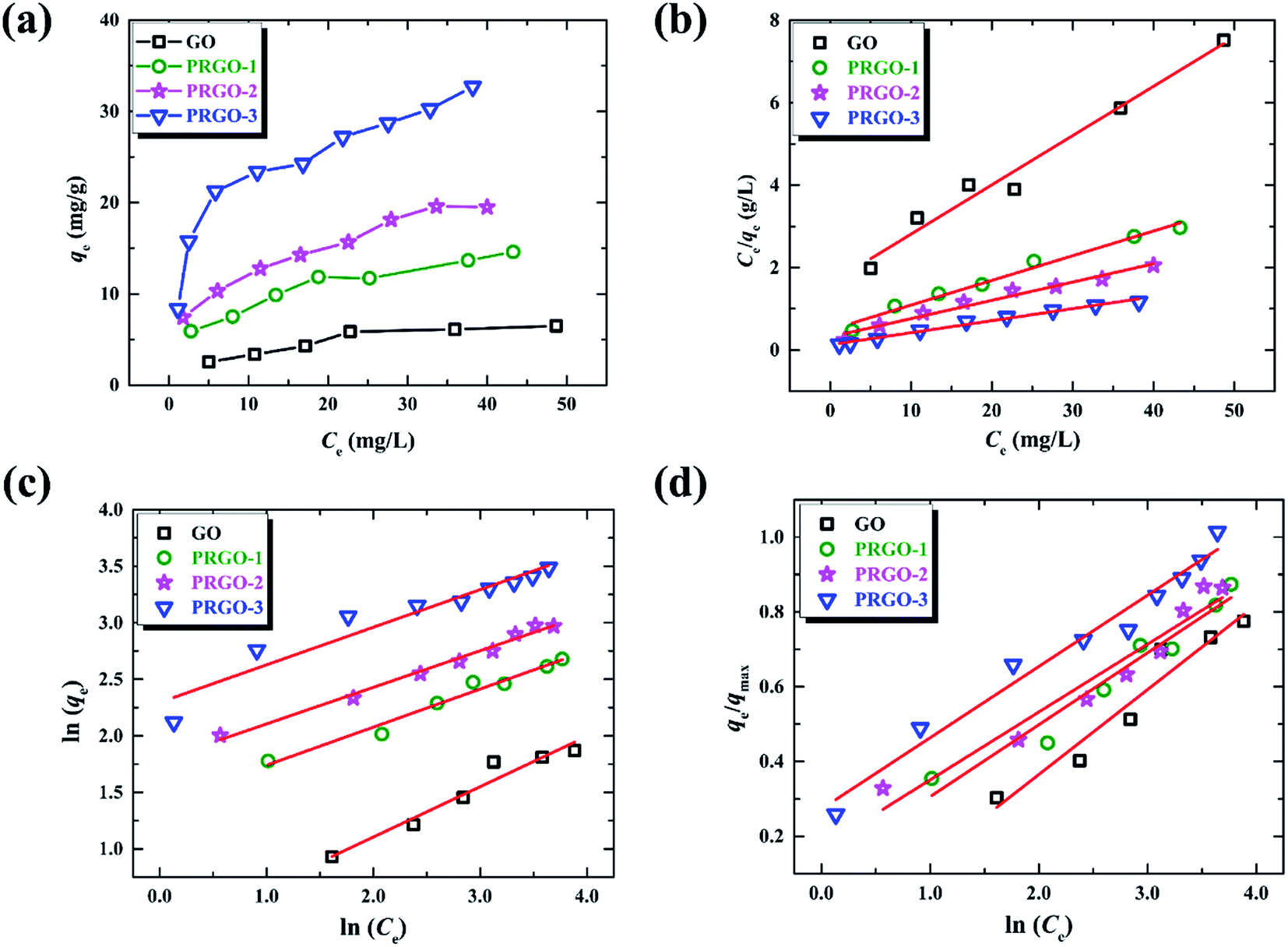
The adsorption isotherm here describes the adsorption capacity of OII dye adsorbed per unit weight of GO or PRGO materials (qe, mg g−1), and also the relationship between the adsorption capacity and the dyes concentrations at equilibrium condition (Ce, mg L−1). Adsorption isotherm can provide in-depth information about adsorption mechanisms, surface properties and affinity of an adsorbent towards an adsorbate. The adsorption of OII dye on GO or PRGO with different initial concentrations have been investigated. The adsorption capacities as well as the equilibrium concentrations have been determined after shaking the mixed aqueous with 24 h under room temperature (22 °C), as shown in Fig. 5a. Apparently, PRGO displays much larger adsorption capacity than GO. Furthermore, as the increasing of reduction degree, the capacity was distinctly improved. On the other hand, as the increasing of the equilibrium concentrations (associating with the increasing of initial concentrations), the adsorption capacities tend to saturation. To determine the maximum adsorption capacity and the mechanistic parameters, three most commonly used models are adopted to analyze the experimental data: Langmuir, Freundlich and Temkin isotherm models.
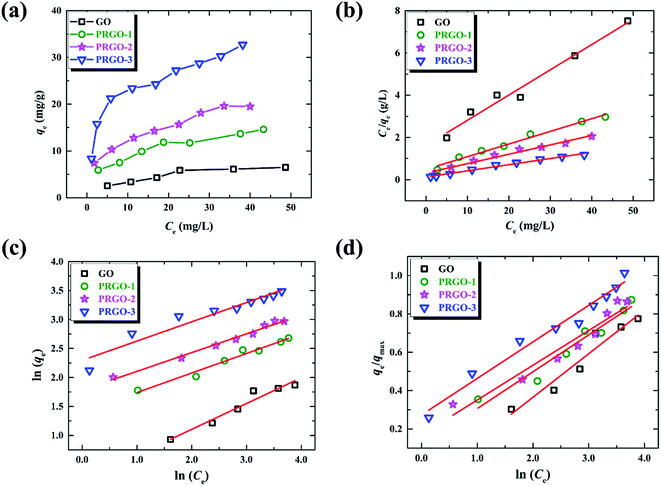 |
| | Fig. 5 (a) Adsorption isotherms of OII on GO and PRGO-1, 2, 3, respectively. (b)–(d) are the plots of Langmuir, Freundlich and Temkin isotherm models for the OII adsorption on GO and PRGO, respectively. | |
The Langmuir model depicts the monolayer adsorption process on a uniform surface with no interaction among the adsorption molecules (dyes).39 It can be expressed as equation as following:
| |
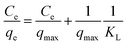 | (7) |
where
qmax is the maximum adsorption capacity in mg g
−1, corresponding to the complete monolayer coverage,
KL is the Langmuir constant in L mg
−1, related to the affinity of the binding sites and energy of adsorption. All the parameters of Langmuir model are presented in
Table 3, which are determined by linearly fitting the plots of
Ce/
qe against
Ce, as shown in
Fig. 5b. The correlation coefficients,
R2, in the range of 0.969 to 0.985 indicate that the experimental data are in good agreement with the Langmuir model. The maximum adsorption capacity for PRGO-3 is 32.2 mg g
−1, which is about 4-fold larger than GO with that of 8.36 mg g
−1. The increased
KL values hint that the deeper the reduction, the stronger the dye adsorbed on materials. Another fundamental characteristic of Langmuir model is the separation factor
RL, given by:
| |
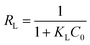 | (8) |
where
C0 is the highest initial dye concentration used in the study (67 mg L
−1).
RL represents the isotherm to be either irreversible (
RL = 0), favorable (0 <
RL < 1), linear (
RL = 1), or unfavorable (
RL > 1).
40 The small
RL values in the
Table 3 indicate that the adsorption of OII dye on both GO and PRGO are the favorable processes. The smallest
RL for PRGO-3 strongly suggests that increasing the initial concentration will improve the adsorption.
Table 3 Parameters of the Langmuir, Freundlich and Temkin models
| |
Parameters |
GO |
PRGO-1 |
PRGO-2 |
PRGO-3 |
| Langmuir |
qmax (mg g−1) |
8.36 |
16.7 |
22.6 |
32.2 |
| kL (L mg−1) |
7.4 × 10−2 |
1.2 × 10−1 |
1.4 × 10−1 |
2.3 × 10−1 |
| RL |
0.17 |
0.11 |
0.10 |
0.061 |
| R2 |
0.970 |
0.979 |
0.969 |
0.985 |
| Freundlich |
1/n |
0.445 |
0.337 |
0.325 |
0.243 |
| KF (L mg−1) |
1.24 |
4.07 |
5.94 |
13.0 |
| R2 |
0.938 |
0.969 |
0.985 |
0.910 |
| Temkin |
ΔQ (kJ mol−1) |
10.8 |
12.8 |
12.9 |
13.5 |
| KT (L mg−1) |
0.677 |
1.81 |
2.55 |
4.19 |
| R2 |
0.923 |
0.944 |
0.931 |
0.969 |
On the contrast to the monolayer adsorption of Langmuir model, the Freundlich model is based on the assumption of multilayer adsorption,41 described as:
| |
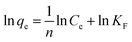 | (9) |
here
KF is an indicator of the adsorption capacity, and 1/
n is an empirical heterogeneity factor, providing an indication of the deviation from linearity of adsorption. 1/
n quantifies the favorability of adsorption and the degree of heterogeneity of the surface. When 1/
n < 1, it suggests favorable adsorption process and also a normal Langmuir isotherm.
34 The corresponding values have been displayed in
Table 3 by fitting the experimental data, as shown in
Fig. 5c. The values of 1/
n lower than 1 further evidence the adsorption of OII on GO and PRGO are favorable Langmuir adsorption processes.
The Temkin is also used to analyze the data to uncover thermodynamic information, which assumes that the heat of adsorption of dyes in the layer decreases linearly with coverage,42 represented by:
| |
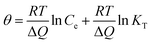 | (10) |
where
θ =
qe/
qmax, is the fraction coverage.
R is the universal gas constant and equals to 8.314 J (mol
−1 K
−1),
T is the temperature and equals to 295 K (22 °C) in the experiments. Δ
Q is the vitiation of adsorption energy in J mol
−1 and
KT is the Temkin equilibrium constant in L mg
−1. The Temkin parameters have been determined by linearly fitting the plots of
qe/
qmax against ln
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ce
Ce, as shown in
Fig. 5d, and the results have been presented in
Table 3. The applicable
R2 illustrate the isotherm data can be fitted with the Temkin model. The values of Δ
Q are all positive, suggesting that the adsorption reaction is exothermic in nature.
42 Therefore high temperature is not favorable for anionic dye adsorption. Both Δ
Q and
KT increase with the reduction degree, revealing that the adsorption is more favorable for the PRGO materials.
3.4 Desorption and recycle of PRGO
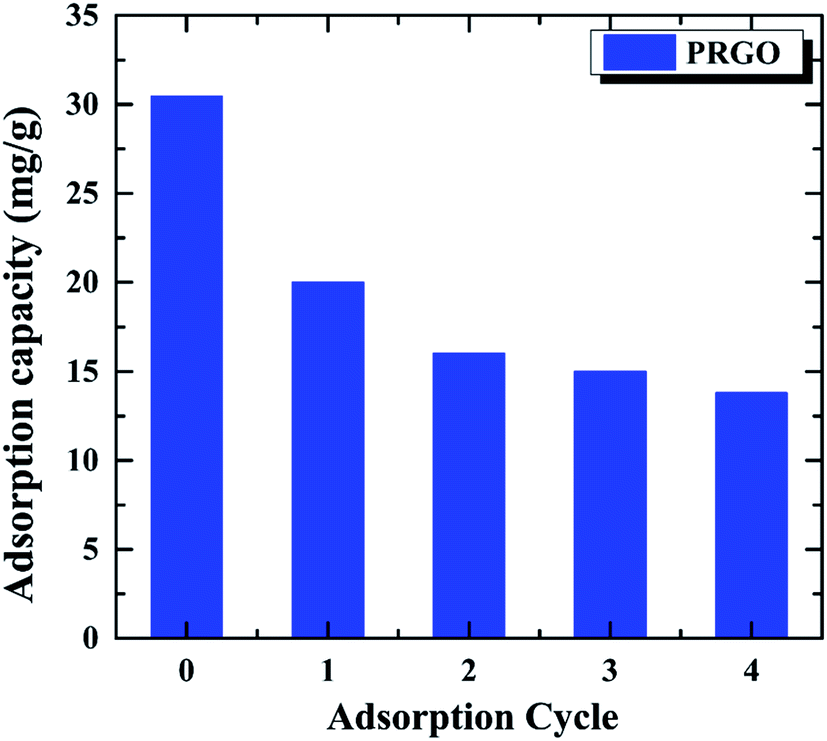
The recycling and regenerating ability of an adsorbent is crucial for its practical application, an excellent adsorbent not only has the high adsorption capacity, but also can be recycling to save energy. In order to investigate the recycling ability of PRGO as the adsorbent, the OII solution with the initial concentration of 60 mg L−1 was used to study the adsorption–desorption cycle experiment for five times. After the OII adsorbed on PRGO, it was washed by EG and deionized water for three times, respectively. The recycled adsorbed behavior are shown in Fig. 6. It can be found that the adsorption capacity of 30.4 mg g−1 for the initial adsorption decreases to 13.8 mg g−1 after four cycles, without considering the loss of PRGO materials during the cycling. Although the adsorption decrease rapidly, it is tending towards stability and keeping at ∼13.5 mg g−1 at last. The rapid decrease during recycling may be attributed to the strong π–π stacking interaction between the aromatic structure of dye and the sp2 framework of PRGO surface, preventing desorption of dyes from the PRGO during simple washing processes.15,43 After the adsorption sites with strong interaction have been saturated by dyes, the adsorption capacity stands at a plateau level. The still adsorption may arise from the adsorption sites with remaining oxygen-containing functional groups surrounding, resulting in the relatively weak adsorption and the facile desorption.
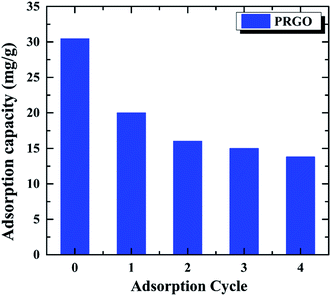 |
| | Fig. 6 Recycling of PRGO on the removal of OII dye. The initial OII concentration was 60 mg L−1. | |
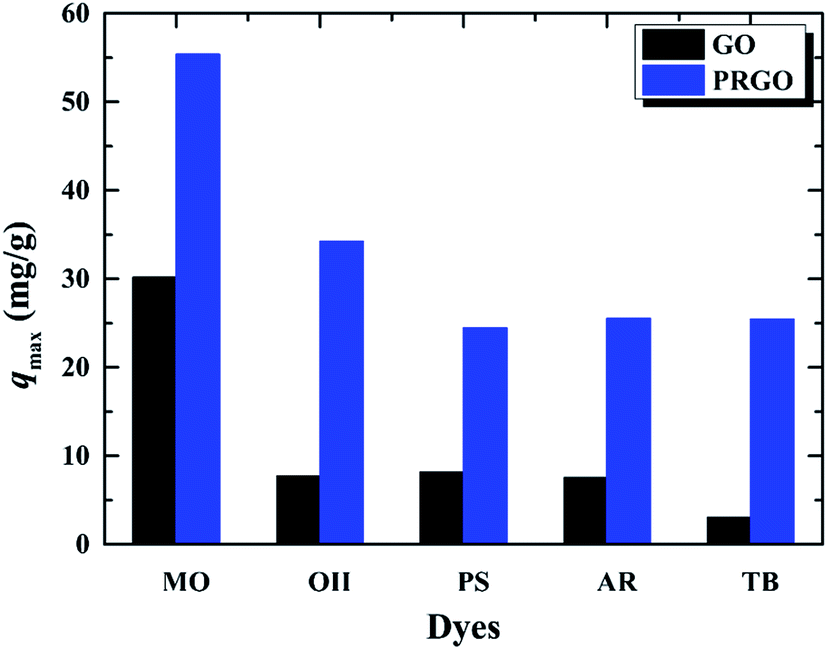
3.5 Adsorption of other anionic dyes
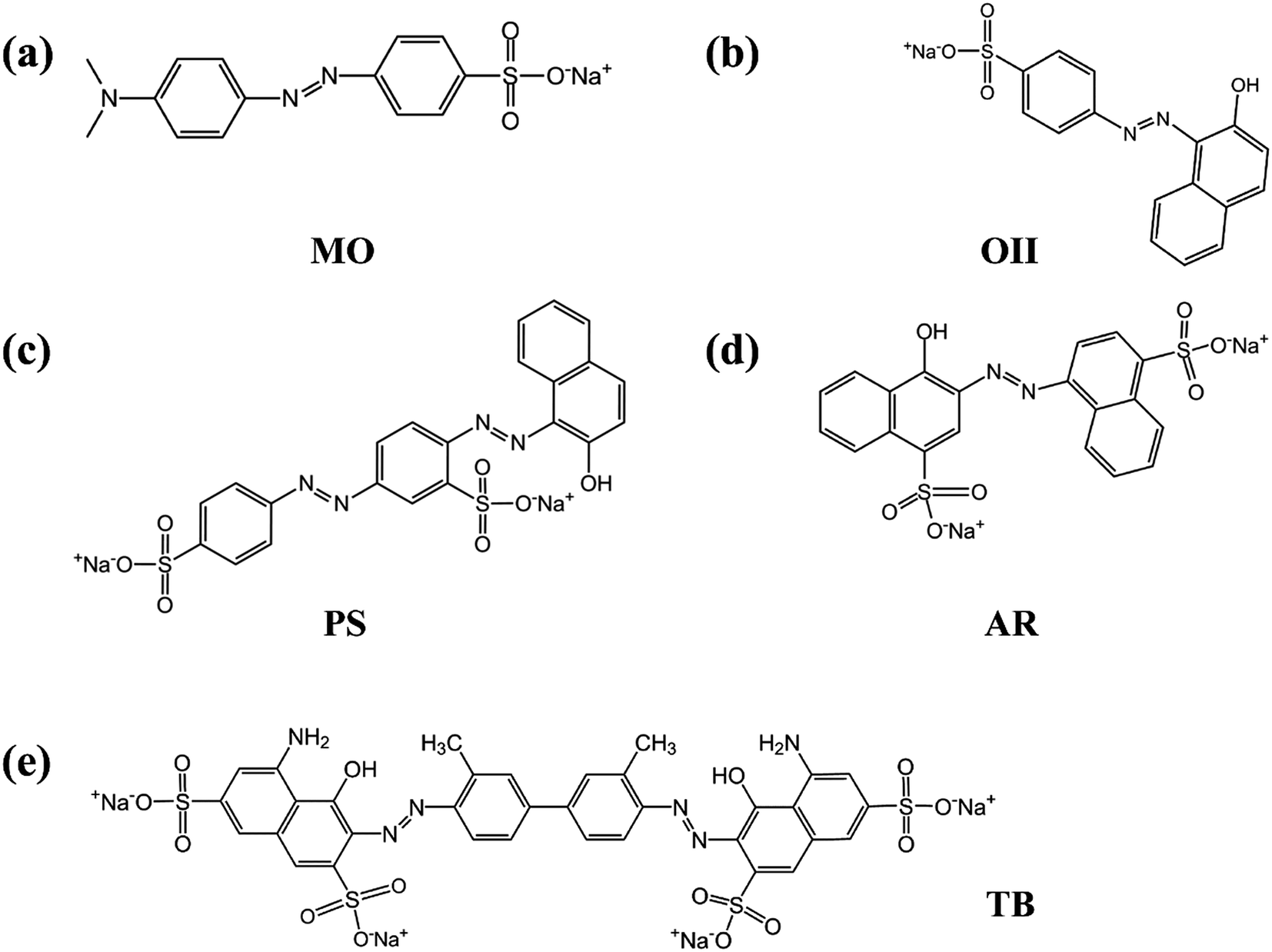
In order to verify whether the PRGO can be applied as a broad and efficient adsorbent to remove the anionic dyes with different aromatic structures and negative charges, the adsorption capacities of GO and PRGO have also been measured towards to MO, PS, AR, and TB, respectively, as shown in Fig. 7. Apparently, for all these anionic dyes, the PRGO shows significantly improved adsorption capacities. Among them, TB presents the maximum enhanced folds of ∼8. The reason may be distinguished into two aspects. The TB with four negative charges, as presented in Fig. 8, will emerge the strongest repulsion interaction between TB and negative GO surface, leading to the weakest absorption capacity on GO. On the other hand, the TB has six aromatic rings, which makes strong π–π stacking interaction between TB and PRGO surface, resulting in large adsorption capacity on PRGO. The PS and AR with similar chemical structures and negative charges perform almost the same results. The enhanced folds for them are 3 and 3.4, respectively. The repulsion interaction between them and GO, and also the π–π stacking interaction between them and PRGO are both moderate. In the experiments, MO shows the largest adsorption capacity on GO among dyes, and also the smallest enhanced folds of ∼2. It can be attributed to the weak repulsion interaction between MO and GO, as well as the relative simple chemical structure makes MO adsorbing on the residual sp2 framework of GO efficiently.
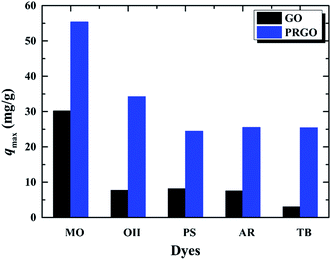 |
| | Fig. 7 Adsorption capacities of five anionic dyes on GO and PRGO with irradiation time of 72 h, respectively. | |
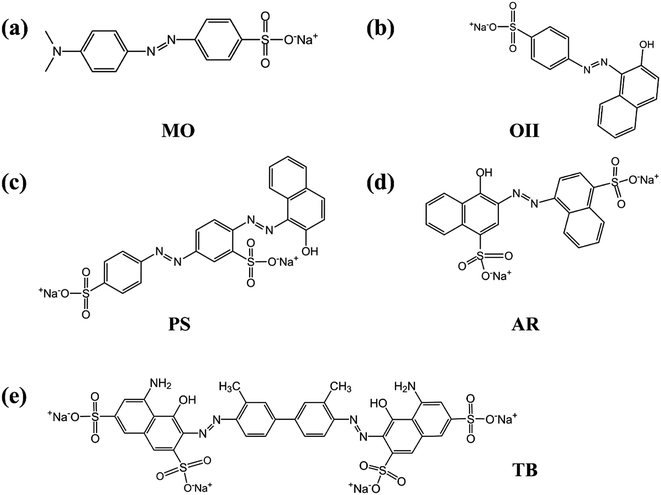 |
| | Fig. 8 (a)–(e) are the chemical structures of MO, OII, PS, AR, and TB anionic dyes used in the experiments. | |
4. Conclusion
In this study, a green and cost-free method based on the solar light irradiation was developed to prepare PRGO suspension in order to improve the adsorption capacity on anionic dyes in water treatment. Characterizations by UV-visible absorption, Raman, AFM, SEM, FTIR and XPS proved the successful reduction of GO suspension under solar light irradiation. Three commonly used kinetic models and three isotherm models were applied to reveal the adsorption mechanisms of OII dye adsorbed on GO and PRGO, respectively. The well-fitted pseudo-second order kinetic model as well as simultaneously adopted Langmuir, Freundlich and Temkin models reveal that the adsorption is a monolayer and exothermic process. The maximum adsorption capacities of PRGO for five anionic dyes, MO, OII, PS, AR, and TB are determined, which is much higher than that of the pristine GO, with the enhanced folds of ∼2, 4, 3, 3.4, and 8, respectively. The significant improved adsorption is attributed to the decrease of charge density of GO surface by removing the oxygen-containing functional groups, and the increase of π–π stacking interaction between aromatic dyes and PRGO by restoring the sp2 π-conjugation frameworks during solar reduction. The enhanced adsorption capacity strong suggests that PRGO reduction by solar irradiation has great potential to be used as a promising adsorbent for the high efficiency removal of anionic dyes in the wastewater treatment.
Conflicts of interest
The authors declare that no competing financial conflicts exist.
Acknowledgements
The project is sponsored by the National Key Research and Development Program of China (Grant No. 2017YFA0304203), the Natural Science Foundation of China (Grant No. 11404200, 61527824, 11374196, 61605104 and U1510133), the Program for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT13076).
References
- J. Eliasson, Nature, 2015, 517, 6 CrossRef CAS PubMed.
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed.
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172–184 CrossRef CAS PubMed.
- H. Sun, L. Cao and L. Lu, Nano Res., 2011, 4, 550–562 CrossRef CAS.
- M. R. S. Kebria, M. Jahanshahi and A. Rahimpour, Desalination, 2015, 367, 255–264 CrossRef CAS.
- J. Labanda, J. Sabaté and J. Llorens, J. Membr. Sci., 2009, 340, 234–240 CrossRef CAS.
- G. C. Silva, V. S. T. Ciminelli, A. M. Ferreira, N. C. Pissolati, P. R. P. Paiva and J. L. López, Mater. Res. Bull., 2014, 49, 544–551 CrossRef CAS.
- C. Li, H. Zhu, X. She, T. Wang, F. She and L. Kong, RSC Adv., 2016, 6, 67242–67251 RSC.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- R. K. Upadhyay, N. Soin and S. S. Roy, RSC Adv., 2014, 4, 3823–3851 RSC.
- S. Chowdhury and R. Balasubramanian, Adv. Colloid Interface Sci., 2014, 204, 35–56 CrossRef CAS PubMed.
- J.-H. Deng, X.-R. Zhang, G.-M. Zeng, J.-L. Gong, Q.-Y. Niu and J. Liang, Chem. Eng. J., 2013, 226, 189–200 CrossRef CAS.
- G. Z. Kyzas, E. A. Deliyanni and K. A. Matis, J. Chem. Technol. Biotechnol., 2014, 89, 196–205 CrossRef CAS.
- J. Wang and B. Chen, Chem. Eng. J., 2015, 281, 379–388 CrossRef CAS.
- X. Wang, S. Huang, L. Zhu, X. Tian, S. Li and H. Tang, Carbon, 2014, 69, 101–112 CrossRef CAS.
- G. K. Ramesha, A. V. Kumara, H. B. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS PubMed.
- L. Sun, H. Yu and B. Fugetsu, J. Hazard. Mater., 2012, 203–204, 101–110 CrossRef CAS PubMed.
- J. Xiao, W. Lv, Z. Xie, Y. Tan, Y. Song and Q. Zheng, J. Mater. Chem. A, 2016, 4, 12126–12135 CAS.
- V. Eswaraiah, S. S. Jyothirmayee Aravind and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 6800 RSC.
- M. Mohandoss, S. S. Gupta, A. Nelleri, T. Pradeep and S. M. Maliyekkal, RSC Adv., 2017, 7, 957–963 RSC.
- W. He, C. Qin, Z. Qiao, G. Zhang, L. Xiao and S. Jia, Carbon, 2016, 109, 264–268 CrossRef CAS.
- Z. Qiao, C. Qin, Y. Gao, G. Zhang, R. Chen, L. Xiao and S. Jia, Sci. Rep., 2015, 5, 14441 CrossRef CAS PubMed.
- H. T. Zhou, C. B. Qin, R. Y. Chen, G. F. Zhang, L. T. Xiao and S. T. Jia, Appl. Phys. Lett., 2014, 105, 153301 CrossRef.
- R. Y. N. Gengler, D. S. Badali, D. Zhang, K. Dimos, K. Spyrou, D. Gournis and R. J. D. Miller, Nat. Commun., 2013, 4, 2560 Search PubMed.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 2, 032183 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- H. Feng, R. Cheng, X. Zhao, X. Duan and J. Li, Nat. Commun., 2013, 4, 1539 CrossRef PubMed.
- V. Le Borgne, H. Bazi, T. Hayashi, Y. A. Kim, M. Endo and M. A. El Khakani, Carbon, 2014, 77, 857–867 CrossRef CAS.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, J. Phys. Chem. C, 2011, 115, 19280–19286 CAS.
- Q. Zhang, H. Zheng, Z. Geng, S. Jiang, J. Ge, K. Fan, S. Duan, Y. Chen, X. Wang and Y. Luo, J. Am. Chem. Soc., 2013, 135, 12468–12474 CrossRef CAS PubMed.
- J. N. Tiwari, K. Mahesh, N. H. Le, K. C. Kemp, R. Timilsina, R. N. Tiwari and K. S. Kim, Carbon, 2013, 56, 173–182 CrossRef CAS.
- X. H. Li, J. S. Chen, X. Wang, M. E. Schuster, R. Schlogl and M. Antonietti, ChemSusChem, 2012, 5, 642–646 CrossRef CAS PubMed.
- Y. Matsumoto, M. Koinuma, S. Y. Kim, Y. Watanabe, T. Taniguchi, K. Hatakeyama, H. Tateishi and S. Ida, ACS Appl. Mater. Interfaces, 2010, 2, 3461–3466 CAS.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- R. J. W. E. Lahaye, H. K. Jeong, C. Y. Park and Y. H. Lee, Phys. Rev. B, 2009, 79, 125435 CrossRef.
- Y. Qi, M. Yang, W. Xu, S. He and Y. Men, J. Colloid Interface Sci., 2017, 486, 84–96 CrossRef CAS PubMed.
- X. Mi, G. Huang, W. Xie, W. Wang, Y. Liu and J. Gao, Carbon, 2012, 50, 4856–4864 CrossRef CAS.
- M. Heidarizad and S. S. Şengör, J. Mol. Liq., 2016, 224, 607–617 CrossRef CAS.
- Y. Wang, Y. Xie, Y. Zhang, S. Tang, C. Guo, J. Wu and R. Lau, Chem. Eng. J., 2016, 114, 258–267 CAS.
- S. Bai, X. Shen, X. Zhong, Y. Liu, G. Zhu, X. Xu and K. Chen, Carbon, 2012, 50, 2337–2346 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Wenjun Heab,
Zhixing Qiaoab,
Guofeng Zhangab,
Ruiyun Chenab,
Yan Gaoab,
Liantuan Xiao*ab and
Suotang Jiaab
*ab,
Wenjun Heab,
Zhixing Qiaoab,
Guofeng Zhangab,
Ruiyun Chenab,
Yan Gaoab,
Liantuan Xiao*ab and
Suotang Jiaab