DOI:
10.1039/C7RA10016G
(Paper)
RSC Adv., 2017,
7, 50546-50554
Sulfur-doped CoFe2O4 nanopowders for enhanced visible-light photocatalytic activity and magnetic properties†
Received
8th September 2017
, Accepted 18th October 2017
First published on 30th October 2017
Abstract
Magnetically recoverable sulfur-doped CoFe2O4 nanopowders were used as photocatalyst for degradation of oxytetracycline (OTC) under visible light in aqueous solution. S doped CoFe2O4 nanopowders synthesized by a simple calcining process were characterized by XRD, TEM, VSM, FT-IR, UV-vis DRS and XPS. The results showed that introducing of S element in CoFe2O4 crystal structure narrowed its band gap and increased visible light absorption. 0.3 S-CoFe2O4 exhibited the highest magnetic property and photocatalytic activity. The maximum photocatalytic conversion ratio of OTC reached 90.4% at pH = 8.5 after 5 h irradiation. During photocatalytic process, seven intermediate products were detected, and based on that a detailed degradation path way of OTC was proposed.
1. Introduction
Antibiotics have been widely used as antibacterial agents in human and veterinary medicine for the treatment of bacterial infections.1,2 Because of their poor adsorption by human beings or live stocks and chemical stability, some of these antibiotics, such as tetracyclines (TCs), have been accumulating in the aquatic environment.3,4 Antibiotic residues are potentially dangerous to ecosystems, especially to human health through drinking water and the food chain. Among the TCs family, oxytetracycline (OTC) is one of the most frequently detected tetracyclines in water bodies and sediments in many countries, which is difficult to removal by conventional water treatment process.5,6
Photocatalytic oxidation technology has been considered as an effective way to remove the organic pollutants in aqueous solution, due to its energy saving, environmental friendly performance and no secondary pollution.7 Many photocatalysts have been developed for antibiotic degradation under solar and visible light.8,9 However, the recoverability and photocatalytic activity under visible light of common photocatalysts need to be further improved for practical application.
Spinel ferrites (MFe2O4, M = metal cation) can be easily collected due to their great magnetic property,10–12 and excited under visible light irradiation due to their relatively narrow bandgap.11,13 Therefore, it is considered as potential photocatalyst in eliminating various organic pollutants.14,15 Among these materials, CoFe2O4 attracts more attention due to its moderate magnetic performance at room temperature and good chemical stability.16 It is often composited with other semiconductor materials such as TiO2, WO3 and C3N4, or conducting materials such as graphene, oxidized graphene and carbon nanotube, in order to improve the photocatalytic degradation performance.17–19 However, the synthetic process of these composites is complex, and the magnetism may be reduced due to introduce of nonmagnetic components. Non-metal doping is an effective way to modify common photocatalysts, such as TiO2, which has been reported in many literatures.20,21 But there are few references about improving photocatalytic performance of CoFe2O4 by non-metal doping. Although Cao etal. have reported that the magnetic property of CoFe2O4 is improved by doped nonmetal sulfur,22 the effects of doped S element on photocatalytic performance has not been involved.
Therefore, this work attempted to realize photocatalytic degradation of OTC under visible light using magnetically recoverable S doped CoFe2O4 nanopowders as photocatalysts. The S doped CoFe2O4 nanopowders were synthesized and characterized by XRD, TEM, VSM, FT-IR, UV-vis DRS and XPS. The effects of S on photocatalytic activity and magnetic property were investigated to optimize the concentration of S. The photocatalytic degradation of OTC at different initial pH was studied, as well as the photocatalytic stability. Then the intermediate products during photocatalytic process were determined to deduce the degradation path way of OTC. At last, the photocatalytic mechanism was further investigated based on research results.
2. Experiment section
2.1 Synthesis
CoFe2O4 and sulfur-doped CoFe2O4 (S-CoFe2O4) nanoparticles have been synthesized as reported in literature.22 To obtain CoFe2O4 nanopowders, 4.848 g Fe(NO3)3·9H2O, 1.746 g Co(NO3)2·6H2O were dissolved in dimethyl formamide (DMF, 15 mL), then this solution was put into an alumina crucible and calcined at 700 °C for 2 hours with the heating rate of 1 °C min−1 in air. S-CoFe2O4 nanopowders were synthesized using the same method by adding thiourea in the DMF solution. The concentration of thiourea was in a range of 0.1–0.5 mol L−1 to adjust S content in S-CoFe2O4.
2.2 Characterization
The phases of obtained CoFe2O4 and S doped CoFe2O4 nanopowders were identified by X-ray diffraction (XRD, Rigaku, D/MAX2500V) using Cu-Kα (λ = 0.15418 nm) radiation. The XRD data were collected in the range of 20–80° with a scanning rate of 8° min−1. Vibrating sample magnetometer (VSM, China Trend Limited, Lakeshore 7407) was used to measure the magnetic properties of all samples.
The morphologies of both CoFe2O4 and 0.3 mol L−1 S doped CoFe2O4 (0.3S-CoFe2O4) nanopowders were observed using the transmission electron microscope (TEM, Jeol, JEM-2100F) equipped with energy-dispersive X-ray (EDX). The Fourier transfer infrared spectroscopy (FT-IR) of S-CoFe2O4 nanopowders was recorded using an infrared spectrometer (FT-IR, Nicolet, AVATAR370), and compared with CoFe2O4 nanopowders. The UV-vis diffuse reflectance spectra of both nanopowders were recorded by UV-vis spectrometer (DRS, Persee, TU-1901) with an integrating sphere.
The binding energies of Co, Fe, S of CoFe2O4 and 0.3S-CoFe2O4 were determined by X-ray photoelectron spectroscopy (XPS, Thermofisher Scientific, ESCALAB 250Xi). The zeta potential (Zeta) of sample was measured with a nanoparticles and potential analyzer (Zeta, Malvern lnstruments, Zetasizer 3000HS).
2.3 Photocatalytic degradation measurement
The photocatalytic experiments were carried out as described in our previous work.5 The experimental procedure was as follows: 70 mg of photocatalyst was first suspended in 70 mL OTC aqueous solution (80 mg L−1, C22H24N2O9·HCl: 95% purity, Aladdin Chemistry Co., Ltd, Shanghai, China) in a quartz tube, then stirred at 900 rpm continuously for 1 h before light irradiation to ensure that the photocatalyst surface was saturated with OTC molecules. The mixture was irradiated under a 300 W iodine tungsten lamp, which located 25 cm away from the quartz tube. All the tests were performed at room temperature. At given time intervals of irradiation, 5 mL of the suspension was filtered and the residual concentration of OTC was monitored using a UV-Vis spectrophotometer (Cary 5000 UV-Vis-NIR, Agilent) at 365 nm. The photocatalytic conversion ratio (PCR) of OTC was estimated according to the formula:| | |
PCR = (1 − Ct/C0) × 100%
| (1) |
where Ct (mg L−1) is the solution concentration of OTC after irradiation at any time, and C0 is the initial concentration of OTC solution. C can be calculated through the corresponding absorbance according to the standard curve. The total organic carbon (TOC) in OTC aqueous solution after photocatalytic was measured using TOC Analyzer Total (TOC, Analytik Jena Ag, N/C 2100).
The photocatalytic stability of 0.3S-CoFe2O4 nanopowders was investigated by recycling tests at room temperature. In these experiments, 70 mg of photocatalysts were suspended in 70 mL OTC (initial concentration of 80 mg L−1) aqueous solution, and the pH of solution was adjusted to 8.5 for best performance. After each cycle of test, 0.3S-CoFe2O4 photocatalyst was separated from OTC solution in an external magnetic field and washed with water, dried at 120 °C for 4 h, then used for the next cycle. The process was repeated five times.
2.4 Photocatalytic degradation mechanism of OTC
The photocatalyst degradation intermediate products of OTC were detected by high performance liquid phase chromatography coupled with low resolution electrospray ionization mass spectrometry (LC/MSD SL, Agilent 1100 Series G1956B LC/MSD SL).
In order to determine ˙OH production, 200 mM NaOH and 60 mM terephthalic acid (TA) were added in the photocatalytic suspended solution. This solution was stirred in the dark for 1 h, then illuminated under a high pressure 300 W iodine tungsten lamp for 3 h. After centrifuged, the solution was analyzed on a fluorescence spectrometer (LS 55, PerkinElmer, UK) to record the fluorescence signal of the generated highly fluorescent product TAOH (excitation, λexc = 312 nm; the scanning speed, v = 200 nm min−1; widths of the excitation slit and the emission slit, d = 5 nm).
The trapping experiment for hydroxyl radicals and holes on the photocatalytic photodegradation of OTC were carried out by using tert-butanol (t-BuOH, 0.1 M, radical scavenger) and ethylenediaminetetraacetic acid disodium salt (EDTA-2Na, 0.1 M, hole scavenger) as scavenger. For study the role of superoxide radicals, carbon tetrachloride (CCl4 1 mM) was added under sparging N2. The followed experimental process was carried out as mentioned above.
3. Results and discussions
3.1 Characterization of S-CoFe2O4 nanopowders
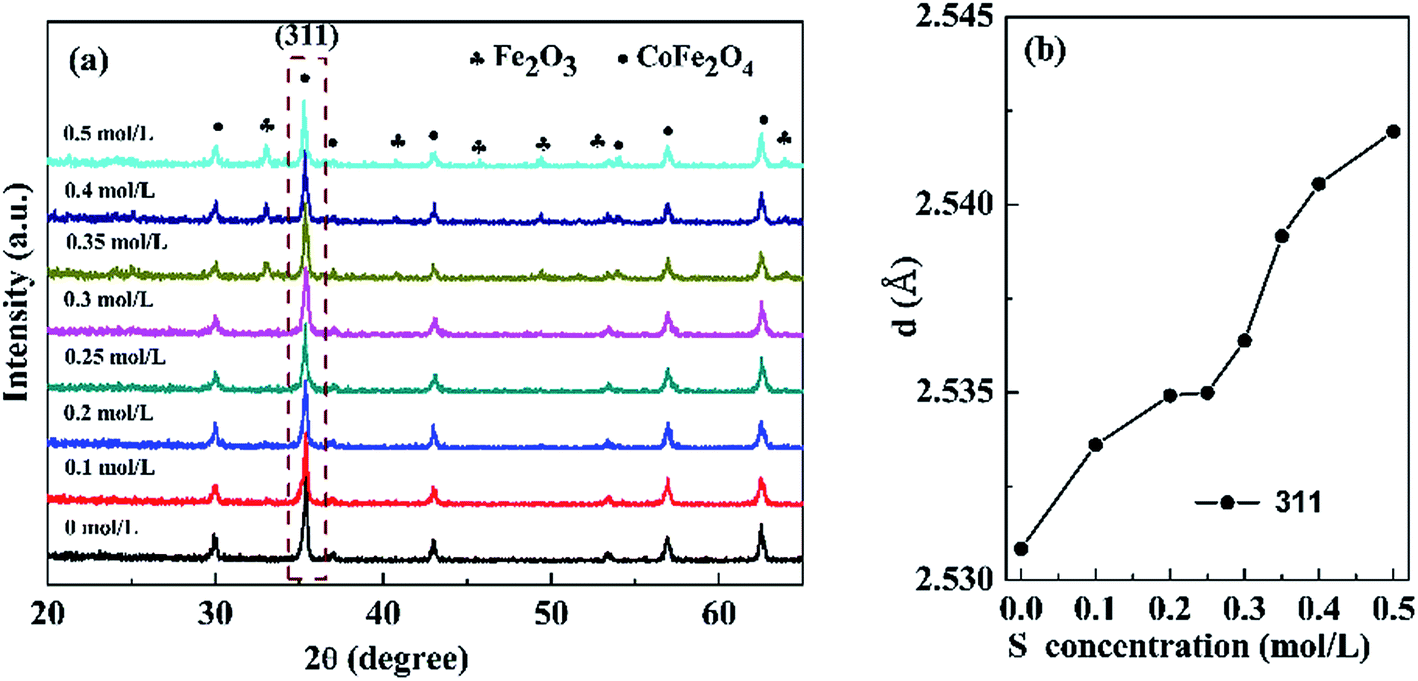
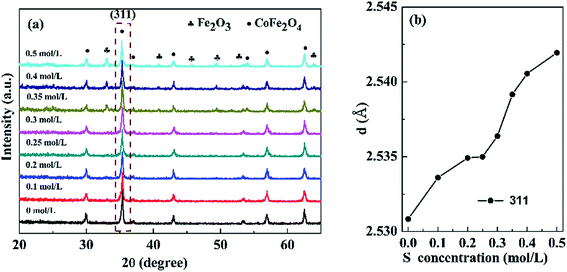
3.1.1 XRD analysis. Fig. 1(a) presents the XRD patterns of the nanopowders with different S concentrations. When the S concentration increased for 0 to 0.3 mol L−1, the samples exhibited single cubic phase of CoFe2O4 (JCPDS card no. 22-1086). However, further increasing the S concentration to 0.5 mol L−1, an extraneous α-Fe2O3 (JCPDS card no. 33–0664) impurity phase was observed. Fig. 1(b) shows that the d value of (311) diffraction peak slightly increases with increasing S concentration due to expansion of the lattice. The results indicated that S was effectively incorporated into the CoFe2O4 nanopowders.
 |
| | Fig. 1 (a) XRD patterns of the sample with different S concentrations, (b) the d value of (311) diffraction peaks varied with S concentrations. | |
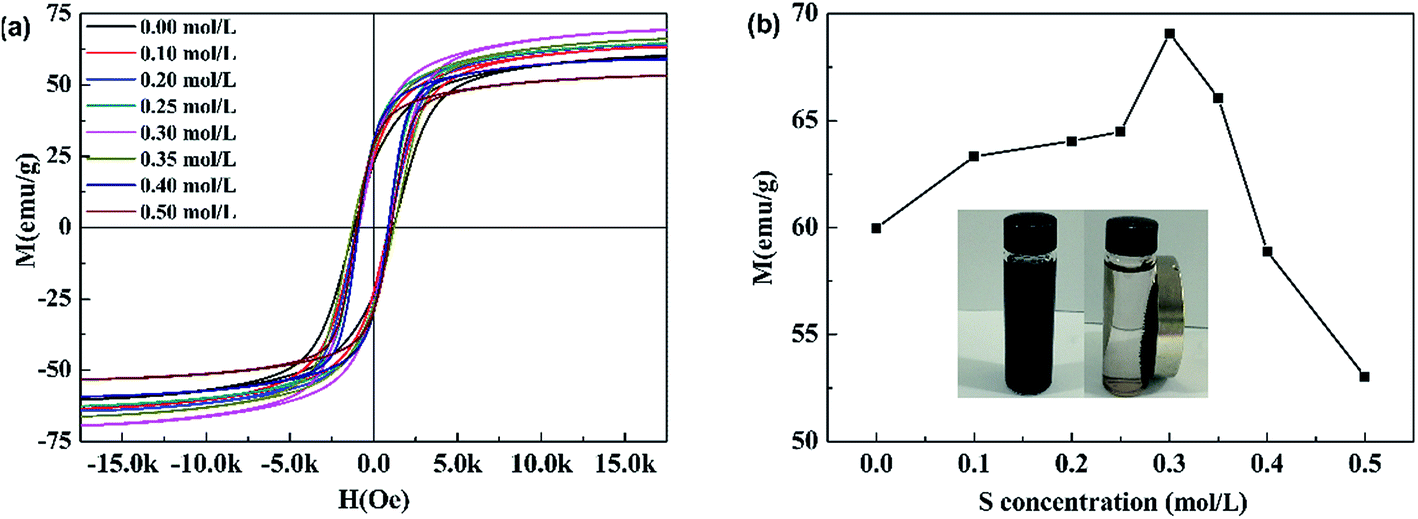
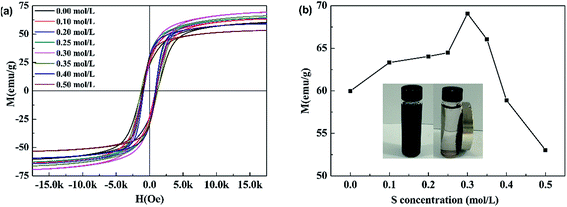
3.1.2 Magnetism. Fig. 2(a) shows the magnetic hysteresis (M − H) loops of pure CoFe2O4 and S doped CoFe2O4. All samples followed typical room-temperature ferromagnetic behavior. The S concentration dependence of the magnetic saturation (MS) of all products is given in Fig. 2(b). The MS monotonically increased with S concentration rising from 0 to 0.3 mol L−1, then decreased with further rising S concentration to 0.5 mol L−1 0.3S-CoFe2O4 exhibited the highest magnetic saturation (MS) of 69.1 emu g−1. The inset camera photo shows that 0.3S-CoFe2O4 is easily separated from the solution by a magnet.
 |
| | Fig. 2 (a) Magnetization curves at 296 K, (b) S concentration-dependence of MS for the corresponding samples, inset shows the magnetic separation process of S-CoFe2O4. | |
As reported in literature,22 the increase of MS was attributed to the migration of Co2+ ions from the B site to the A site, and hence the migration of Fe ions from the A site to the B site in small amounts caused by the inclusion of thiourea during the calcining process. In addition, the increase of average crystalline size (calculated using Scherrer equation form XRD, shown in Table 1) might also cause the increase of MS. The decrease of MS with S concentration above 0.3 mol L−1 was mainly caused by the presence of non-magnetic α-Fe2O3, which led to the relative reduction of magnetic CoFe2O4.
Table 1 The average crystalline size calculated by Scherrer equation with different S concentration
| S content (mol L−1) |
0 |
0.1 |
0.2 |
0.25 |
0.3 |
| Average crystalline size (nm) |
29.06 |
32.39 |
35.65 |
39.54 |
42.09 |
3.1.3 TEM analysis. Fig. 3(a) and (b) shows the representative TEM image of CoFe2O4 and 0.3S-CoFe2O4 nanopowders, respectively. It can be seen that the pure CoFe2O4 and 0.3S-CoFe2O4 are composed of uniform microspheres with size of 20–40 nm, but agglomerations also existed.
 |
| | Fig. 3 TEM image of (a) CoFe2O4, (b) 0.3S-CoFe2O4, HTEM image of (c) 0.3S-CoFe2O4, (d) EDX-mapping of 0.3S-CoFe2O4 nanoparticles. | |
Fig. 3(c) shows a typical HAADF-STEM image of 0.3S-CoFe2O4 nanopowders, the distribution of each element has been studied by STEM mapping (shown in Fig. 3(d)). Co, Fe, O were all distributed uniformly throughout the 0.3S-CoFe2O4 nanoparticle. However, the signal points of S element are fewer, because of the lower concentration of S than other elements. It seems that elemental S may thus have been absorbed near gap or edge of CoFe2O4.
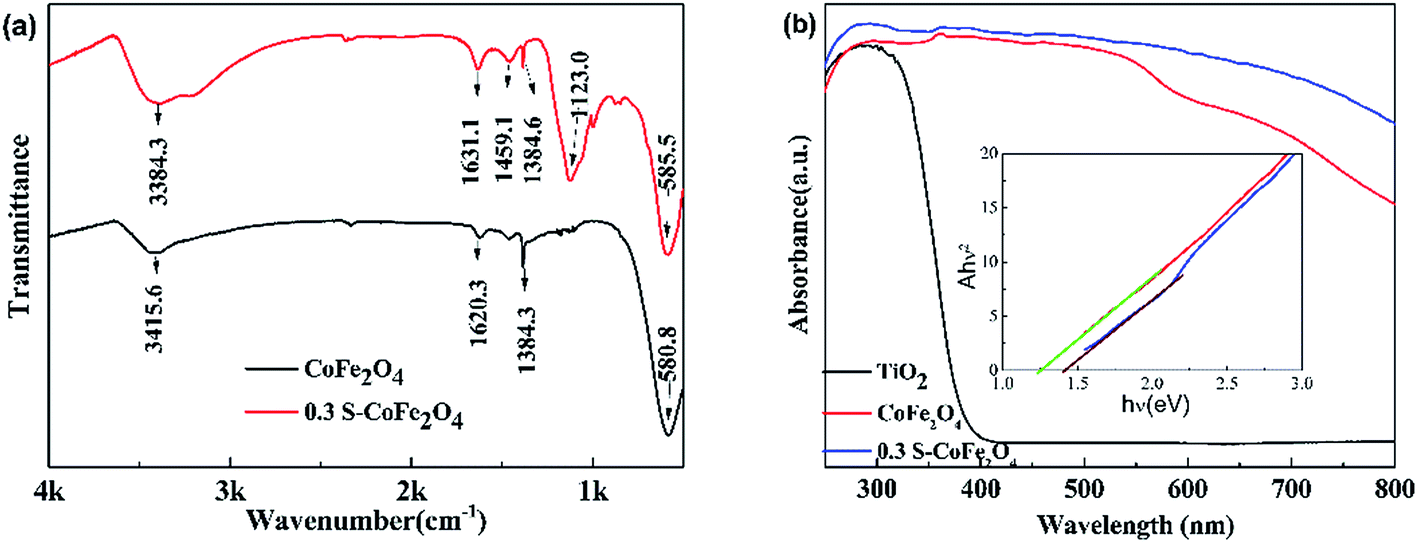
3.1.4 FT-IR and UV-vis absorption spectra. FT-IR spectra and UV-vis absorption spectra of pure CoFe2O4 and 0.3S-CoFe2O4 are shown in Fig. 4(a) and (b), respectively. In Fig. 4(a), compared with pure CoFe2O4, the spectrum of 0.3S-CoFe2O4 shows a distinguishing divisive band at about 1123 cm−1, which assigns to S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or S–O bond in sulfated oxides.23,24 In Fig. 4(b), it can be seen that both CoFe2O4 and 0.3 S-CoFe2O4 exhibit stronger absorption in visible light region compared with TiO2 nanopowders (P25). As expected, 0.3S-CoFe2O4 has stronger light absorption intensity than pure CoFe2O4 at the range of 540–800 nm. From the inset in Fig. 4(b), the band gap of S-CoFe2O4 is around 1.25 eV, bit less than that of CoFe2O4 (1.4 eV). The stronger absorption in visible light area and narrowed bandgap of 0.3S-CoFe2O4 were attributed to the introduction of S atoms in the lattice of CoFe2O4.
O or S–O bond in sulfated oxides.23,24 In Fig. 4(b), it can be seen that both CoFe2O4 and 0.3 S-CoFe2O4 exhibit stronger absorption in visible light region compared with TiO2 nanopowders (P25). As expected, 0.3S-CoFe2O4 has stronger light absorption intensity than pure CoFe2O4 at the range of 540–800 nm. From the inset in Fig. 4(b), the band gap of S-CoFe2O4 is around 1.25 eV, bit less than that of CoFe2O4 (1.4 eV). The stronger absorption in visible light area and narrowed bandgap of 0.3S-CoFe2O4 were attributed to the introduction of S atoms in the lattice of CoFe2O4.
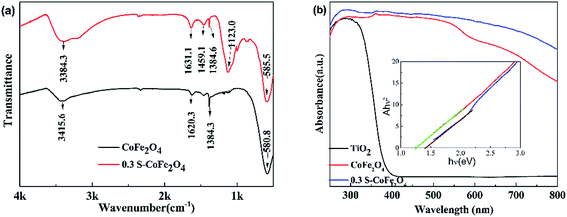 |
| | Fig. 4 (a) FT-IR spectra (b) UV-vis DRS of CoFe2O4 and 0.3S-CoFe2O4, the inset shows plot of transferred Kubelka–Munk vs. energy of the light absorbed. | |
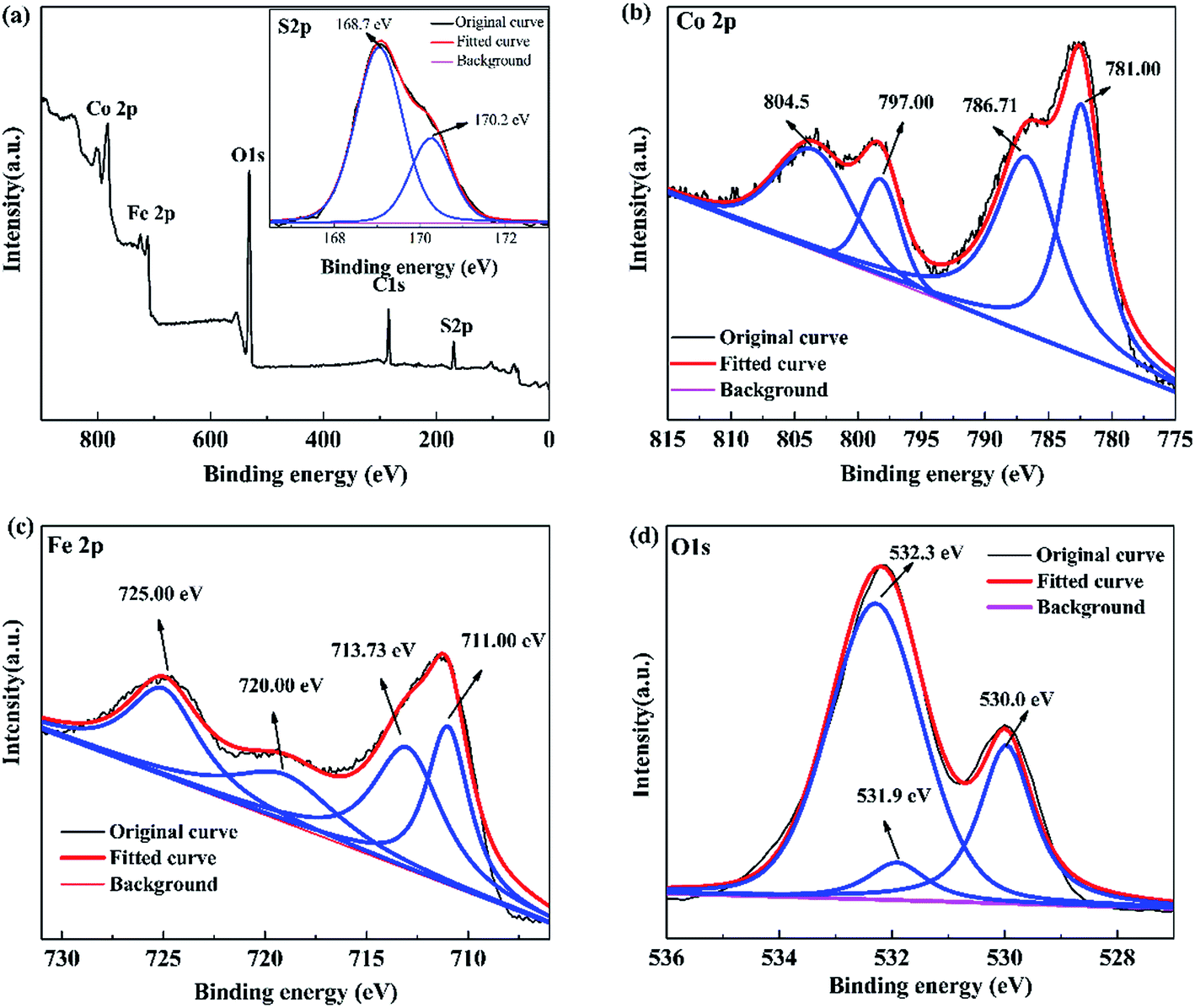
3.1.5 X-ray photoelectron spectroscopy. XPS is used to analyze the elemental composition of the prepared 0.3S-CoFe2O4. The full scanned XPS spectra demonstrate the existing of elements of Co, Fe, S and O (Fig. 5(a)), which is corresponding to the EDX analysis. The high-resolution XPS spectrum of S 2p is shown inset in Fig. 5(a), two individual peaks at 168.7 eV and 170.2 eV, which can be attributed to the oxidized S species,22,23 corresponding to the result from FT-IR. Combined with the increase of the interplanar spacing from XRD patterns, it suggested that S atoms might enter into the octahedral gap of CoFe2O4 lattice instead of replacing O atoms as expected.
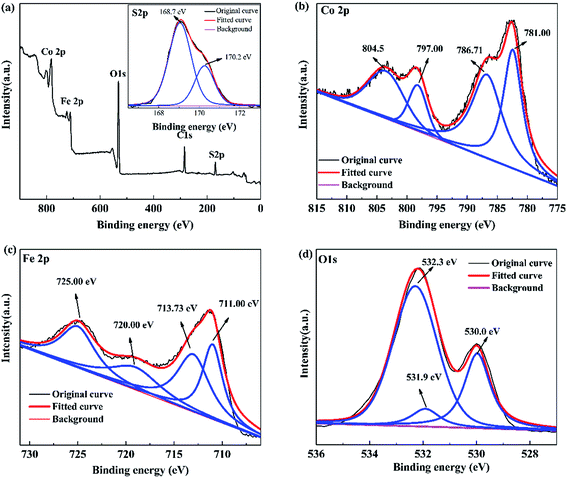 |
| | Fig. 5 XPS spectra of 0.3S-CoFe2O4: (a) XPS survey, inset shows S 2p XPS spectra; (b) Co 2p, (c) Fe 2p, and (d) O 1s XPS spectra. | |
As discerned from the XPS spectrum of Co 2p (Fig. 5(b)), two main peaks at 781.00 eV and 797.00 eV correspond to Co 2p3/2 and Co 2p1/2, respectively.25 Besides, two satellite peaks at 786.71 eV and 804.50 eV relate to shake-up type of Co 2p3/2 and Co 2p1/2 edge, confirming the +2 oxidation state of the Co elements in 0.3S-CoFe2O4 nanopowders. In the XPS spectrum of Fe 2p (Fig. 5(c)), the peaks at 710.88 eV and 724.28 eV correspond to Fe 2p3/2 and Fe 2p1/2, respectively. The characteristic satellite peak between these two peaks at 717.73 eV suggests the +3 oxidation state of the Fe element.26 The corresponding O 1s spectrum is given in Fig. 5(d), peaks at 530.0 and 532.3 eV represent the lattice oxygen of metal oxides and chemisorbed oxygen (M − OH).27,28
3.2 Photocatalytic degradation activity tests
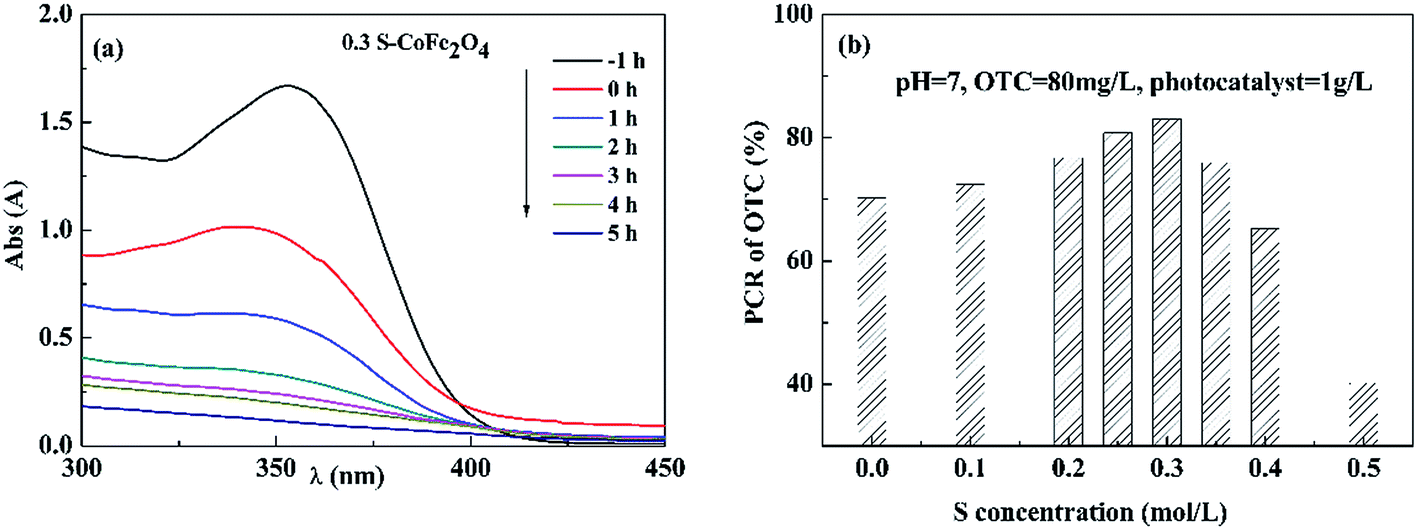
3.2.1 Effect of S concentration on photocatalytic activity. Fig. 6(a) shows typical UV-vis spectra changes of OTC solution with reaction time using 0.3S-CoFe2O4 as photocatalyst. The result showed that the adsorption peak of OTC around 354 nm significantly reduced with increasing irradiation time in 5 h. Fig. 6(b) gives the dependence of S concentration on PCR of OTC under visible light. The PCR firstly increased when S concentration was increased from 0 to 0.3 mol L−1, then decreased with further increasing S concentration to 0.5 mol L−1 due to the formation of α-Fe2O3. 0.3S-CoFe2O4 exhibited the highest photocatalytic activity under visible light, the PCR of OTC reached to 83% after 5 h irradiation. And in our tests (Fig. 2(b)), the 0.3S-CoFe2O4 also exhibited highest magnetic property. Thus, 0.3S-CoFe2O4 was selected in the subsequent experiments.
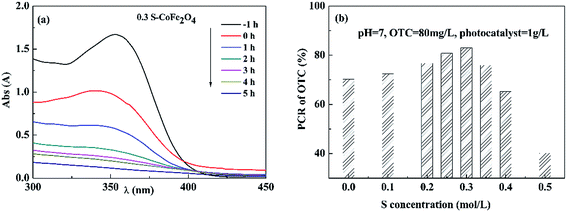 |
| | Fig. 6 (a) Typical UV-vis spectra changes of OTC solution with reaction time using 0.3S-CoFe2O4 as photocatalyst under visible light; (b) effect of S content on PCR of OTC after 5 h irradiation under visible light. | |
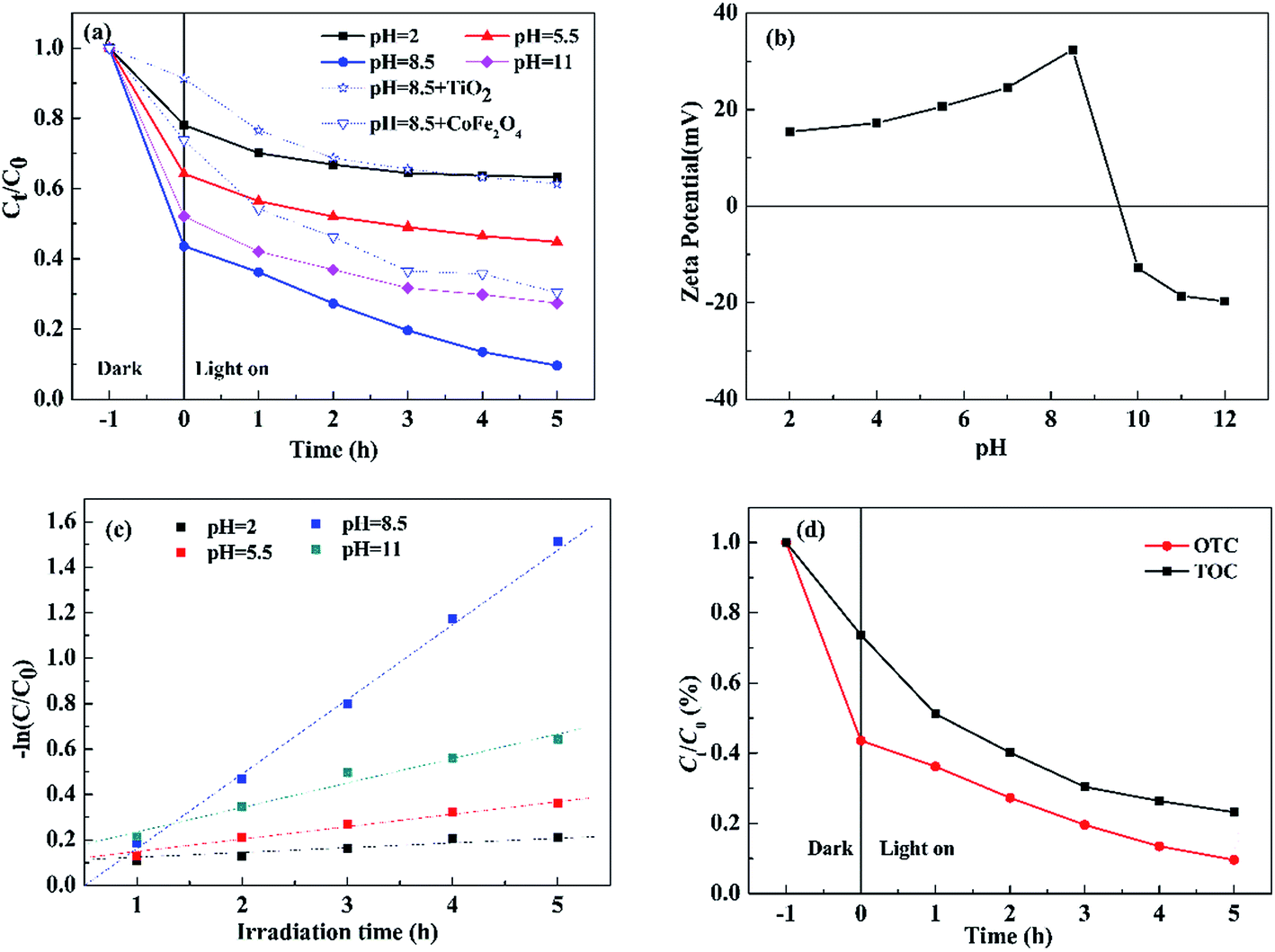
3.2.2 Effect of pH on photocatalytic degradation of OTC. Since the species of OTC are changed critically with pH values (pH = 2: H3OTC+; pH = 5.5: H3OTC±; pH = 8.5: HOTC− and pH = 11.0: OTC2−),29 the effect of initial pH on photocatalytic degradation of OTC was investigated under visible light (Fig. 7(a)). For comparison, the results using TiO2 (P25) and pure CoFe2O4 as photocatalysts are also given in Fig. 7(a). The photocatalytic degradation of OTC increased with pH from 2.0 to 8.5, but significantly reduced at pH = 11.0. It could be well explained by pH-dependent zeta potential of 0.3S-CoFe2O4 shown in Fig. 7(b). From the figure, the PZC of 0.3S-CoFe2O4 is between 9 to 10. At pH = 11 or at pH = 2, 5.5, electro-static repulsion is produced due to the similarly charged nanoparticle surface and species of OTC, which reduces surface absorption and leads to lower degradation efficiency. At pH = 8.5, OTC acts as HOTC−, which can be easily adsorbed on the positive charged surface, and increases the conversion of OTC.
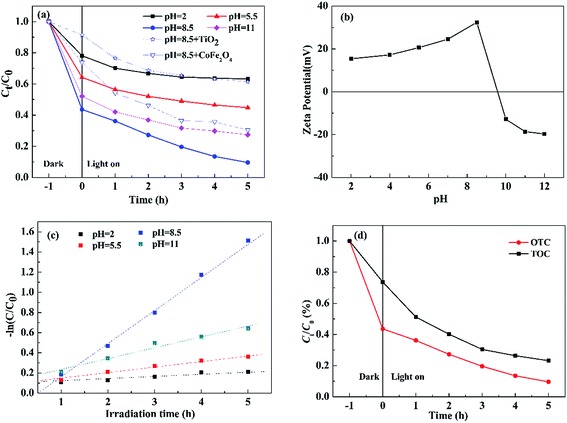 |
| | Fig. 7 (a) Effect of pH on photocatalytic degradation of OTC over 0.3S-CoFe2O4; (b) zeta-potential of aqueous dispersions of 0.3S-CoFe2O4 at different pH values; (c) Pseudo-first-order kinetic model fits the degradation kinetic curves of OTC; (d) remove efficiency changes of TOC and OTC with irradiation time in the presence of 0.3S-CoFe2O4 under visible light. | |
Moreover, the photocatalytic degradation kinetic process of OTC could be well described by the pseudo-first-order kinetic model (−ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C/C0 = kt) as shown in Fig. 7(c), where C0 is the initial concentration of OTC, C is the concentration of OTC at time t, and k is the rate constant. As listed in Table 2, at pH = 8.5 the highest rate constant is 0.3358 h−1, while the maximum conversion ratio of OTC reached to 89.4%.
C/C0 = kt) as shown in Fig. 7(c), where C0 is the initial concentration of OTC, C is the concentration of OTC at time t, and k is the rate constant. As listed in Table 2, at pH = 8.5 the highest rate constant is 0.3358 h−1, while the maximum conversion ratio of OTC reached to 89.4%.
Table 2 The pseudo-first order kinetic equations, rate constants (k) and regression coefficients (R2) of photocatalytic degradation of OTC over S-CoFe2O4 at different pH
| pH |
The first order kinetic equation |
k (h−1) |
R2 |
| 2 |
−ln(C/C0) = 0.0284t |
0.0284 |
0.955 |
| 5.5 |
−ln(C/C0) = 0.0572t |
0.0572 |
0.974 |
| 8.5 |
−ln(C/C0) = 0.3358t |
0.3358 |
0.997 |
| 11 |
−ln(C/C0) = 0.1073t |
0.1073 |
0.964 |
Fig. 7(d) presents the variation of the total organic carbon (TOC) in aqueous solution with irradiation time, it can be observed that the TOC concentration declined with the degradation of OTC, and dropped to 23.2% after 5 h irradiation. The result suggested that the complete degradation of OTC to mineral end-products may be achieved using photocatalytic technology.
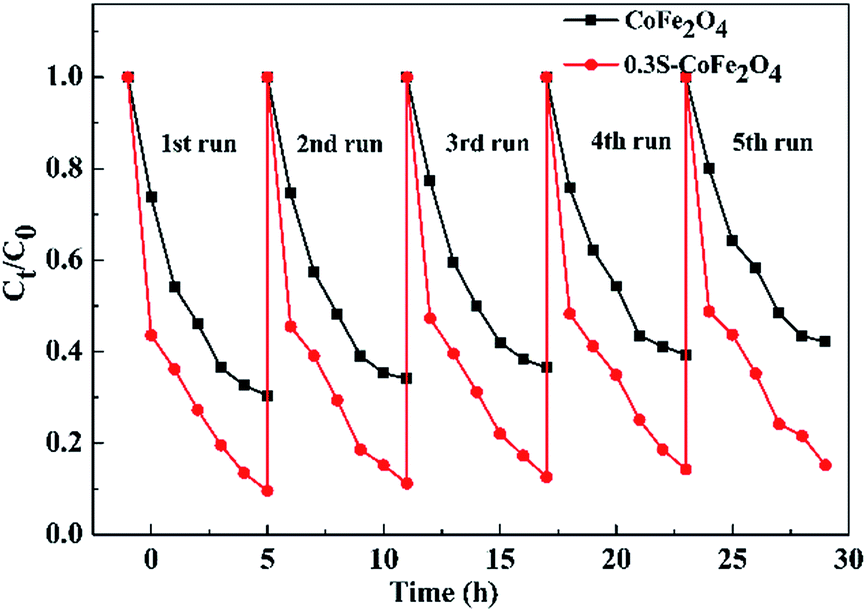
3.2.3 Reusability of 0.3S-CoFe2O4. The catalytic stabilities of CoFe2O4 and 0.3S-CoFe2O4 are tested upon separation of the photocatalysts magnetically, and their performance for each cycle is shown in Fig. 8. The PCR of OTC decreased from 90.4% to 84.8% after 5 cycles for 0.3S-CoFe2O4, while from 70.6% to 57.8% for CoFe2O4. This result showed that 0.3S-CoFe2O4 exhibited better photocatalytic stability and recoverability, because less photocatalyst lost during the separation progress due to its stronger magnetism. The slight decrease in PCR might attribute to the residual catalytic products absorbed on the surface of photocatalyst, which reduced the contact area between catalyst and pollutant.
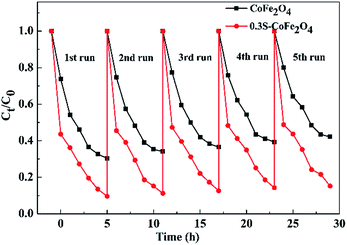 |
| | Fig. 8 Recyclability of 0.3S-CoFe2O4 for OTC degradation. | |
3.3 Photocatalytic degradation pathway of OTC
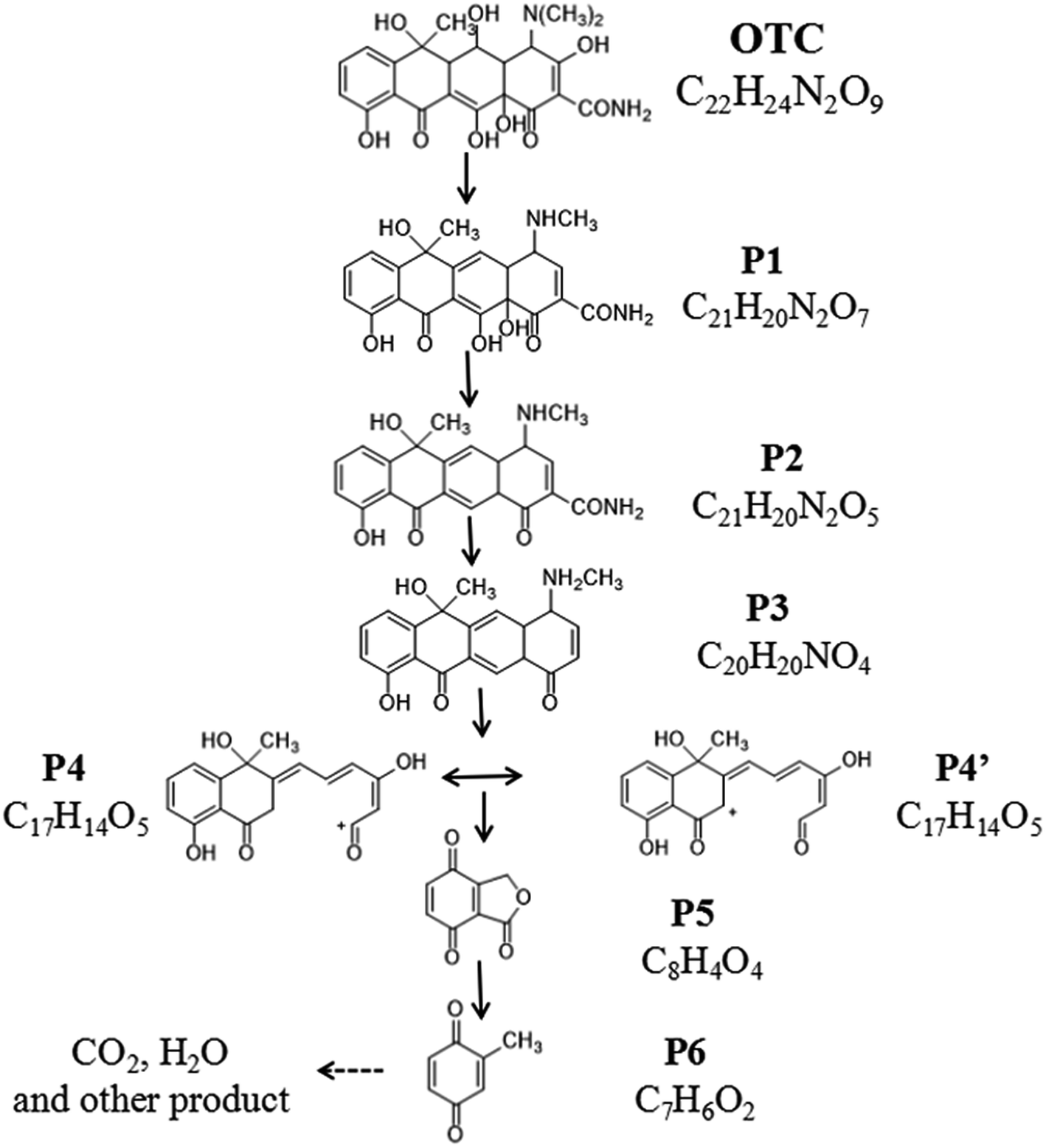
The identification of intermediate products are carried out by LC/MSD SL, and iron spectra at different retention time (RT) of OTC solution after photocatalytic degradation for 1 h, 3 h and 5 h are presented in ESI Scan (Fig. S1†).
The iron spectra at RT = 5.527 and 1.455 min were detected in the solution after 1 h photocatalytic degradation. The main product ions at m/z 461.2 ([M + H]+), m/z 145.0 ([M + Na]+) might be identified as C22H24N2O9 (OTC) and C7H6O4 (P6).30 After 3 h photocatalytic degradation, three irons spectra at RT = 14.749, 13.066 and 1.455 min were detected in the solution. The main product ions at 381.3 ([M + H]+), m/z 339.2 ([M + H]+) and m/z 145.0 ([M + Na]+) might be identified as C21H20N2O5 (P2), C20H20NO4 (P3) and C7H6O4 (P6).31 At RT = 15.444, 12.073 and 1.505 min, four main products at m/z 413.3 ([M + H]+), m/z 299.2 ([M + H]+), m/z 187.0 ([M + Na]+) were detected after 5 h photocatalytic degradation, which might be identified as C21H20N2O7 (P1), C17H15O5 (P4 or P4′) and C8H4O4 (P5).32 Based on the molecular weight and previous report. The possible photocatalytic degradation pathways of OTC are proposed and shown in Fig. 9.
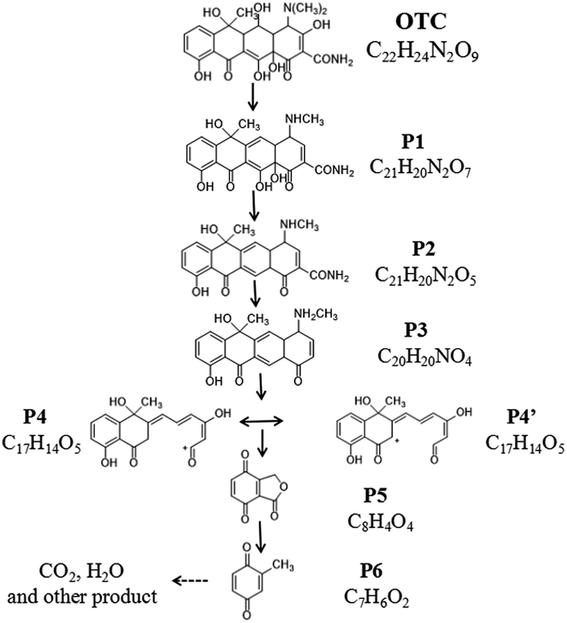 |
| | Fig. 9 Proposed degradation pathways of OTC. | |
3.4 Mechanism of photocatalysis
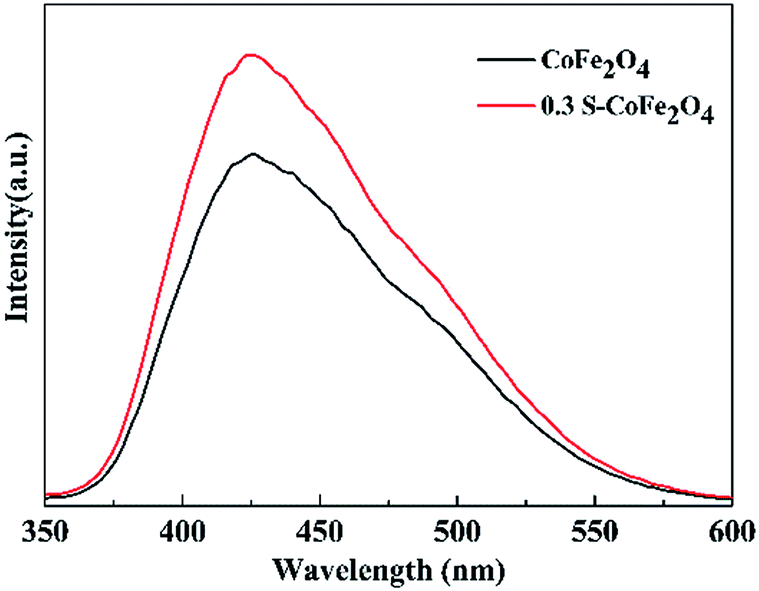
Fluorescence signal intensity of TAOH over CoFe2O4 and 0.3S-CoFe2O4 after visible light irradiation are presented in Fig. 10. The stronger fluorescence signal for 0.3S-CoFe2O4 meant lower level of recombination and the longer lifetimes of the carriers than CoFe2O4 nanoparticles,33 then more valid e−–h+ pairs participated in the radical-chain reaction and more ˙OH groups were generated.
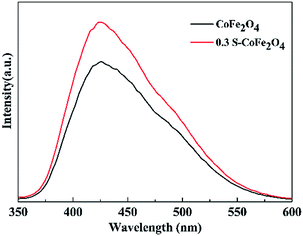 |
| | Fig. 10 Fluorescence signal intensity of TAOH over CoFe2O4 and 0.3S-CoFe2O4 after visible light irradiation. | |
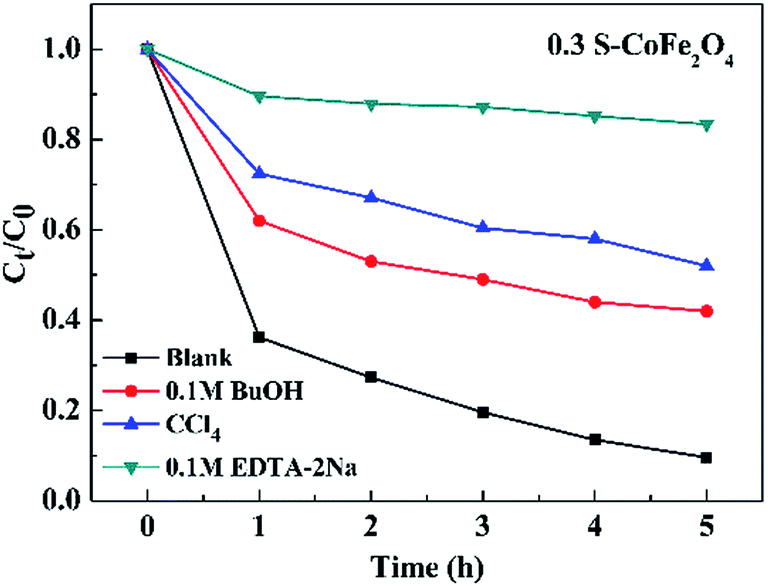
In order to investigate the photocatalytic degradation mechanism of OTC, the trapping experiments of 0.3S-CoFe2O4 were performed to explore the main reactive species. CCl4 (under sparging N2), EDTA-2Na and t-BuOH served as O2˙−, hole (h+) and hydroxyl radical (˙OH) scavenger, respectively. The different influences on photocatalytic degradation were observed in Fig. 11. CCl4 and t-BuOH showed the significant reduce in PCR of OTC, dropped by 42.5% and 32.4% compared with the blank value, while EDTA-2Na almost inhibited the photocatalytic degradation process. The results suggested that photogenerated holes were the key active oxidizing species involved in the photocatalytic process, however O2˙− and ˙OH also participated in the photocatalytic reaction.
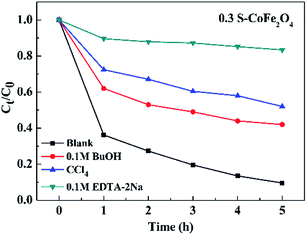 |
| | Fig. 11 Effect of different scavengers on the OTC degradation in the presence of 0.3S-CoFe2O4. | |
The mechanism of OTC degradation can be described as the eqn (2)–(6). Firstly, in the solution, OTC molecules were adsorbed onto the surface of 0.3S-CoFe2O4. Under the visible light irradiation, conduction band electron (e−) and valence band holes (h+) are generated in 0.3S-CoFe2O4 bulk, then diffuse to the surface and react with H2O and O2 molecules adsorbed there to form reactive oxygen species (ROS), such as ˙OH radical and ˙O2−. The ROS and h+ can oxidize most of TC antibiotics to the mineral-end products.
| | |
0.3S-CoFe2O4 + OTC → (0.3S-CoFe2O4) − OTC
| (2) |
| | |
0.3S-CoFe2O4 + hν → 0.3S-CoFe2O4 (e− + h+)
| (3) |
| | |
OTC + ˙OH/˙O2−/h+ → intermediate products → CO2↑ + H2O
| (6) |
Previous experiments showed that S might be doped in the bulk of CoFe2O4 lattice as S6+ and S2−. The substitution of metal ion by S6+ may create an intra-band-gap state between VB and near to the CB edges that encourages the visible-light absorption,20 while the anionic S doping into the lattice structure generates more oxygen defects and shifts the absorption edge in the visible-light region.34 Thus, 0.3S-CoFe2O4 had narrower band gap and higher absorption rate for visible light than pure CoFe2O4. Besides, this impurity band hold on photoinduced e− for more time to reduce the recombination rate, promoting more photoinduced h+ to produce active radicals (hydroxyl and peroxide).
4. Conclusion
Sulfur-doped CoFe2O4 nanopowders with various content of S were synthesized using a simple calcining process. The effects of S doping on magnetism and photocatalytic activity under visible light have been studied, and 0.3S-CoFe2O4 exhibited the best comprehensive performance. The characterization results showed that introduction of S narrowed the band gap of 0.3S-CoFe2O4 and increased the absorption of the visible light. The optimized pH for OTC degradation was 8.5 due to the surface adsorption, and the maximum PCR could reach to 90.4% after 5 h visible light irradiation. 0.3S-CoFe2O4 also showed excellent recoverability, the PCR reached 84.8% at the fifth cycle. Seven intermediate products were detected after photocatalytic degradation, among these, P5 and P6 might suggest a partial ring-ruptured process, indicating the complete degradation of OTC to mineral end-products. Hole isolated from photocatalyst played key role in oxidative degradation of OTC, while ˙O2− and ˙OH also participated the oxidative reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Instrumental Analysis and Research Center of Shanghai University for the experimental assistance. This work was supported by the National Natural Science Foundation of China (No. 51472156, 51702206) and the Science and technology project of Guangdong Province (2013B090600025).
References
- Y. Hong, C. Li, G. Zhang, Y. Meng, B. Yin, Y. Zhao and W. Shi, Chem. Eng. J., 2016, 299, 74–84 CrossRef CAS.
- J. B. Chen, Y. Wang, Y. J. Qian and T. Y. Huang, J. Hazard. Mater., 2017, 335, 117–124 CrossRef CAS PubMed.
- A. J. Watkinson, E. J. Murby, D. W. Kolpin and S. D. Costanzo, Sci. Total Environ., 2009, 407, 2711–2723 CrossRef CAS PubMed.
- N. Barhoumi, H. O. Vargas, N. Oturan, D. Huguenot, A. Gadrie, S. Ammare, E. Brillasd and M. A. Oturanb, Appl. Catal., B, 2017, 209, 637–647 CrossRef CAS.
- R. Li, Y. Jia, J. Wu and Q. Zhen, RSC Adv., 2015, 5, 40764–40771 RSC.
- F. L. Zhang, Q. Y. Yue, Y. Gao, B. Gao, X. Xu, Z. Ren and Y. Jin, Chemosphere, 2017, 182, 483–490 CrossRef CAS PubMed.
- A. Y. Zhang, Y. Y. He, T. Lin and N. H. Huang, J. Hazard. Mater., 2017, 330, 9–17 CrossRef CAS PubMed.
- Q. Chen, S. Wu and Y. Xin, Chem. Eng. J., 2016, 302, 377–387 CrossRef CAS.
- J. Xue, S. Ma, Y. Zhou and Z. Zhang, RSC Adv., 2015, 5, 18832–18840 RSC.
- D. Zhang, X. Pu, Y. Gao and C. Su, Mater. Lett., 2013, 113, 179–181 CrossRef CAS.
- K. Tezuka, M. Kogure and Y. J. Shan, Catal. Commun., 2014, 48, 11–14 CrossRef CAS.
- S. Sun, X. Yang, Y. Zhang, F. Zhang, J. Ding, J. Bao and C. Gao, Prog. Nat. Sci.: Mater. Int., 2012, 22, 639–643 CrossRef.
- F. Miao, Z. Deng, X. Lv and G. Gu, Solid State Commun., 2010, 150, 2036–2039 CrossRef CAS.
- D. Moitra, M. Chandel, B. K. Ghosh and R. K. Jani, RSC Adv., 2016, 6, 76759–76772 RSC.
- M. Saha, S. Mukherjee, S. Kumar and S. Dey, RSC Adv., 2016, 6, 58125–58136 RSC.
- T. Prabhakaran, R. V. Mangalaraja, J. C. Denardin and J. A. Jiménez, Ceram. Int., 2017, 43, 5599–5606 CrossRef CAS.
- Y. Yao, G. Wu, F. Lu, S. Wang, Y. Hu, J. Zhang, W. Huang and F. Wei, Environ. Sci. Pollut. Res., 2016, 23, 21833–21845 CrossRef CAS PubMed.
- S. Singh and K. Neeraj, Mater. Lett., 2015, 161, 64–67 CrossRef CAS.
- H. Y. He and J. Lu, Sep. Purif. Technol., 2017, 172, 374–381 CrossRef CAS.
- S. A. Bakar and C. Ribeiro, J. Mol. Catal. A: Chem., 2016, 421, 1–15 CrossRef.
- Q. Guo, Z. Zhang, X. Ma, K. Jing, M. Shen, N. Yu, J. Tang and D. D. Dionysiou, Sep. Purif. Technol., 2017, 175, 305–313 CrossRef CAS.
- D. Cao, X. Wang, L. Pan and H. Li, J. Mater. Chem. C, 2016, 4, 951–957 RSC.
- X. J. Guo, H. R. Li and S. G. Zhao, J. Taiwan Inst. Chem. Eng., 2015, 55, 90–100 CrossRef CAS.
- Y. H. Lin, H. T. Hsueh, C. W. Chang and H. Chu, Appl. Catal., B, 2016, 199, 1–10 CrossRef CAS.
- Z. Yang, Z. Zhang, Y. Jiang, M. Chi, G. Nie, X. Lu and C. Wang, RSC Adv., 2016, 6, 33636–33642 RSC.
- S. Singh and N. Khare, RSC Adv., 2015, 5, 96562–96572 RSC.
- Z. P. Zhou, Y. Zhang and Z. Y. Wang, Appl. Surf. Sci., 2008, 254, 6972–6975 CrossRef CAS.
- S. Lee, J. S. Kang, K. T. Leung, W. Lee, D. Kim, S. Han, W. Yoo, H. J. Yoon, K. Nam and Y. Sohn, J. Ind. Eng. Chem., 2016, 43, 69–77 CrossRef CAS.
- S. A. Sassman and L. S. Lee, Environ. Sci. Technol., 2005, 39, 7452–7459 CrossRef CAS PubMed.
- Q. H. Chen, S. N. Wu and Y. J. Xin, Chem. Eng. J., 2016, 302, 377–387 CrossRef CAS.
- Z. Lu, P. Huo, Y. Luo, X. Liu, D. Wu, X. Gao, C. Li and Y. Yan, J. Mol. Catal. A: Chem., 2013, 378, 91–98 CrossRef CAS.
- N. Shao, J. N. Wang, D. D. Wang and P. Corvini, Appl. Catal., B, 2017, 203, 964–978 CrossRef CAS.
- E. Jimenez-Relinque and M. Castellote, Cem. Concr. Res., 2015, 74, 108–115 CrossRef CAS.
- M. Harb, P. Sautet and P. Raybaud, J. Phys. Chem. C, 2013, 117, 8892–8902 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10016g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Chencen Sun,
Jia Liu and
Qiang Zhen*
*,
Chencen Sun,
Jia Liu and
Qiang Zhen*