
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of nanosilica content on the corrosion inhibition of composite coatings of a filled epoxy resin grafted with a hydrophobic fluoroalkylsilane: a dual critical concentrations interpretation
Fuguo Dengabc,
Lishi Wangc,
Yong Zhoud,
Xinghou Gongabc,
Xipo Zhaoabc,
Tao Hu*abc and
Chonggang Wu *abc
*abc
aHubei Provincial Key Laboratory of Green Materials for Light Industry, Hubei University of Technology, Wuhan, Hubei 430068, China. E-mail: hutao@mail.hbut.edu.cn; cgwu@mail.hbut.edu.cn
bCollaborative Innovation Center of Green Light-weight Materials and Processing, Hubei University of Technology, Wuhan, Hubei 430068, China
cSchool of Materials and Chemical Engineering, Hubei University of Technology, Wuhan, Hubei 430068, China
dKey Laboratory for Green Chemical Process of Ministry of Education, Wuhan Institute of Technology, Wuhan, Hubei 430205, China
First published on 17th October 2017
Abstract
3,3,3-Trifluoropropylmethyldimethoxysilane (TMDMS) was successfully grafted to molecules of an epoxy resin (EPR) of bisphenol-A origin with an epoxy value of 0.440, i.e. EP44, by transesterification (i.e. alcoholysis) of the TMDMS methoxyls with the EP44 hydroxyls under anhydrous dibutyltin-dilaurate catalysis. Meanwhile, a nanosilica powder was modified with 1,1,1,3,3,3-hexamethyldisilazane (HMDS) via electrophilic substitution of the nanosilica hydroxyls by the HMDS trimethylsilyls to significantly decrease its surface hydroxyls for controlled aggregation. Six coatings, onto an NaOH solution-treated tinplate substrate, of EP44, of the TMDMS-grafted EP44, and of the composites of the TMDMS-grafted EP44 filled with 0.5, 1, 3 and 5 wt% of the HMDS-modified nanosilica were then step-cured mildly with an amino-terminated polyamide from a solution. Owing to the introduction of superhydrophobic trifluoropropyls possibly plus a reduction in the hydrophilic hydroxyl concentration present, the hydrophobicity, evaluated by the water contact-angle, of the EP44 coating increased considerably upon its TMDMS grafting, which then changed little with further addition of the nanosilica. Electrochemical impedance spectroscopy data and simulations revealed that the anticorrosive performance of the TMDMS-grafted EP44 coating, upon immersion into an NaCl solution, was significantly improved compared with the EP44 coating, primarily due to its remarkably enhanced hydrophobic barrier to the water-mediated corrosives (water, NaCl, oxygen, other molecules and ions, etc.). However, the anticorrosion behaviour of the composite coatings was dominated by a corrosion inhibition (i.e. physical barrier) mechanism by the nanosilica filling the pores (free volumes, voids, cracks, etc.) susceptible to the corrosives. As the nanosilica content steadily was raised from 0 to 5 wt%, the corrosion inhibition of the composite coatings first intensified, probably thanks to an enhancement of the filling rate of the pores, and subsequently weakened, presumably due to an increase in the porosity from increased size exclusion of aggregated nanoparticles, and finally improved again possibly owing to a densification of the outside-of-pore barriers of greatly aggregated, size excluded particles, which constituted dual critical concentrations (DCCs) of the nanosilica at 0–0.5 and ∼3 wt%, respectively, that gave rise to a maximum followed by a minimum in the corrosion inhibition. To our knowledge, this has been the first work reporting this unique DCCs behaviour for EPR/nanosilica coatings, which, dictated by a hydrophobic EPR matrix of small porosity as well as a modified nanosilica of controlled aggregation, may universally be extended to the corrosion inhibition of other polymer/nanoceramic coatings.
Introduction
Metals have found extensive applications in construction, microelectronics, aerospace, automotives, etc. industries.1–3 However, owing to their relatively high chemical activity, metals are subject to corrosion, which has resulted in their increased consumption causing great economic losses and may even lead to catastrophic accidents.4 The first practical and widely used method to inhibit metal corrosion was based on self-healing chromate conversion coatings, which, nevertheless, have been restricted or entirely banned in many countries due to the presence of carcinogenic and environmentally detrimental hexavalent chromium ions (Cr6+).2,5–11 Great efforts have been made to seek alternative candidates of promising and durable corrosion protection including organic coatings, more friendly inorganic conversion coatings, organic–inorganic hybrid coatings, conducting polymer coatings, etc.Organic coatings12,13 have increasingly become an effective solution for corrosion protection, of which the coatings of epoxy resin (EPR) are the most popular instance applied due to its excellent chemical resistance, strong adhesion to metallic substrates, low curing shrinkage, etc.14 However, due to its rich presence of hydrophilic hydroxyl groups,14,15 a cured EPR displays such poor ageing16 and heat17 resistance as well as high brittleness that its coatings are difficult to produce long lasting corrosion protection. To overcome this, silicone resin (SR), i.e. polysiloxane, by virtue of its superior weather resistance, high thermal stability, good toughness, and strong hydrophobicity, has most typically been incorporated to improve the anticorrosive performance of EPR coatings by application of an SR primer underneath an EPR finish,8,18 by blending as a dispersed gel phase with an EPR,9,19 by formation of an interpenetrating polymer network with an EPR,20 by (block or graft) copolymerisation with an EPR during its cure,21,22 etc. In the last decade, organosilane small molecules,14,23 e.g. those of (3-glycidoxypropyl)trimethoxysilane (GPTMS),14 have interestingly been grafted onto EPR molecules prior to their cure via transesterification (i.e. alcoholysis) reaction of the alkoxy (e.g. methoxy) groups of the former with the hydroxyls of the latter anhydrously catalysed by dibutyltin dilaurate (DBTDL); accordingly, Ji et al.14,23 claimed that the absorption of water, medium for corrosives, into the cured EPRs was decreased as a result of a consumption of their preexisting hydrophilic hydroxyls by the reaction, which favoured the anticorrosion of the EPR coatings. Nevertheless, this effect of the pre-cure decrease in the hydroxyl concentration presumably was insignificant since more than 80 mol% of the hydroxyls present in the EPRs, which were obtained from graft modification with various organosilanes of the bisphenol-A based EPR of an epoxy value of 0.440 (i.e. EP44), were newly formed upon their cure. The above organosilane-grafted EPRs approach,14,23 although reported by a single research group, has provided a promising strategy for the chemical modification of the EPRs molecular structures.
Alternatively, inorganic ceramic (IC) nanofillers, at no more than 10 wt% loading levels, have been added to EPR matrices to form composite coatings for the corrosion inhibition of metallic substrates; this generally is because the nanosized particles, at their low concentrations and thus small sizes, effectively fill the as many as possible pores (free volumes, voids, cracks, etc.) of the EPRs, which leads to the significantly detoured diffusion channels, present in the composite coatings, that conduce to improved corrosion prevention of the metallic substrates.24–28 It has further been found that, as the content of IC nanoparticles is increased steadily, the corrosion inhibition of EPR/IC composite coatings first improves and then deteriorates, exhibiting the maximum at an optimum (i.e. critical) IC concentration beyond which the porosity (from the failure of aggregated IC nanoparticles to fill the smaller pores) of the composite coatings begins to increase gradually.24,29 For instance, Ghanbari and Attar30 reported that the composite coatings of an EPR and a nanosilica powder treated with GPTMS demonstrated the maximum corrosion resistance at a critical nanosilica concentration of ∼4 wt%.
In this work, we have accomplished successfully a novel modification of EPRs for anticorrosive coatings, which is a combination of the grafting of fluoroalkyl-containing organosilane onto and the incorporation of surface-treated IC nanofiller into EP44 molecules. Specifically, a 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) grafted EP44 is studied against EP44 for improved corrosion protection, based on its hydrophilic hydroxyl concentration decrease from the graft reaction and the hydrophobic trifluoropropyl introduction jointly for hydrophobic barrier anticorrosion, as well as on its water consumption from hydrolysis of the residual TMDMS methoxy groups for water-digestive barrier anticorrosion. Further, the TMDMS-grafted EP44 is filled with different contents of 1,1,1,3,3,3-hexamethyldisilazane (HMDS) treated nanosilica for maximisation of the corrosion inhibition of the composite coatings.
Experimental
Materials
A bisphenol-A based EPR of an epoxy value of 0.440 (component A), i.e. EP44, and its curing agent (component B), an amino-terminated polyamide-based compound (i.e. G650), were supplied by Nanjing Haozhuo Materials Technology Co., Ltd., China. 3,3,3-Trifluoropropylmethyldimethoxysilane (TMDMS) (≥99%), dibutyltin dilaurate (DBTDL) (95%), and 1,1,1,3,3,3-hexamethyldisilazane (HMDS) (nitrogen flushed, 98%), respectively, were obtained from Zhejiang Research Institute of Chemical Industry, China, Sigma-Aldrich, and Acros Organics. A nanosilica powder of 30 nm in mean diameter (99.5% metals basis), potassium bromide (KBr) (specpure reagent), and deuterated chloroform (99.8%) were purchased from Aladdin Industrial Corp., China. Tinplate (120 × 50 × 2.8 mm3) and sand papers of increasing meshes (in the order of #240, #600, and #1000) were supplied by Guangzhou DMY Instruments Co., Ltd., China and Hubei Yuli Abrasive Belts Group, China, respectively. Poly(vinyl chloride) (PVC) tubes of 40 mm in outer diameter and 36.6 mm in inner diameter, obtained from Yingda Plastic Building Materials Co., Ltd., China, were cut into 50 mm lengths. Sodium hydroxide (NaOH) (analytical reagent, ≥96%), sodium chloride (NaCl) (analytical reagent, ≥99.5%), potassium chloride (KCl) (analytical reagent, ≥99.5%), toluene (analytical reagent, ≥99.5%), acetone (analytical reagent, ≥99.5%), n-butanol (analytical reagent, ≥99.5%), xylene (analytical reagent, ≥99%), and molecular sieves (Type 4A) all were purchased from Sinopharm Chemical Reagents Co., Ltd., China. Distilled water was home made in our laboratory using a stainless steel water distiller. All the chemicals were used as received without further purification.Pre-cure grafting of 3,3,3-trifluoropropylmethyldimethoxysilane onto epoxy resin molecules
A modified EPR was synthesised by reaction of an EPR with a fluorosilane monomer under anhydrous, DBTDL catalyst conditions, for which a process of chemical grafting of TMDMS to EP44 molecules is illustrated in Scheme 1a and the proposed chemical reaction in Scheme 1b. A solution of 61.0 g of EP44, 27.2 g of TMDMS, and ∼1.6 g of DBTDL in 60.4 g of toluene fully dried by Molecular sieves 4A was added to a 250 mL three-necked flask equipped with a magnetic stirrer hotplate as well as a condenser. The mixture was stirred and refluxed at 95 °C (oil bath) for 8 h, which was then rotary-evaporated, starting at 30 °C slowly heated to 75 °C within ∼1.5 h, subsequently transferred to a beaker, and finally dried in vacuo at 50 °C overnight followed by at 80 °C for at least 48 h, to thoroughly remove toluene, unreacted TMDMS, any by-product(s), etc. A colourless, viscous liquid product was thus obtained, which supposedly was a TMDMS-grafted EP44.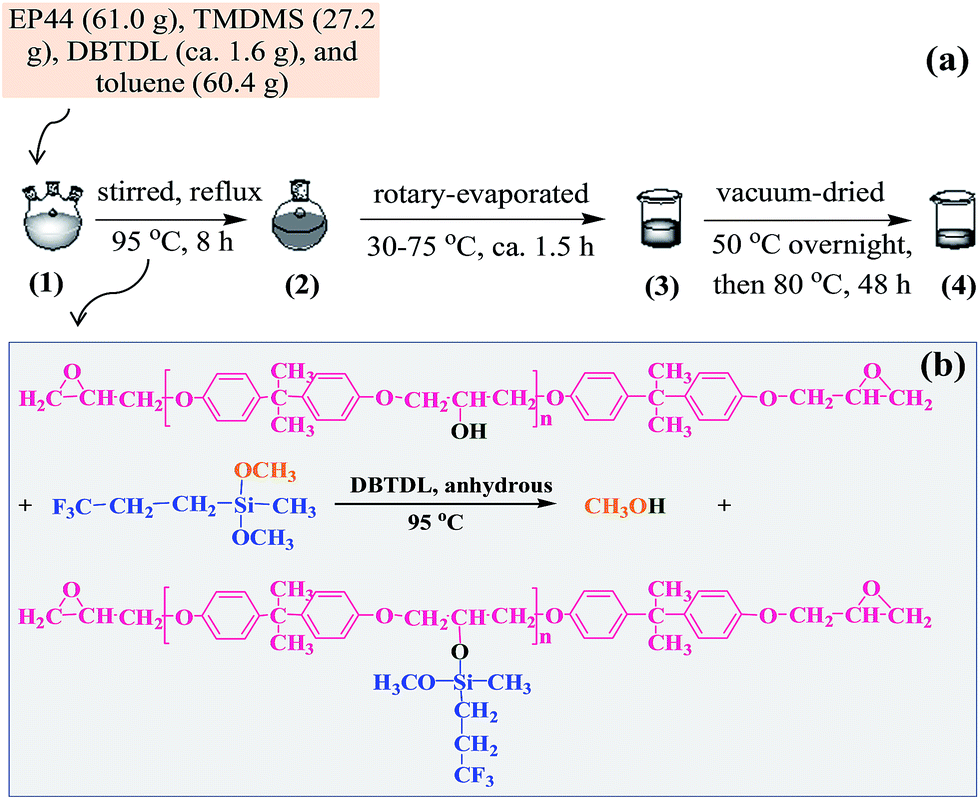 | ||
| Scheme 1 (a) Illustration of a grafting process of 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) onto the molecules of a bisphenol-A based epoxy resin of an epoxy value of 0.440 (i.e. EP44) prior to its cure, in toluene solution under anhydrous catalysis by dibutyltin dilaurate (DBTDL); (b) schematic of the graft reaction of TMDMS onto EP44 molecules (i.e. alcoholysis of TMDMS by EP44) to form a fluoroalkyl-containing EP44 of a significantly decreased hydroxyl concentration, which occurs during the process given in (a) from step 1 to 2. | ||
Surface modification of a nanosilica powder with 1,1,1,3,3,3-hexamethyldisilazane
A schematic of the surface modification process of the nanosilica powder with HMDS is shown in Scheme 2a, and the supposed reaction mechanism in Scheme 2b. 5.2 g of the white nanosilica powder was vacuum-dried at 150 °C for 3 h, which was then transferred, along with 35.8 g of HMDS, to a 250 mL three-necked flask equipped with a hotplate and a condenser. The resulting suspension was vigorously stirred mechanically and refluxed at 50 °C (oil bath) for 20 h, which was subsequently transferred to a beaker, and finally allowed to stand (i.e. be air-dried) in fume hood at 50 °C (oil bath) for at least 1.5 h, followed by vacuum drying at 80 °C for at least 48 h, to thoroughly remove unreacted HMDS molecules, any by-product(s), etc. An HMDS-modified nanosilica was thus obtained, still as a white powder.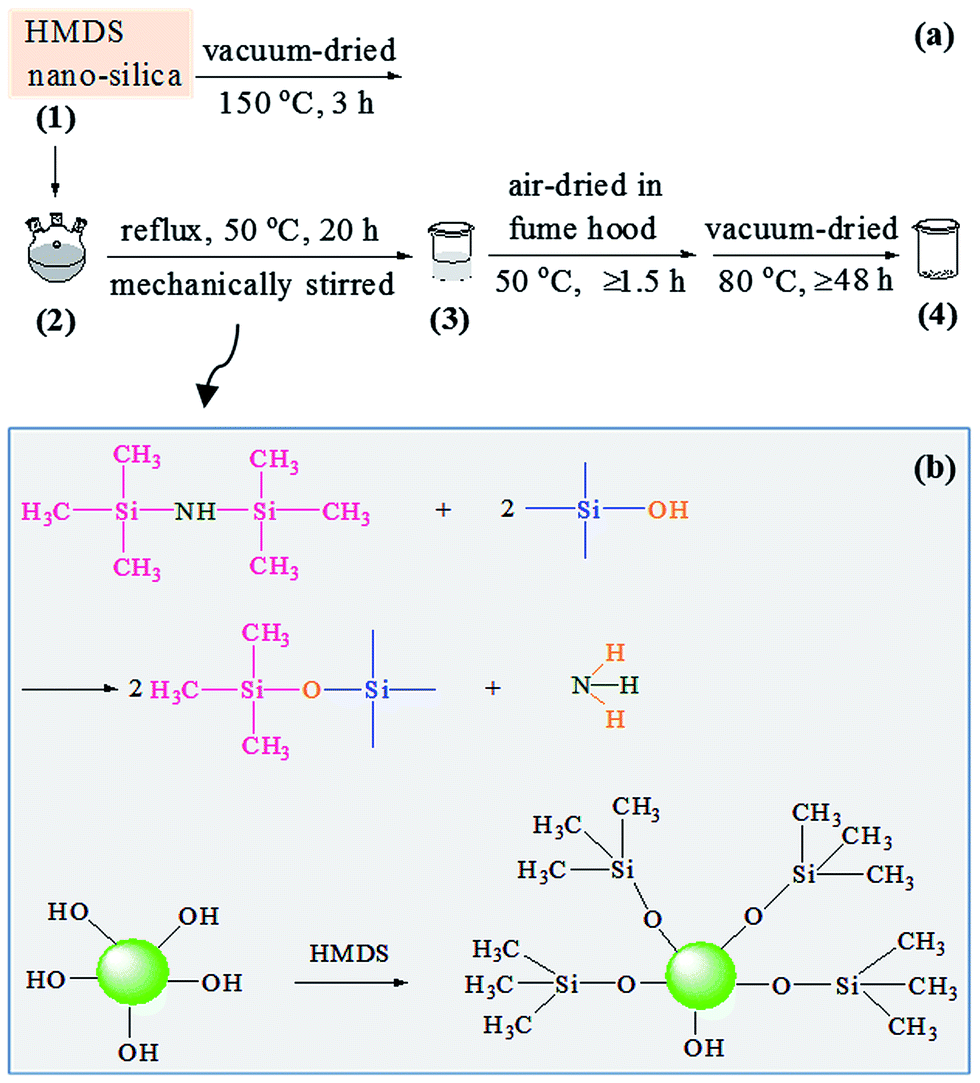 | ||
| Scheme 2 (a) Illustration of the surface modification process of a nanosilica powder with 1,1,1,3,3,3-hexamethyldisilazane (HMDS); (b) schematic of the reaction of (the imino groups of) HMDS with (the hydroxy groups of) the nanosilica to modify the polar hydroxyls into non-polar methyls on the nanosilica powder surfaces, which occurs during the process given in (a) from step 2 to 3. | ||
Fourier transform infrared (FTIR) spectroscopy
An FTIR spectrometer (Bruker, Tensor II) was used to collect the absorption spectra, of a 4 cm−1 resolution at room temperature (RT), of EP44, TMDMS, the TMDMS-grafted EP44, HMDS, the nanosilica, and the HMDS-modified nanosilica, based on 16 scans between 4000–600 cm−1 in the attenuated total reflection (ATR) mode for the former four liquids and 24 scans between 4000–400 cm−1 in the transmission mode for the latter two solids. For the runs, a drop (∼15–20 mg) of each of the liquids samples was pipetted onto the center of the ATR attachment, while the solids samples were prepared as KBr pellets by grinding together, using a pestle and mortar, ∼2.0 mg of either solid powder and ∼50 mg of KBr dried with an infrared baking lamp, followed by compression of the mixture powder on a hydraulic press. Prior to the spectroscopic measurements, samples of EP44, the TMDMS-grafted EP44, and the HMDS-modified nanosilica were all dried in vacuo at 80 °C for at least 48 h, and that of the nanosilica was vacuum-dried at 150 °C for 5 h.1H nuclear magnetic resonance (NMR) spectroscopy
An NMR spectrometer (Varian, INOVA 600 MHz) was employed to collect the 1H spectra of EP44, TMDMS, and the TMDMS-grafted EP44 with respect to a tetramethylsilane internal standard. For the runs, two drops (∼30–40 mg) of each of the liquid compounds fully were dissolved in 1.0 g of deuterated chloroform in an NMR tube to form a uniform solution sample of ∼30–40 mg g−1. The spectra of the 1H peak chemical shifts (δ's) of the solution samples all were simulated using Chemdraw to aid the assignment analysis.Thermogravimetric analysis (TGA)
A thermogravimetric-differential-thermal simultaneous analyzer (TA Instruments, SDT Q600) was applied to study the weight decreasing behaviour, until 800 °C, of the HMDS-modified nanosilica against that of the nanosilica. For the runs, ∼5.0 mg of either sample of the nanosilica powder and the HMDS-modified nanosilica powder, upon vacuum drying at 150 °C for 5 h and at 80 °C for at least 48 h respectively, was placed into an alumina crucible, which was then loaded onto the alumina sample tray of the internal balance. TGA thermograms were recorded from RT to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere (i.e. purge gas) of 100 mL min−1.Surface treatment of a tinplate substrate with alkaline solution
The as-purchased tinplate substrates (120 × 50 × 2.8 mm3) were first polished in turn by abrasive Sand-papers #240, #600 and #1000 of increasing meshes, then degreased in RT acetone for 5 min, subsequently treated in a 60 °C, 2 wt% NaOH aqueous solution for 2 min, and finally vacuum-dried at 100 °C for at least 48 h.Preparation of epoxy resin based coatings against the surface-treated tinplate substrate
First, the formulae of the six coating compositions investigated are listed in Table 1, which, respectively, were based on EP44, the TMDMS-grafted EP44, and the mixtures of the TMDMS-grafted EP44 and different contents (0.5, 1, 3 and 5 wt%) of the HMDS-modified nanosilica powder, in the presence of an amino-terminated polyamide-based curing agent (i.e. G650) and a mixed solvent of n-butanol/xylene (7/3 w/w). In each composition of Table 1, the loading levels of all the other ingredients relative to that of the base resin (EP44 or TMDMS-grafted EP44) were determined according to the rules indicated in the footnote to Table 1.| EPR-based coating no. | EP44 (g) | TMDMS-grafted EP44 (g) | HMDS-modified nanosilica (g) | Curing agent G650 (g) | n-Butanol (g) | Xylene (g) |
|---|---|---|---|---|---|---|
| a In formulae a–f, the mass (8.40 g) of the mixed n-butanol/xylene solvent was determined according to its mass ratio to the (TMDMS-grafted) EP44 (9.00 g) of 1/1.07, which was found to be optimal in the coating viscosity for the thickness uniformity of the coatings applied to the tinplate substrate; the mass of the G650 was equal to that (9.00 g) of EP44 (in Formula a), or to that (7.64 g) of the EP44 moieties of the TMDMS-grafted EP44 (in formulae b–f) under the assumption that the EP44 hydroxyls 100% were consumed upon the graft reaction; and, in formulae c–f, the contents (0.5, 1, 3 and 5 wt% corresponding to 0.084, 0.168, 0.515 and 0.876 g, respectively) of the HMDS-modified nanosilica each were determined as the ratio of its mass to the total mass of the TMDMS-grafted EP44, the G650, and itself. | ||||||
| (a) | 9.00 | 0 | 0 | 9.00 | 5.88 | 2.52 |
| (b) | 0 | 9.00 | 0 | 7.64 | 5.88 | 2.52 |
| (c) | 0 | 9.00 | 0.084 | 7.64 | 5.88 | 2.52 |
| (d) | 0 | 9.00 | 0.168 | 7.64 | 5.88 | 2.52 |
| (e) | 0 | 9.00 | 0.515 | 7.64 | 5.88 | 2.52 |
| (f) | 0 | 9.00 | 0.876 | 7.64 | 5.88 | 2.52 |
Then, the mixtures of the TMDMS-grafted EP44 and the different contents of the HMDS-modified nanosilica were prepared as follows. As illustrated in Scheme 3(1)–(4), predetermined amounts, i.e. 0.084, 0.168, 0.515 and 0.876 g corresponding to 0.5, 1, 3 and 5 wt% respectively (cf. Table 1), of the HMDS-modified nanosilica powder each were added into a 50 mL beaker along with ∼40 g of acetone. The suspension was sonicated at a power of 270 W for 10 min under the protection of an iced water bath to form an organosol, which was then poured into a 250 mL beaker containing 9.00 g (according to Table 1(c–f) of the TMDMS-grafted EP44. The pristine mixture was stirred at RT for ∼1 min, subsequently air-dried in fume hood at RT for at least 6 h, and finally vacuum-dried at 80 °C overnight to obtain the acetone-free mixture of the TMDMS-grafted EP44 and the HMDS-modified nanosilica.
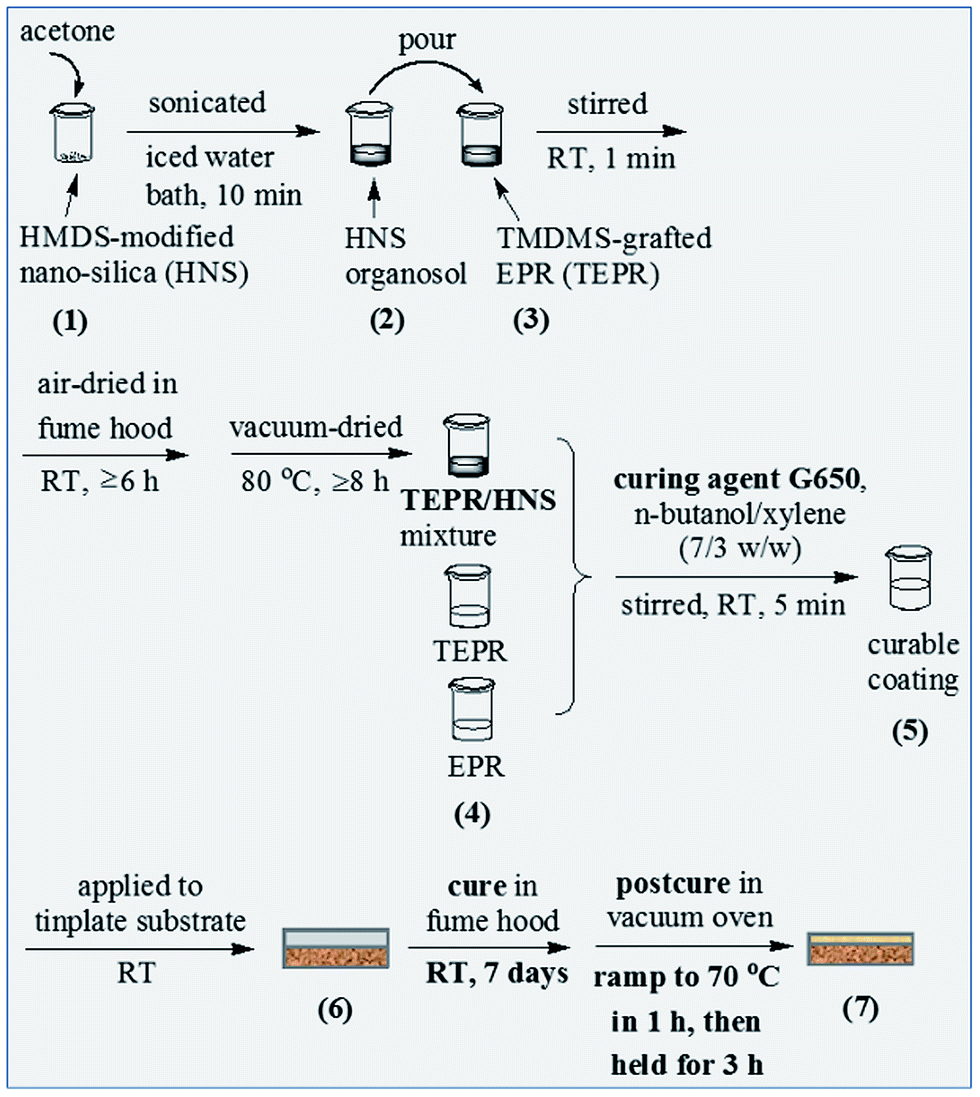 | ||
| Scheme 3 Illustration of the preparation and application process, to a 2 wt% NaOH solution treated tinplate substrate, of the corrosion preventive coating based on an epoxy resin (EPR) of bisphenol-A origin with an epoxy value of 0.440, i.e. EP44, on a 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) grafted EP44, or on the mixtures of the TMDMS-grafted EP44 and a 1,1,1,3,3,3-hexamethyldisilazane (HMDS) modified nanosilica, in the presence of an amino-terminated polyamide-based curing agent (i.e. G650) and a mixed solvent of n-butanol/xylene (7/3 w/w). | ||
Finally, the application and cure of the six coating compositions to the surface-treated tinplate substrate was completed. As illustrated in Scheme 3(4)–(7), 7.64 g of the curing agent G650 and 8.40 g of the n-butanol/xylene (7/3 w/w) mixed solvent, read from Table 1(c–f), together were stirred with each of the obtained (TMDMS-grafted EP44)/(HMDS-modified nanosilica) mixtures, in the beaker, at RT for ∼5 min to form a curable coating composition (basically solution), which was then applied uniformly to the surface-treated tinplate substrate at RT, subsequently cured in fume hood at RT for 7 days, and finally postcured in vacuo, through a ramp from RT to 70 °C within ∼1 h followed by a hold there for 3 h, to obtain a composite coating completely cured against the tinplate substrate. This procedure (i.e. Scheme 3(4)–(7)) was repeated for the preparation of two base resin coatings, respectively, by curing the solutions of EP44 and the TMDMS-grafted EP44 in the mixed solvent with the G650 present, according to the formulae of Table 1(a and b). In the cure, the formed cross-linked structure and typical cross-linking reaction, respectively, presumably are represented in Scheme 4a(1) and b, while those during the postcure in Scheme 4a(2) and c respectively. Measured by a coating pachymeter (ARCO Science & Technology Ltd., China, Smart Sensor® AR932), the thicknesses of all the coatings, onto the substrate, made from the six compositions were maintained at 52 ± 4 μm, which were then stored in a desiccator for later use.
 | ||
| Scheme 4 (a) Representation of the cross-linked structures formed during the curing process of a (3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) grafted) epoxy resin (EPR) with an amino-terminated polyamide-based curing agent (i.e. G650): (1) cure at room temperature for 7 days to form V-shaped cross-links; (2) subsequent postcure via a ramp from room temperature to 70 °C in 1 h followed by a hold there for 3 h further to form X-shaped cross-links. (b) Schematic of the typical cross-linking reaction during the cure (i.e. step 1 in (a)), in which the epoxide groups of the (TMDMS-grafted) EPR react with the amino groups of the G650 via ring-opening addition to produce hydroxy and imino groups. (c) Schematic of the typical cross-linking reaction during the postcure (i.e. step 2 in (a)), in which the epoxide groups of the (TMDMS-grafted) EPR react with the imino groups of the G650 via ring-opening addition to produce hydroxy and nitrilo groups. | ||
Contact angle measurements
The contact angles (θ's), at RT (∼25 °C), of water against the six compositions of coatings fully cured onto the tinplate substrate were measured on an optical contact angle goniometer (Shanghai Zhongchen Digital Technic Apparatus Co., Ltd., China, JC-2000D). First, a syringe filled with 100 μL of distilled water was made to slowly extrude a pendant water droplet of ∼0.1 μL attached to its needle tip. Then, the stage loaded with a tinplate coating sample, below the tip, was raised to allow the surface of the coating to contact the droplet, and subsequently lowered slowly to detach the droplet from the tip, thus forming a sessile water droplet onto the coating. After aligning the baseline right with the coating surface, a software program of digital video image processing (JC-2000D) was used to analyse the droplet contour, from which the water θ of the coating was calculated. For each composition of coating, at least three parallel samples were measured to obtain three θ values, respectively, from which the median value was taken as the θ for analysis.Electrochemical impedance spectroscopy (EIS)
A sealant for insulation of the coatings cured onto the tinplate substrate was prepared from a curable EP44/G650 (1/1 w/w) composition as follows: 20 g of the composition in a 250 mL beaker was stirred for 2 min, then deaerated in vacuo for 30 min, and subsequently allowed to stand (i.e. pre-cure) overnight, all at RT, to ensure that the flowability of the gelling composition (i.e. sealant) considerably was reduced compared with the pristine composition. Immediately, the PVC tubes (of 40 mm in outer diameter, 36.6 mm in inner diameter, and 50 mm in length) each, by their end cross sections, were dipped in a predetermined dosage of the sealant and then attached (i.e. glued) to the surface of each of the tinplate coatings, which was followed by pipetting an appropriate amount of the sealant around the seam between the coating and the PVC tube. At last, the PVC tube-attached coatings (i.e. EIS specimens) were cured (with respect to the sealant) at RT for at least 48 h and then stored in a desiccator at all times prior to EIS experiments.The anticorrosive performance of the six compositions of coatings was therefore measured at RT using EIS, a test mode of an electrochemical workstation (CH Instruments, Inc., China, CHI660E). Each of the (PVC tube-attached) coating specimens was immersed into an NaCl aqueous solution of 3.5 wt% at RT (∼25 °C) by adding ∼47 mL of the solution into the tube that was positioned vertically against the levelled coating, with an immersion area of ∼10.5 cm2 calculated from the tube inner-diameter of 36.6 mm. The EIS runs of each coating specimen across a frequency range of 105 to 10−2 Hz were conducted, respectively, upon its increasing immersion times of 1, 24, 240, 480, 720 and 960 h. For the experiments, a three-electrode arrangement was used, which comprised the tinplate substrate working electrode, a reference electrode of Ag/AgCl in saturated KCl aqueous solution at RT, and a counter electrode of platinum; the latter two electrodes were both immersed into the NaCl solution of the coating specimens, with the reference electrode bridged by another electrolytic aqueous solution of saturated KCl at RT. Each composition of coating was measured using five coating specimens, from which the EIS spectra (i.e. Bode plots) upon the increasing immersion times of the specimen having a median spectrum upon 1 h of immersion were taken as the experimental data for analysis with the aid of the Zsimpwin software.
Results and discussion
Evidence for successful grafting of 3,3,3-trifluoropropylmethyldimethoxysilane onto epoxy resin molecules
Fig. 1 shows the FTIR spectra of (1) EP44, (2) TMDMS, and (3) the TMDMS-grafted EP44 in the wavenumber ranges of (a) 4000–2400 cm−1 and (b) 2400–600 cm−1. In spectrum 2 of TMDMS, the absorption peaks (and shoulder) at 2967, 2915, 2946, 2841 and 1080 cm−1, respectively, were attributable to the C–H antisymmetric and symmetric stretches of its –CH3 groups, the C–H antisymmetric and symmetric stretches of its –CH2– groups, and the C–O stretching of its –OCH3 groups. Since the 827 cm−1 absorption band, ascribed to the C–H out-of-plane bending of the bisphenol-A phenylenes, was supposed to be unchanged in absorbance upon the possible graft reaction, Spectra 1 and 3, respectively, of EP44 and the TMDMS-grafted EP44 both were normalised with respect to it for a typical comparison. In spectrum 1 of EP44: the bands at 2965, 2871, 2926, 2838, 1230 and 1031 cm−1, respectively, were assigned to the C–H antisymmetric and symmetric stretches of its –CH3 groups, the C–H antisymmetric and symmetric stretches of the –CH2– neighbouring its epoxide groups, and the C–O–C antisymmetric and symmetric stretches of its methyleneoxyphenylene groups; those at 1606, 1580 and 1507 cm−1 probably were attributed to the skeletal vibrations of its phenylenes; those at 1384 and 1364 cm−1 were the split, weak double bands from the C–H symmetric bending of its –C(CH3)2– groups; and that at 911 cm−1 likely was characteristic of the C–O deformational vibration of its epoxide groups. All these bands (including that at 827 cm−1) appeared as well in spectrum 3 of the TMDMS-grafted EP44, at exactly the same wavenumbers and comparable absorbances, indicating that the graft reaction, if any, might not involve the aforementioned groups of EP44.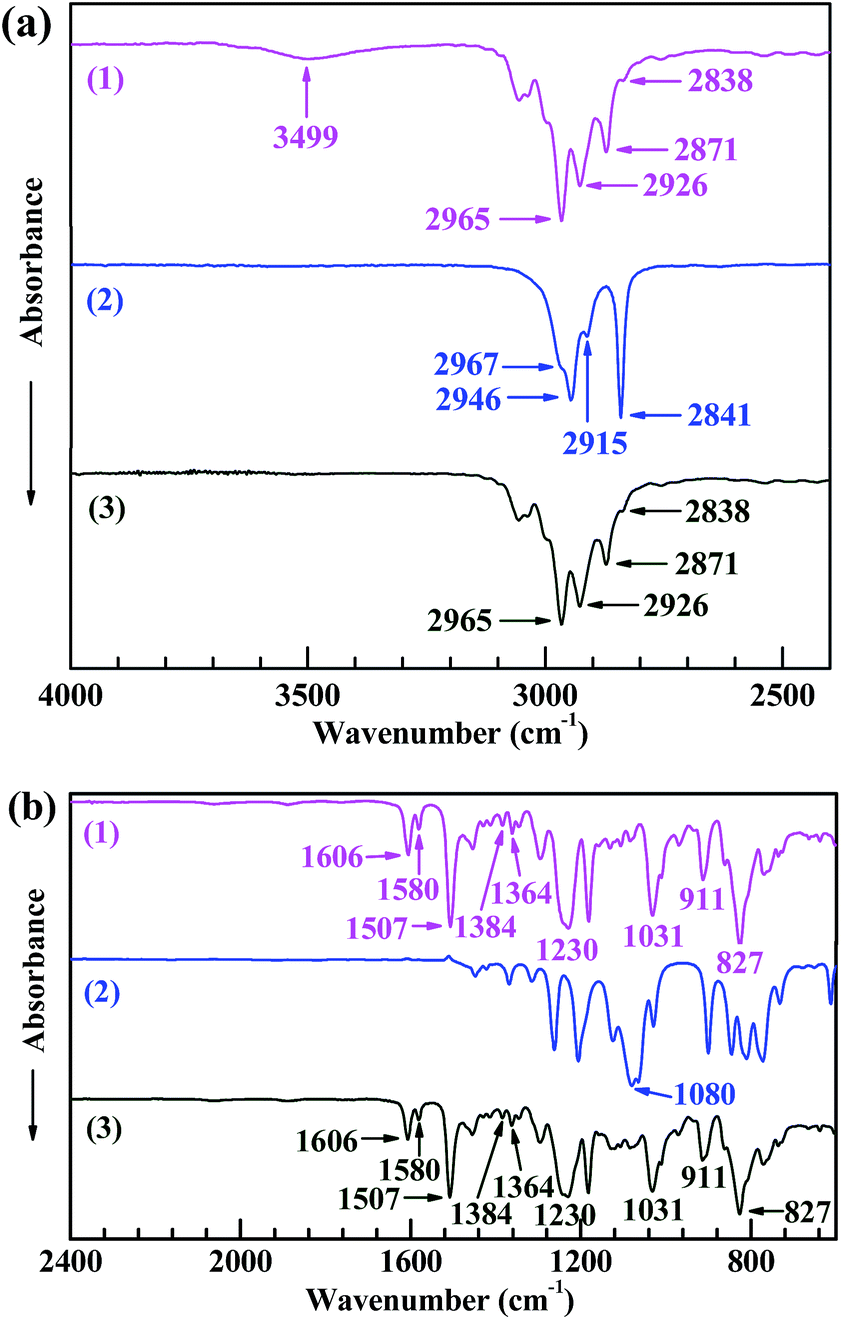 | ||
| Fig. 1 Fourier transform infrared (FTIR) absorption spectra of (1) a bisphenol-A based epoxy resin of an epoxy value of 0.440 (i.e. EP44), (2) 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS), and (3) a TMDMS-grafted EP44, (a) between 4000 and 2400 cm−1 and (b) between 2400 and 600 cm−1, at room temperature in the attenuated total reflection (ATR) mode. In graph a, the 3499 cm−1 absorption band in spectrum 1 characteristic of the O–H stretching of the –OH groups present in EP44 molecules nearly disappears in spectrum 3 of the TMDMS-grafted EP44 molecules, suggesting the significant occurrence of a graft reaction that consumes dramatically the EP44 –OH groups. | ||
In Fig. 1a, the 3499 cm−1 absorption band in spectrum 1 of EP44 molecules characterised the O–H stretching of their –OH groups, which, however, nearly disappeared in spectrum 3 of the TMDMS-grafted EP44 molecules, revealing a dramatic consumption of the –OH groups of EP44 through its significant graft modification; in other words, the –OH groups participated as a leading role in the graft reaction. It is worth mentioning that, since both EP44 and the TMDMS-grafted EP44 were well dried (in vacuo at 80 °C for at least 48 h) prior to their FTIR analysis, the peak disappearance should result largely from a sharp decrease in the –OH concentration, but not primarily from a considerably decreased content of adsorbed water. Therefore, under the anhydrous, DBTDL-catalytic conditions, the –OCH3 groups of TMDMS, most reactive among all its groups, presumably were reacted markedly with the –OH groups of EP44, via transesterification (i.e. alcoholysis), to produce a TMDMS-grafted EP44 plus a methanol by-product, as schematically shown in Scheme 1b; mechanistically, the –OH groups of EP44 molecules were substituted electrophilically by TMDMS molecules under anhydrous catalysis of DBTDL. These FTIR results agree with other reports14,23 in the literature and confirm that TMDMS successfully was grafted to EP44 molecules, to a significant extent, at the –OH group sites of EP44.
Fig. 2 shows the 1H NMR spectra of (a, d) EP44, (b, e) TMDMS, and (c, f) the TMDMS-grafted EP44. The supposed H atoms of EP44, TMDMS and the TMDMS-grafted EP44 are numbered in Fig. 2(d–f), respectively, to which the 1H-peak δ values in Fig. 2(a–c) were assigned as given in Table 2. In Fig. 2a of EP44 (cf. Fig. 2d): the peaks of the H atoms of its epoxide CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) (no. 2), phenylenes (no. 4 and 5) and –C(CH3)2– (no. 6) appeared at δ values of 3.28, 7.11, 6.81 and 1.61 ppm, respectively; the peak of the H atoms of its epoxide –CH2– (no. 1) split in two at 2.69 and 2.83 ppm, and that of the –CH2– neighbouring its epoxides (no. 3) at 3.89 and 4.14 ppm, either of which might be caused by a variation in the respective steric configuration;31 the peak of its –OH hydrogens (no. 9) was missing presumably due to their high activity, which possibly repressed as well the peaks of their neighbouring H atoms (no. 7 and 8). In Fig. 2b of TMDMS (cf. Fig. 2e), the peaks of the H atoms of its CF3–CH2– (no. 1′), –CH2–Si (no. 2′), Si–CH3 (no. 3′) and –Si(OCH3)2– (no. 4′), respectively, occurred at 2.09, 0.83, 0.14 and 3.51 ppm. As shown in Fig. 2c (cf. Fig. 2f): the spectrum of the TMDMS-grafted EP44 completely resembled that of EP44 (Fig. 2a) with the additional H-atom peaks at 2.11, 0.81, 0.15 and 3.52 ppm, which obviously were attributed, respectively, to the CF3–CH2– (no. 1′), –CH2–Si (no. 2′), Si–CH3 (no. 3′) and Si–O–CH3 (no. 4′) groups of the TMDMS graft (cf. Fig. 2b and Table 2), despite their sharply weakened intensities due to the significantly decreased group concentrations (note that n ∼0.4 in Fig. 2f, for EP44); moreover, the peaks of H-atoms no. 7 and 8 of EP44 moieties emerged upon the graft reaction at 2.33 and 1.26 ppm, respectively, suggesting a pronounced elimination of the active –OH hydrogens (no. 9 in Fig. 2d) of EP44 that released the NMR of their neighbouring H atoms. In addition to the above FTIR data, these NMR results further corroborate that TMDMS indeed was grafted significantly onto EP44 molecules, likely through a transesterification (i.e. alcoholysis) reaction under anhydrous, DBTDL catalyst conditions between the –OCH3 groups of TMDMS and the –OH groups of EP44, as schematically shown in Scheme 1b or Fig. 2(d–f). Particularly worth noting is that the TMDMS-grafted EP44 still had the H-atom (no. 4′) peak of the –OCH3 groups at 3.52 ppm (Fig. 2c vs. f), disclosing that a single TMDMS molecule reacted with the EP44 –OH largely by only one –OCH3, with the other –OCH3 remaining in the TMDMS graft possibly due to a steric hindrance effect.
(no. 2), phenylenes (no. 4 and 5) and –C(CH3)2– (no. 6) appeared at δ values of 3.28, 7.11, 6.81 and 1.61 ppm, respectively; the peak of the H atoms of its epoxide –CH2– (no. 1) split in two at 2.69 and 2.83 ppm, and that of the –CH2– neighbouring its epoxides (no. 3) at 3.89 and 4.14 ppm, either of which might be caused by a variation in the respective steric configuration;31 the peak of its –OH hydrogens (no. 9) was missing presumably due to their high activity, which possibly repressed as well the peaks of their neighbouring H atoms (no. 7 and 8). In Fig. 2b of TMDMS (cf. Fig. 2e), the peaks of the H atoms of its CF3–CH2– (no. 1′), –CH2–Si (no. 2′), Si–CH3 (no. 3′) and –Si(OCH3)2– (no. 4′), respectively, occurred at 2.09, 0.83, 0.14 and 3.51 ppm. As shown in Fig. 2c (cf. Fig. 2f): the spectrum of the TMDMS-grafted EP44 completely resembled that of EP44 (Fig. 2a) with the additional H-atom peaks at 2.11, 0.81, 0.15 and 3.52 ppm, which obviously were attributed, respectively, to the CF3–CH2– (no. 1′), –CH2–Si (no. 2′), Si–CH3 (no. 3′) and Si–O–CH3 (no. 4′) groups of the TMDMS graft (cf. Fig. 2b and Table 2), despite their sharply weakened intensities due to the significantly decreased group concentrations (note that n ∼0.4 in Fig. 2f, for EP44); moreover, the peaks of H-atoms no. 7 and 8 of EP44 moieties emerged upon the graft reaction at 2.33 and 1.26 ppm, respectively, suggesting a pronounced elimination of the active –OH hydrogens (no. 9 in Fig. 2d) of EP44 that released the NMR of their neighbouring H atoms. In addition to the above FTIR data, these NMR results further corroborate that TMDMS indeed was grafted significantly onto EP44 molecules, likely through a transesterification (i.e. alcoholysis) reaction under anhydrous, DBTDL catalyst conditions between the –OCH3 groups of TMDMS and the –OH groups of EP44, as schematically shown in Scheme 1b or Fig. 2(d–f). Particularly worth noting is that the TMDMS-grafted EP44 still had the H-atom (no. 4′) peak of the –OCH3 groups at 3.52 ppm (Fig. 2c vs. f), disclosing that a single TMDMS molecule reacted with the EP44 –OH largely by only one –OCH3, with the other –OCH3 remaining in the TMDMS graft possibly due to a steric hindrance effect.
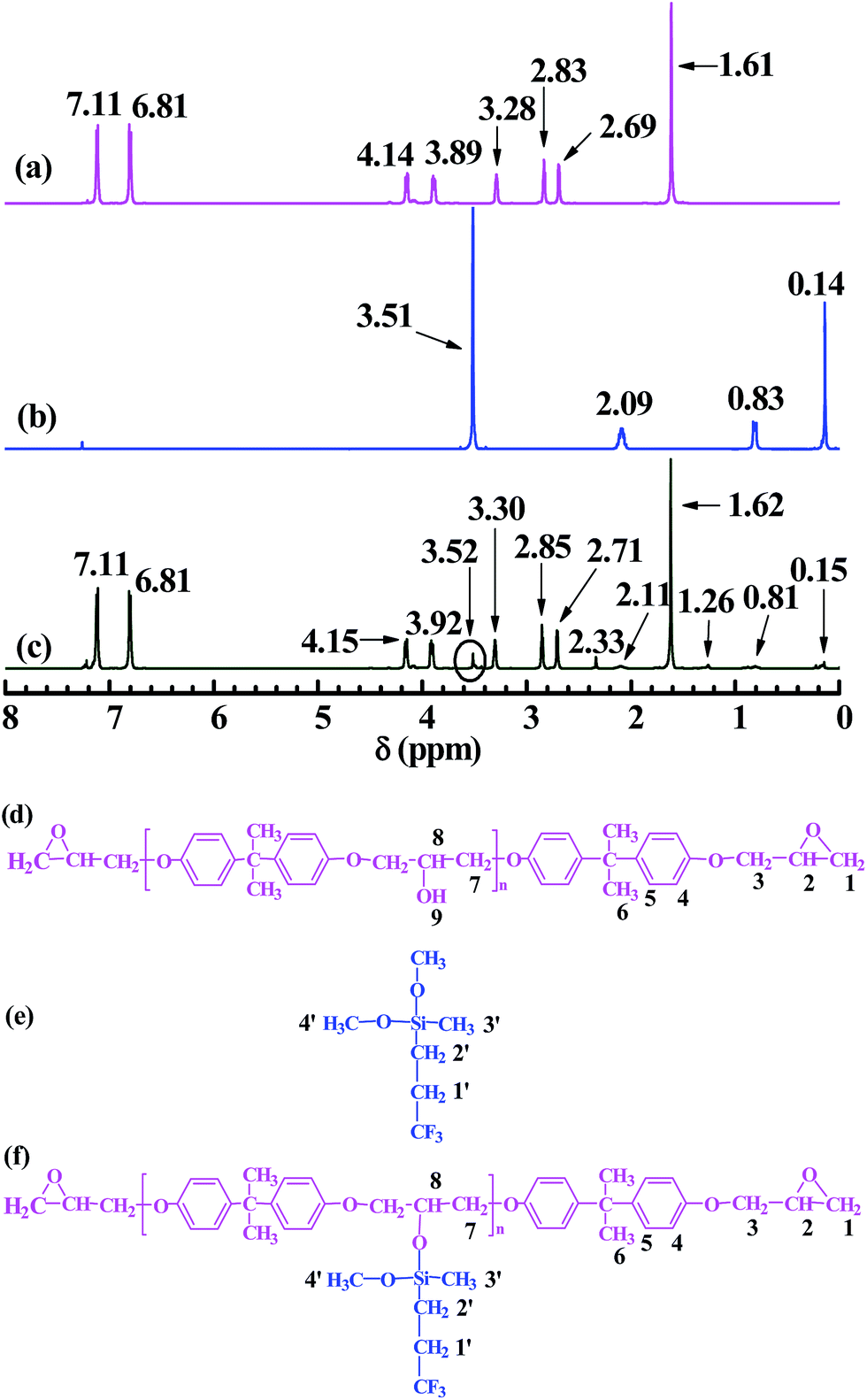 | ||
| Fig. 2 1H nuclear magnetic resonance (NMR) spectra of (a) a bisphenol-A based epoxy resin of an epoxy value of 0.440 (i.e. EP44), (b) 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS), and (c) a TMDMS-grafted EP44, respectively, for H-atoms no. 1–6 shown in (d), 1′–4′ shown in (e), and 1–8 and 1′–4′ shown in (f); the assignments of (a) to (d), (b) to (e), and (c) to (f) are given in Table 2, where the chemical shift (δ) values of 2.09, 0.83, 0.14 and 3.51 ppm, respectively, for H-atoms no. 1′–4′ of TMDMS molecules all are present in the 1H NMR spectrum of the TMDMS-grafted EP44 molecules, confirming a successful grafting of TMDMS onto EP44. | ||
| H atom no. | δ (ppm) | ||
|---|---|---|---|
| EP44 | TMDMS | TMDMS-grafted EP44 | |
| a Owing to the high activity of the EP44 –OH groups, the δ values of their H atoms (i.e. H-atoms no. 9) and neighbouring H atoms (i.e. H-atoms no. 7 and 8) are not subject to detection.b These H atoms do not belong to EP44, TMDMS and the TMDMS-grafted EP44 molecules, respectively. | |||
| 1 | 2.69, 2.83 | —b | 2.71, 2.85 |
| 2 | 3.28 | —b | 3.30 |
| 3 | 3.89, 4.14 | —b | 3.92, 4.15 |
| 4 | 7.11 | —b | 7.11 |
| 5 | 6.81 | —b | 6.81 |
| 6 | 1.61 | —b | 1.62 |
| 7 | —a | —b | 2.33 |
| 8 | —a | —b | 1.26 |
| 9 | —a | —b | —b |
| 1′ | —b | 2.09 | 2.11 |
| 2′ | —b | 0.83 | 0.81 |
| 3′ | —b | 0.14 | 0.15 |
| 4′ | —b | 3.51 | 3.52 |
Confirmation of successful modification of nanosilica with 1,1,1,3,3,3-hexamethyldisilazane
The reaction between the nanosilica and HMDS was characterised by FTIR spectroscopy as shown in Fig. 3. In spectrum 2 of HMDS, the absorption bands at 3381, 2955, 2898, 1250 and 834 cm−1, respectively, were ascribed to its N–H stretching, the C–H antisymmetric and symmetric stretches of its –CH3 groups, and the Si–C deformation and rocking vibrations of its –Si(CH3)3 groups.32–34 To make a typical comparison, both spectra 1 and 3 of the nanosilica and HMDS-modified nanosilica, respectively, were normalised with respect to their band at 473 cm−1, which, likely characteristic of the Si–O–Si bending of the silica gel networks, presumably was unchanged in absorbance upon the surface modification. Thus, the 1108 and 803 cm−1 bands, respectively, from the Si–O–Si antisymmetric and symmetric stretches of the silica gel networks35 both changed little as well in absorbance despite the modification of the nanosilica surfaces (spectrum 1 vs. 3). However, the 3420 cm−1 band in spectrum 3 of the HMDS-modified nanosilica, which was attributed to an O–H stretching, weakened considerably compared with that in spectrum 1 of the nanosilica; as both the samples were well dried (in vacuo, at 80 °C for at least 48 h and at 150 °C for 5 h, respectively) prior to the FTIR runs, this band weakening essentially should originate from a significant decrease in the surface –OH concentration upon the modification, but not from a marked reduction in the water absorption, which may be verified by the fact that the 1629 cm−1 band in spectrum 3, assigned to the H–O–H bending of the bound water, varied little in absorbance compared with that in spectrum 1. Moreover, the 967 cm−1 band presumably characterising the bending of theSi–OH at the nanosilica surfaces weakened notably after the HMDS modification (spectrum 1 vs. 3), inferring again a pronounced reduction in the surface –OH groups. Finally, a new band at 2965 cm−1, feature of the C–H antisymmetric stretch of –CH3, emerged, although small, in spectrum 3 of the HMDS-modified nanosilica, indicating the formation of –CH3 groups from the nanosilica and HMDS reaction. All these observations confirm the occurrence of a significant surface modification of the nanosilica with HMDS, by which a lot of –CH3 groups successfully were grafted to the nanosilica surfaces via electrophilic substitution of its surface –OH groups with the –Si(CH3)3 groups of HMDS, as schematically shown in Scheme 2b.
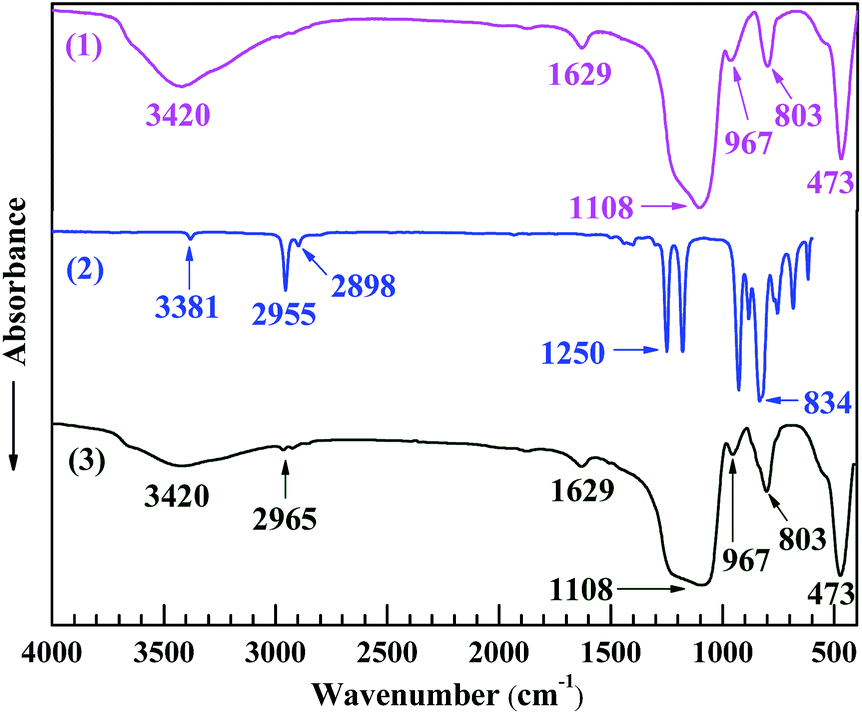 | ||
| Fig. 3 Fourier transform infrared (FTIR) absorption spectra of (1) a nanosilica powder in the transmission mode, (2) 1,1,1,3,3,3-hexamethyldisilazane (HMDS) in the attenuated total reflection (ATR) mode, and (3) an HMDS-modified nanosilica powder in the transmission mode, between 4000 and 400 cm−1 wavenumbers at room temperature. In spectrum 3 of the HMDS-modified nanosilica powder, the absorption band at 3420 cm−1 characteristic of the O–H stretching of its surface –OH groups weakens significantly compared with that in spectrum 1 of the nanosilica powder, which, along with the emergence of a new band at 2965 cm−1 characteristic of the C–H antisymmetric stretching of –CH3 groups, indicates the occurrence of a significant surface modification of the nanosilica powder with HMDS that converts –OH to –CH3 groups. | ||
TGA also was used to corroborate the generation of the methyl-attached organic nanosilica from the surface modification. As shown in Fig. 4, the weight losses of both the nanosilica (curve 1) and HMDS-modified nanosilica (curve 2) occurred sequentially in two stages.3,36 The first stage occurred from RT to 100 °C due mainly to an evaporation of their bound (i.e. adsorbed) water and also possibly to a condensation reaction of the surface Si–OH groups during their thermal aggregation to release water molecules, both of which would increase with increasing the surface –OH concentration. It should be noted that, although they both were well dried in vacuo (at 150 °C for 5 h and at 80 °C for at least 48 h, respectively), the nanosilica and modified nanosilica samples would quickly adsorb water during the short time period of after taken out of the vacuum oven and before subjected to the TGA. It is seen that the nanosilica lost its weight more than did the modified nanosilica by ∼0.7% in the first stage, indicating that the –OH concentration was decreased on the nanosilica surfaces upon its modification presumably owing to the substitution of part of the hydroxyls by the trimethylsilyl groups. The second stage occurred from 100 to 800 °C due to the burning of their surface groups: for the nanosilica, the burning primarily involved the further condensation between its surface Si–OH groups themselves; while, for the organomodified nanosilica, the burning might cause more weight loss in that both its surface hydroxyls and organic-groups participated. In a typical 200–700 °C temperature range, it is observed from Fig. 4 that the modified nanosilica lost 3.8% of its weight, greater than 2.5% lost by the nanosilica, which verifies again the formation of grafted multiple –CH3 groups to the nanosilica surfaces by HMDS (cf. Scheme 2b) that burned off more in weight than did the nanosilica hydroxyls.
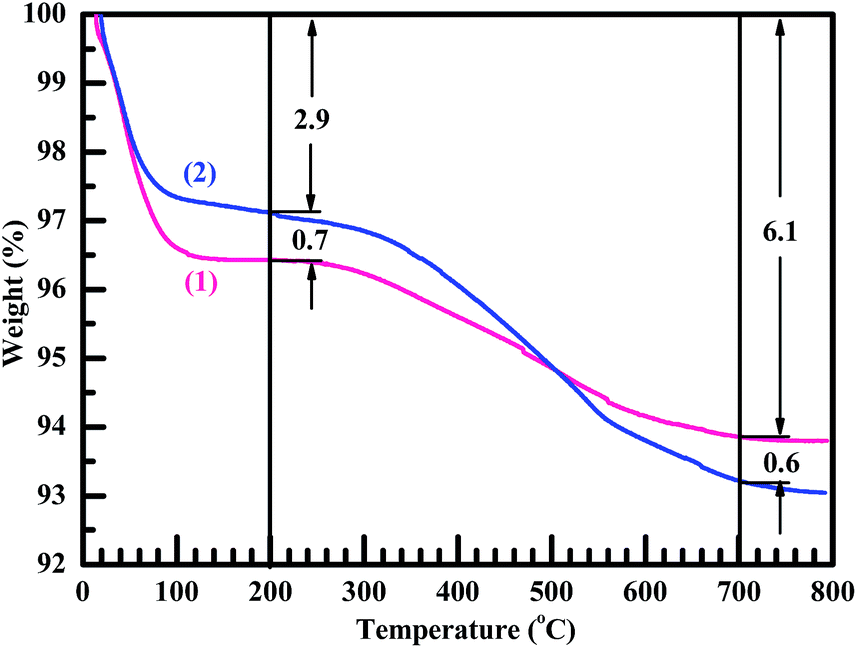 | ||
| Fig. 4 (1) Thermogravimetric analysis (TGA) thermogram of a nanosilica powder against (2) that of the nanosilica powder surface-modified with 1,1,1,3,3,3-hexamethyldisilazane (HMDS), from room temperature until 800 °C at a rate of 10 °C min−1. From 200 to 700 °C, the weight loss of the HMDS-modified, organic nanosilica from the burning of its surface multiple –CH3 groups is 3.8%, larger than that (2.5%) of the nanosilica from the burning of its surface –OH groups, which verifies a successful grafting of –CH3 groups onto the nanosilica surfaces (i.e. conversion of the surface –OH to –CH3 groups) through an HMDS modification reaction. | ||
Hydrophobicity enhancement of epoxy resin coatings upon their grafting with a fluoroalkyl-containing organosilane
The contact angle (θ) values of water against all the EP44-based resin coatings cured (whose compositions were given in Table 1(a–f)) were measured as presented in Table 3. It is seen that the water θ of the cured EP44 coating was 83.8°, revealing that it slightly was hydrophilic essentially from a concentration of hydroxy groups present. However, upon the pre-cure chemical grafting of TMDMS to EP44 molecules, the cured TMDMS-grafted EP44 coating had a considerably larger water θ of 105.5°, which suggests a significant enhancement of its hydrophobicity. Further, when the different contents (0.5, 1, 3 and 5 wt%) of the HMDS-modified nanosilica were added to the uncured TMDMS-grafted EP44, the water θ's of the cured composite coatings were 101.3°, 101.7°, 105.9° and 104.4°, respectively, indicating that their hydrophobicity changed little (or only slightly) with the modified nanosilica concentration despite the presence of unreacted hydrophilic hydroxyls in the added nanosilica even upon its surface modification with HMDS, as discussed in Fig. 3 and 4. From these results, it is summarised that, once grafted with TMDMS, the cured EP44 coating became significantly more hydrophobic regardless of the content of the modified nanosilica incorporated after the graft modification.| EPR-based coating no. | Base resin | θ (degree) | Image |
|---|---|---|---|
| (a) | EP44 | 83.8 |  |
| (b) | TMDMS-grafted EP44 | 105.5 |  |
| (c) | TMDMS-grafted EP44/0.5 wt% HMDS modified nanosilica | 101.3 |  |
| (d) | TMDMS-grafted EP44/1 wt% HMDS modified nanosilica | 101.7 |  |
| (e) | TMDMS-grafted EP44/3 wt% HMDS modified nanosilica | 105.9 |  |
| (f) | TMDMS-grafted EP44/5 wt% HMDS modified nanosilica | 104.4 |  |
The rationale behind this hydrophobic improvement may lie in a combination of the introduction of hydrophobic trifluoropropyl groups and a decrease in the hydrophilic hydroxyl concentration both from the graft reaction, with the effect of the former predominating over that of the latter. In the molecular structure of EP44 shown in Scheme 1b or Fig. 2d, the value of n is equivalent to ∼0.4, while the two end epoxides, respectively, are converted to two hydroxyls upon EP44's cure. This contrast infers that, even though the EP44–TMDMS reaction was 100% adequate, the consumed hydroxyls merely accounted for ∼1/6 the total hydroxyls of the cured EP44; in other words, upon the grafting, the –OH concentration of the cured EP44 coating was reduced at the most by 17%, which should contribute little to such a large increase in its hydrophobicity (i.e. water θ) from 83.8° to 105.5°. Instead, owing to their superhydrophobic character, the trifluoropropyl groups present in the TMDMS grafts of the cured EP44 coating likely were much more responsible for the significant enhancement of its hydrophobicity.
Improvement in the anticorrosive performance of epoxy resin coatings upon their hydrophobic modification
Although they act as a barrier protection to retard the corrosion of their underneath metallic substrate, organic polymeric coatings are far from perfect for corrosion prevention. For instance, within the EP44-based coatings investigated in this work, there might develop a number of defective pores (free volumes, voids, cracks, etc.) or channels made of them, during their preparation processes especially upon solvent evaporation, that were accessible to the corrosives (i.e. water, electrolyte (NaCl), oxygen, other molecules and ions) by infiltration. Once the NaCl aqueous solution penetrated the coatings to reach the tinplate substrate, the defective pores or channels, constituting the cathodes of electrochemical galvanic cells, were populated with reactive “residents” (e.g. water, oxygen, etc.) that were subject to cathodic reduction reaction (reactions (2) and (3) below), in response to the anodic oxidation reaction (reaction (1) below) on the tinplate anode. Meanwhile, the anodic Fe3+ cations from reaction (1) migrated across the substrate–coating interface to react with the cathodic OH− anions from reactions (2) and (3), resulting in a deposition of Fe(OH)3 and Fe2O3, respectively, into the coating channels by cathodic, non-redox precipitation- and decomposition reactions (reactions (4) and (5) below).Anodic reaction:
| Fe − 3e− → Fe3+ | (1) |
Cathodic reaction:
| 2H2O + 2e− → 2OH− + H2 | (2) |
| O2 + 2H2O + 4e− → 4OH− | (3) |
Precipitation reaction:
| Fe3+ + 3OH− → Fe(OH)3 | (4) |
Decomposition reaction:
| 2Fe(OH)3 → Fe2O3 + 3H2O | (5) |
The EIS Bode plots (scatter plots), i.e. plots of the logarithm of impedance magnitude (log|Z|) vs. that of frequency (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f), of the six compositions of coating specimens upon their increasing immersion times (1, 24, 240, 480, 720 and 960 h) in the NaCl solution are shown in Fig. 5, from which their Nyquist plots (scatter plots), i.e. plots of the opposite of capacitive reactance (−Z′′) vs. resistance (Z′), are drawn in Fig. 6 and plots (bar plots) of the |Z| at 1 × 10−2 Hz, a measure of the anticorrosive performance of the coating specimens (i.e. coatings interfacing with both the electrolyte solution and the substrate), vs. immersion time are made in Fig. 7a. To better understand the anticorrosion mechanism of the coatings, electrochemical equivalent-circuit models were employed to simulate the EIS experimental data. It turned out that all the Bode and Nyquist plots on 1 and 24 h of immersion of the six compositions of coatings were well fitted preferably by a three-impedance model, illustrated in Chart 1a, consisting of the coating resistance (Rc) and capacitance (CPEc), the charge-transfer resistance (Rct) and double-layer capacitance (CPEdl) at the electrolyte–coating interface, and the charge-transfer resistance (Rsf) and double-layer capacitance (CPEsf) at the coating–substrate interface, but not by a simple model of the Rc in parallel to the CPEc. This infers that, even in an early stage of the corrosion process, none of the coatings followed a classical barrier-type behaviour; instead, all of them were infiltrated by the NaCl solution all the way to the substrate to induce the electrode reactions (reactions (1)–(5)) even upon 1 h of immersion, probably due to their small thicknesses (52 ± 4 μm) that were unique in this work. Nevertheless, the Bode and Nyquist data of all the coatings upon 240 h and longer immersion times were found to be fitted more satisfactorily using a four-impedance model represented in Chart 1b. In addition to those in model a, incorporated into model b is a fourth Rdiff–CPEdiff impedance element, which presumably suggests a significance of the finite-layer diffusion,37 in the later corrosion stage, that was caused by an occlusion of the coating channels for the corrosives infiltration by the corrosion products, basically Fe(OH)3 and Fe2O3 from reactions (4) and (5), respectively. According to models a and b for the respective immersion times of 1–24 h and 240–960 h, added to Fig. 5 and 6 are all the simulated results (fitted curves), from which the evaluated Rc, a measure of the anticorrosive performance of the coating itself without respect to its interfacing with the electrolyte solution as well as with the substrate, is plotted as a function of the immersion time for each composition of coating as shown in Fig. 7b (bar plots).
f), of the six compositions of coating specimens upon their increasing immersion times (1, 24, 240, 480, 720 and 960 h) in the NaCl solution are shown in Fig. 5, from which their Nyquist plots (scatter plots), i.e. plots of the opposite of capacitive reactance (−Z′′) vs. resistance (Z′), are drawn in Fig. 6 and plots (bar plots) of the |Z| at 1 × 10−2 Hz, a measure of the anticorrosive performance of the coating specimens (i.e. coatings interfacing with both the electrolyte solution and the substrate), vs. immersion time are made in Fig. 7a. To better understand the anticorrosion mechanism of the coatings, electrochemical equivalent-circuit models were employed to simulate the EIS experimental data. It turned out that all the Bode and Nyquist plots on 1 and 24 h of immersion of the six compositions of coatings were well fitted preferably by a three-impedance model, illustrated in Chart 1a, consisting of the coating resistance (Rc) and capacitance (CPEc), the charge-transfer resistance (Rct) and double-layer capacitance (CPEdl) at the electrolyte–coating interface, and the charge-transfer resistance (Rsf) and double-layer capacitance (CPEsf) at the coating–substrate interface, but not by a simple model of the Rc in parallel to the CPEc. This infers that, even in an early stage of the corrosion process, none of the coatings followed a classical barrier-type behaviour; instead, all of them were infiltrated by the NaCl solution all the way to the substrate to induce the electrode reactions (reactions (1)–(5)) even upon 1 h of immersion, probably due to their small thicknesses (52 ± 4 μm) that were unique in this work. Nevertheless, the Bode and Nyquist data of all the coatings upon 240 h and longer immersion times were found to be fitted more satisfactorily using a four-impedance model represented in Chart 1b. In addition to those in model a, incorporated into model b is a fourth Rdiff–CPEdiff impedance element, which presumably suggests a significance of the finite-layer diffusion,37 in the later corrosion stage, that was caused by an occlusion of the coating channels for the corrosives infiltration by the corrosion products, basically Fe(OH)3 and Fe2O3 from reactions (4) and (5), respectively. According to models a and b for the respective immersion times of 1–24 h and 240–960 h, added to Fig. 5 and 6 are all the simulated results (fitted curves), from which the evaluated Rc, a measure of the anticorrosive performance of the coating itself without respect to its interfacing with the electrolyte solution as well as with the substrate, is plotted as a function of the immersion time for each composition of coating as shown in Fig. 7b (bar plots).
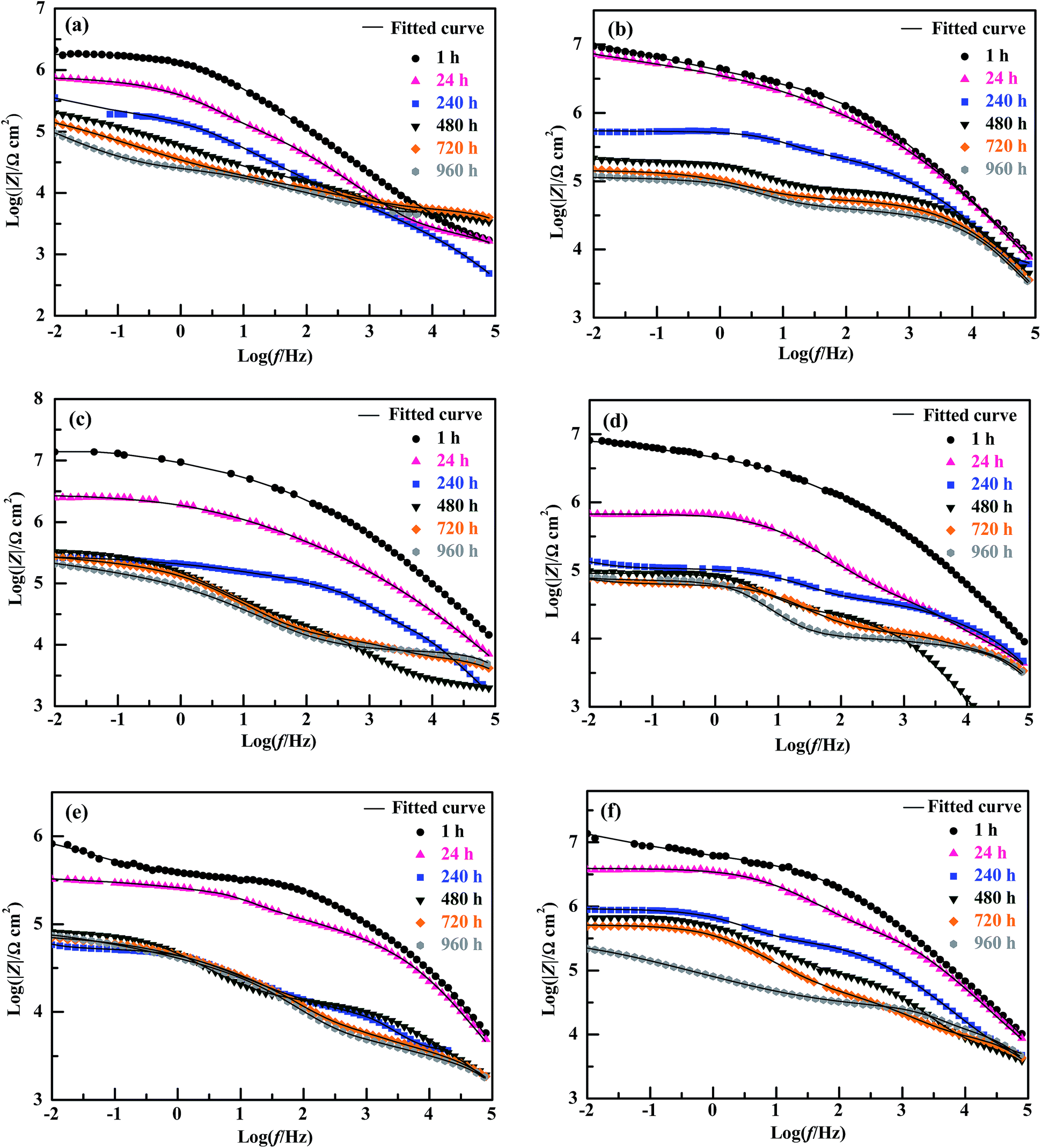 | ||
| Fig. 5 Bode plots (i.e. plots of log|Z| vs. logf) and their fitted curves by electrochemical equivalent circuit models for the corrosion inhibitive coatings, applied onto a 2 wt% NaOH solution-treated tinplate substrate, of (a) a bisphenol-A based epoxy resin of an epoxy value of 0.440 (i.e. EP44), (b) a 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) grafted EP44, and the mixtures of the TMDMS-grafted EP44 filled with different contents (wt%): (c) 0.5; (d) 1; (e) 3; (f) 5 of 1,1,1,3,3,3-hexamethyldisilazane (HMDS) modified nanosilica powder that all are cured in an n-butanol/xylene (7/3 w/w) mixed solution with an amino-terminated polyamide-based curing agent (i.e. G650), upon their different immersion times of 1, 24, 240, 480, 720 and 960 h. | ||
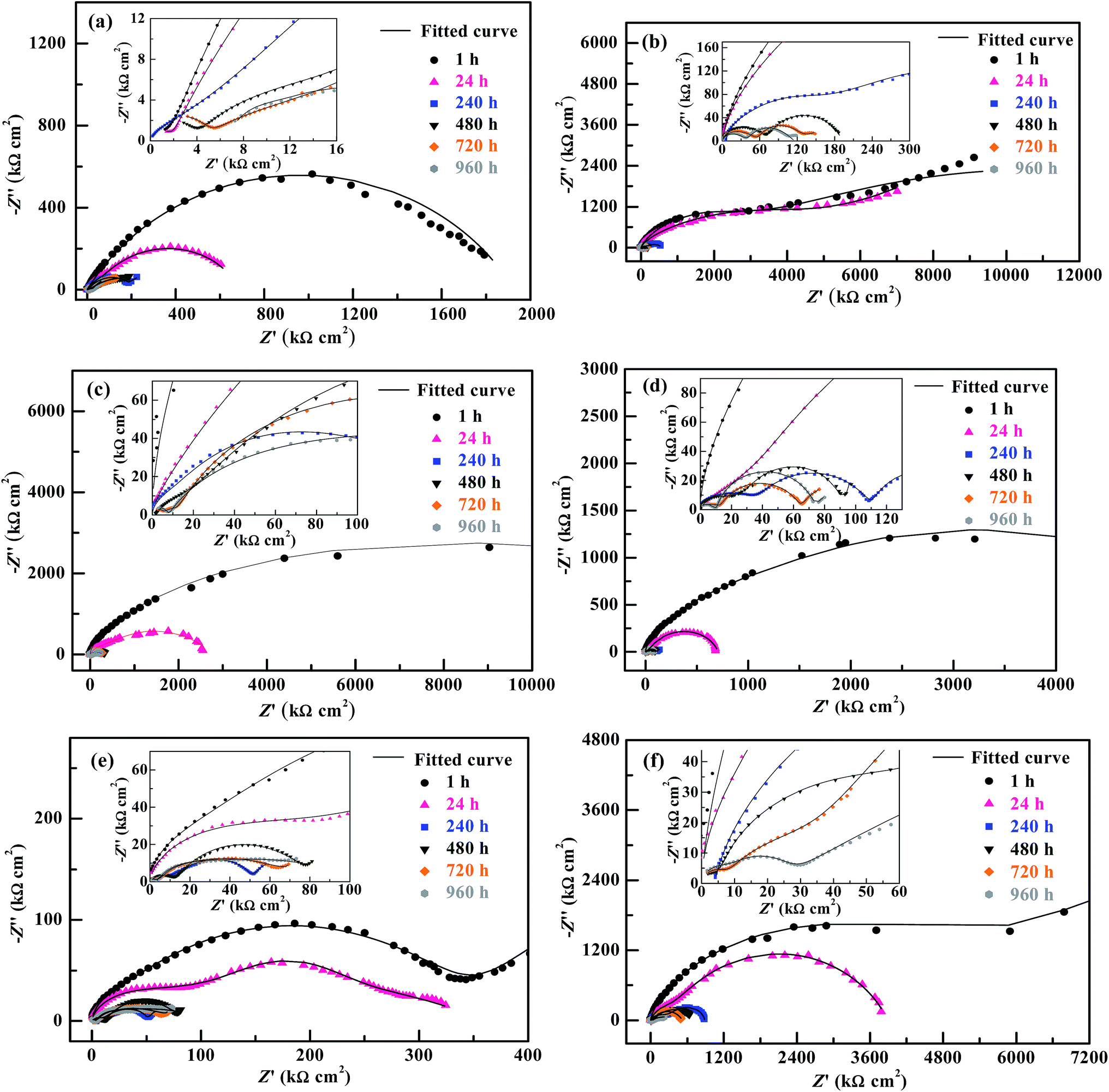 | ||
| Fig. 6 Nyquist plots (i.e. plots of −Z′′ vs. Z′) and their fitted curves by electrochemical equivalent circuit models for the corrosion inhibitive coatings, applied onto a 2 wt% NaOH solution-treated tinplate substrate, of (a) a bisphenol-A based epoxy resin of an epoxy value of 0.440 (i.e. EP44), (b) a 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) grafted EP44, and the mixtures of the TMDMS-grafted EP44 filled with different contents (wt%): (c) 0.5; (d) 1; (e) 3; (f) 5 of 1,1,1,3,3,3-hexamethyldisilazane (HMDS) modified nanosilica powder that all are cured in an n-butanol/xylene (7/3 w/w) mixed solution with an amino-terminated polyamide-based curing agent (i.e. G650), upon their different immersion times of 1, 24, 240, 480, 720 and 960 h. In each of graphs a–f, the inset given is a magnification of the plots and fitted curves in a high frequency range (i.e. in the lower left corner). | ||
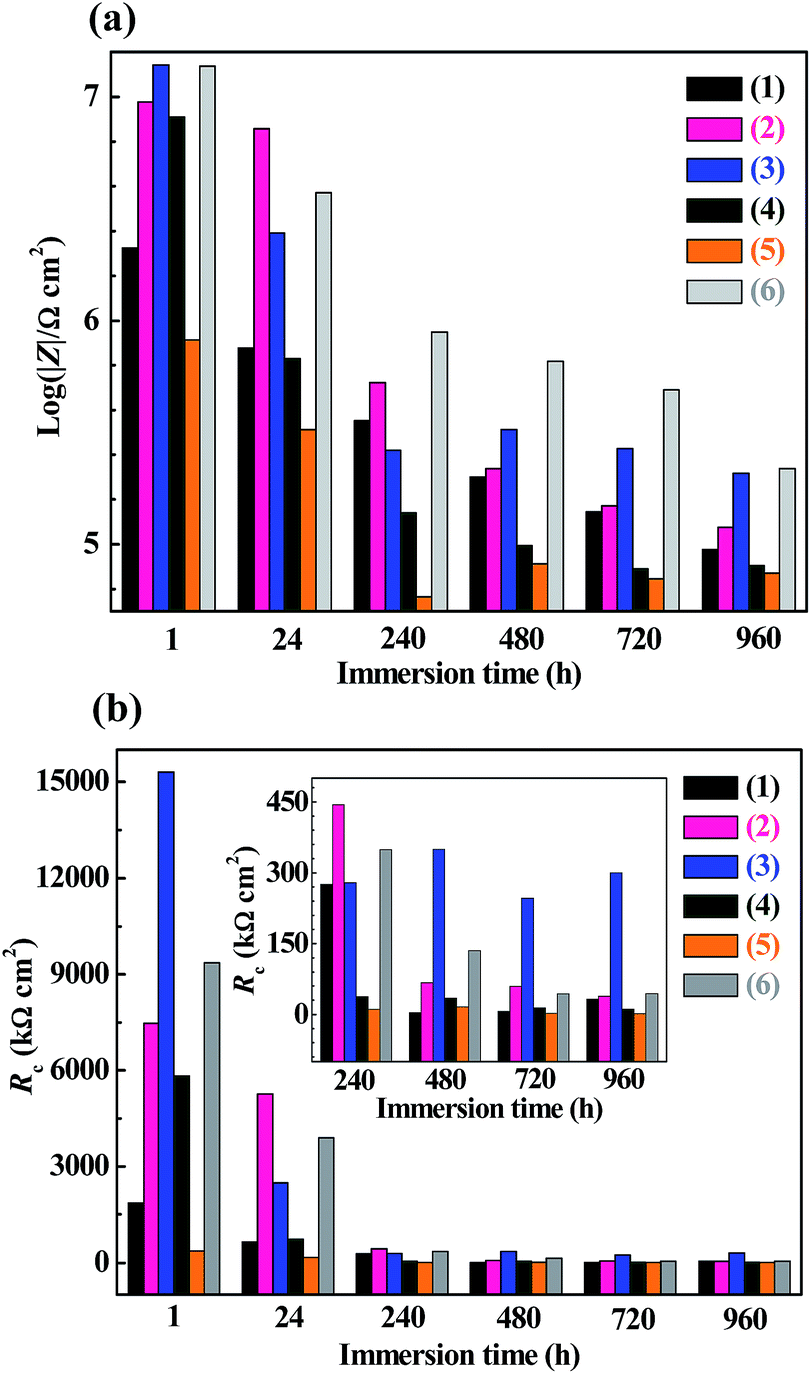 | ||
| Fig. 7 Histograms of (a) the impedance magnitudes (|Z|'s) at a frequency of 1 × 10−2 Hz and (b) the coating resistances (Rc's) both as functions of the immersion time into an NaCl aqueous solution of 3.5 wt%, for the corrosion preventive coatings, against a 2 wt% NaOH solution-treated tinplate substrate, of (1) a bisphenol-A based epoxy resin of an epoxy value of 0.440 (i.e. EP44), (2) a 3,3,3-trifluoropropylmethyldimethoxysilane (TMDMS) grafted EP44, and the mixtures of the TMDMS-grafted EP44 filled with different contents (wt%): (3) 0.5; (4) 1; (5) 3; (6) 5 of 1,1,1,3,3,3-hexamethyldisilazane (HMDS) modified nanosilica powder that all are cured in an n-butanol/xylene (7/3 w/w) mixed solution with an amino-terminated polyamide-based curing agent (i.e. G650). | ||
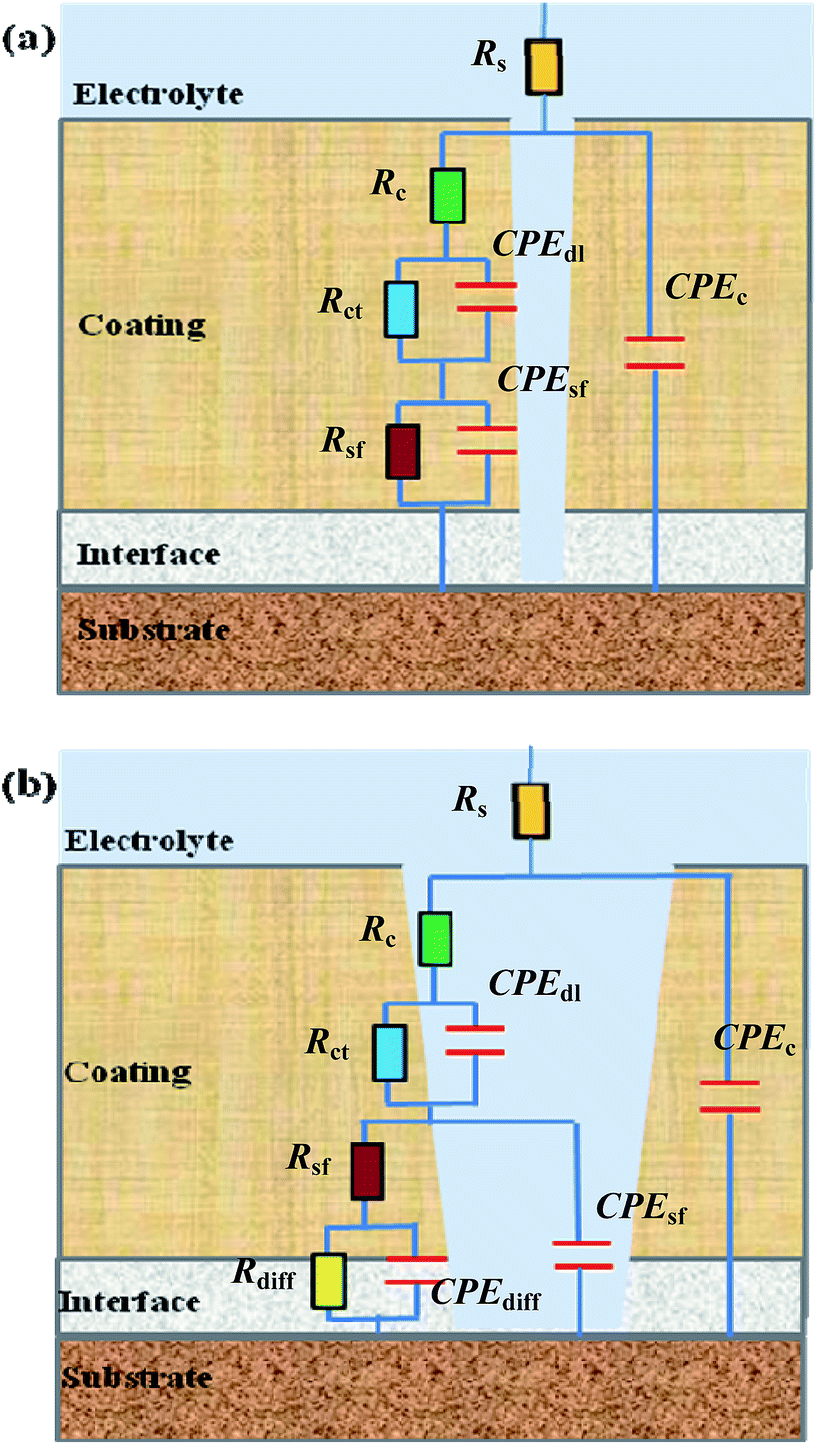 | ||
| Chart 1 Two equivalent-circuit models used to fit the electrochemical impedance spectroscopy (EIS) experimental data presented in Fig. 5 and 6, where model a is appropriate for an early stage of the corrosion process (i.e. at short immersion times) while model b is suitable for the later stage of the corrosion process (i.e. at longer immersion times) when the blockage effect of the corrosion product(s) is significant. In model a, the Rs, Rc, Rct and Rsf, respectively, represent the electrolyte solution resistance, coating resistance, charge-transfer resistances at the electrolyte–coating interface and from the coating to the substrate, while the CPEc, CPEdl and CPEsf, respectively, represent the coating capacitance, double-layer capacitances at the electrolyte–coating and coating–substrate interfaces; in model b, the Rdiff and CPEdiff, respectively, are the resistance and capacitance both associated with the finite-layer diffusion caused by an occlusion of the corrosives channels in the coating by the corrosion product(s), in addition to the presence of all the R's and CPE's shown in model a. | ||
Qualitatively, for the EP44 coating specimen investigated (Fig. 6a), typical, single capacitive arcs of decreasing sizes were observed upon its respective 1 and 24 h immersion times in the NaCl solution, which usually were dictated by a dominant electrode process of reactions (1) and (2) in an initial stage of the corrosion. With an increase in the immersion time to 240 h, the resulting smaller capacitive arc along with a successive short, sleek line probably was associated with the Warburg impedance due to a significance of dissolved oxygen diffusion at that time,38 which, characteristic of an additional electrode process caused by reactions (1) and (3), intensified the Fe3+ diffusion from accelerated tinplate corrosion across the substrate–coating interface.39 When the immersion time further was prolonged beyond 480 h, it seemed that double capacitive arcs occurred upon each of the immersion times, whose combined size further reduced increasingly as the immersion time elapsed from 480 to 960 h. These suggest that, while the immersion time of the EP44 coating specimen steadily was extended from 1 to 960 h, its capacitive arc(s) size and thus anticorrosive performance were decreased monotonously, which arose obviously from progress in (i.e. intensification of) the corrosives diffusion across the coating that enhanced the electrode reactions causing the tinplate corrosion and hence increased the coating conductivity.30 For the TMDMS-grafted EP44 coating specimen (Fig. 6b), despite the observation that an upward tail (in a low frequency region) arose in addition to the capacitive arc (in the higher frequency region) upon its 1, 24 and 240 h of immersion, its anticorrosion behaviour generally resembled that of the EP44 coating specimen (Fig. 6a) in that (1) double capacitive arcs occurred on 480–960 h of immersion, some even with tails, and (2) the capacitive arc(s) size reduced steadily across the entire immersion process. However, upon any of all their immersion times (1–960 h), the capacitive arc(s) size of the TMDMS-grafted EP44 coating specimen (Fig. 6b) essentially was greater than that of the EP44 coating specimen (Fig. 6a) (note that the abscissa and ordinate scales of Fig. 6b both are considerably larger than those of Fig. 6a). This indicates qualitatively that the coating specimen of EP44 was improved in regard to anticorrosive performance upon its hydrophobic modification with the TMDMS grafts.
More quantitatively, it was observed that both the |Z|'s at 1 × 10−2 Hz of the EP44 coating specimen (Fig. 7a(1)) and of the TMDMS-grafted EP44 coating specimen (Fig. 7a(2)) decreased monotonically with their immersion time from 1 to 960 h, which, agreeing with the qualitative results analysed from Fig. 6a and b respectively, corroborate that the anticorrosive performance of both the coating specimens was deteriorated steadily as their immersion proceeded. Nevertheless, with reference to the EP44 and TMDMS-grafted EP44 coatings themselves, although their anticorrosion behaviours (Fig. 7b(1) and (2)) bore general resemblance to those of their respective specimens (Fig. 7a(1) and (2)) in that both the Rc's essentially reduced with increasing the immersion time, the Rc of the TMDMS-grafted EP44 coating decreased little (Fig. 7b(2)) and, even, that of the EP44 coating turned to increase slightly (Fig. 7b(1)) both with increasing their immersion time beyond 480 h. These presumably were ascribed to the significance, in a terminal stage of the corrosion process, of a barrier protection of the corrosion products deposited into the coating.18 More importantly, it is seen from Fig. 7 that increments of the resistive properties of the TMDMS-grafted EP44 coating occurred, when compared with those of the EP44 coating, in terms of both the |Z| read typically at 1 × 10−2 Hz (Fig. 7a(1) vs. (2)) and the Rc extrapolated by the models fits (Fig. 7b(1) vs. (2)), at all the immersion times of 1, 24, 240, 480, 720 and 960 h. These reveal quantitatively that the anticorrosive performance of the coating (i.e. both coating specimen and coating itself) of EP44 was enhanced upon its hydrophobic modification by the TMDMS grafting.
This anticorrosion superiority of the TMDMS-grafted EP44 coating could originate exclusively from the chemical structural changes of EP44 on the graft, which, as discussed in the last subsection, primarily gave rise to a significant improvement in its hydrophobicity (water θ from 83.8° to 105.5°). It generally is held that the protection of coatings against substrate corrosion may tentatively be classified into three mechanisms, barrier, sacrificial, and inhibitive,36 where the barrier capability to water, medium for corrosives, can further be defined by the adsorption ability of water onto coatings, the diffusion rate of water across coatings, and the accessibility of a substrate to water.40 In this work, the notably increased hydrophobicity (or water θ) of the TMDMS-grafted EP44 coating indicates its much lower rates of water adsorption and diffusion that then also caused a well decreased water concentration adsorbed on the substrate, the three of which might effectively raise the Rct, Rc and Rsf of the Chart 1 models, respectively. Hence, it was this significantly stronger ability of hydrophobic barrier41 that primarily was responsible for the improvement, stated above, in the anticorrosive performance of both the coating specimen and the coating itself of the TMDMS-grafted EP44. Besides, the unreacted, residual methoxy groups present in the TMDMS grafts, as evidenced by the NMR result (Fig. 2(c and f)) discussed in the first subsection, were subject to water absorption (i.e. digestion) via their hydrolysis, which decreased as well the water infiltration into the coating; this water-digestive “barrier”, although minor, might also contribute to the anticorrosion enhancement.
An observation can be made from Fig. 6 and 7 as follows: qualitatively, the capacitive arc(s) size of the TMDMS-grafted EP44 coating specimen (Fig. 6b) was significantly greater than that of the EP44 coating specimen (Fig. 6a), and, more quantitatively, the |Z| (Fig. 7a(2)) and Rc (Fig. 7b(2)) of the former were 0.5–1 order of magnitude larger than those (Fig. 7a(1) and b(1)) of the latter, both only upon their 1 and 24 h immersion times; on the longer times of immersion, all the increases in the capacitive arc(s) size, |Z| and Rc were more or less minor from the graft modification. Nevertheless, since the immersion experiments of the two coating specimens virtually were simulated, expedited versions of their practical applications, this observation may not prevent us from judging the anticorrosive performance of the EP44 coating to significantly be improved upon its grafting with TMDMS, basically from the EIS data in the early stages of their experimental corrosion processes (i.e. on their 1 and 24 h immersion times).
Effect of nanosilica content on the corrosion inhibition of composite coatings of a hydrophobic epoxy resin matrix
For the composite coating specimens of the TMDMS-grafted EP44 filled with 0.5, 1, 3 and 5 wt% of the HMDS-modified nanosilica, their respective Nyquist plot profiles (Fig. 6(c–f)) essentially were all similar to those of the EP44 (Fig. 6a) and TMDMS-grafted EP44 (Fig. 6b) coating specimens. These similarities include: (1) in an early stage of the corrosion (upon 1–480 h of immersion), a single capacitive arc occurred, sometimes along with an upward tail, while, in the later stage (on 24–960 h of immersion), double capacitive arcs arose, possibly followed by an upward tail as well, the critical immersion time for the early-to-later stage transition contingent apparently upon the corrosion inhibitive efficiency of the composite coating specimens (i.e. a stronger inhibition of corrosion dictated a longer critical immersion time); and (2) despite its possible fluctuations in the later stage, the capacitive arc(s) size generally reduced as the immersion (i.e. corrosion) progressed steadily. More quantitatively, for all the composite coatings investigated, both the |Z|'s at 1 × 10−2 Hz (Fig. 7a(3)–(6)) of the coating specimens and the Rc's (Fig. 7b(3)–(6)) of the coatings themselves primarily were decreased with the immersion time until 960 h, indicating that the corrosion inhibition (i.e. anticorrosive performance) of the coatings weakened on the whole as the corrosives infiltration into them intensified gradually. However, abnormal variations (e.g. little decrease(s), slight increase(s), or their combination) in the |Z| at 1 × 10−2 Hz and/or Rc occurred in the later stages of after 240, 240, 240 and 720 h immersion times, respectively, for the composite coatings containing 0.5, 1, 3 and 5 wt% of the modified nanosilica; again, these originated probably from the significance of a physical barrier, to the corrosives, provided by the corrosion products, basically Fe(OH)3 and Fe2O3, that were deposited into the composite coatings through cathodic reactions (4) and (5), respectively.To discuss how the loading level (0–5 wt%) of the modified nanosilica affected the corrosion inhibition of the composite coatings, we now focus on comparisons, in Fig. 7, of their |Z| @1 × 10−2 Hz or Rc values (including that of the TMDMS-grafted EP44 coating) upon each of all the immersion times. It is observed from Fig. 7a that, as the modified nanosilica content was raised from 0 wt% (Fig. 7a(2)) steadily to 5 wt% (Fig. 7a(6)), the |Z|'s at 1 × 10−2 Hz upon 1, 480, 720 and 960 h of immersion first increased, then decreased, and finally increased again, going through a maximum followed by a minimum at 0.5 and 3 wt% respectively, while those on 24 and 240 h of immersion first decreased and then increased, giving rise to a minimum still at 3 wt%. These apparently contradictory observations (i.e. double extrema vs. a single extremum in the |Z|) may actually be reconciled if one thinks that, specifically, the optimum nanosilica content for all the maxima in the |Z| lay invariably between 0 and 0.5 wt%. This means that all the |Z|'s at 1 × 10−2 Hz of the composite coating specimens upon their increasing immersion times (1–960 h) presumably followed the above double extrema rule, respectively, at critical 0–0.5 and ∼3 wt% contents of the modified nanosilica, as its concentration was enhanced from 0 wt% all the way to 5 wt%. Interestingly enough, it is seen from Fig. 7b(2)–(6) that the Rc of the composite coatings themselves seemed to exhibit exactly the same double extrema behaviour on each of their 1–960 h of immersion, rising to be maximised at a first critical content of 0–0.5 wt%, then dropping to be minimised at a second, higher critical content of ∼3 wt%, and finally rising again with steady increases in the nanosilica concentration from 0 to 5 wt%. Since the reconciled |Z| @1 × 10−2 Hz and Rc were both measures of the corrosion inhibition of the composite coatings, the established double extrema (or dual critical concentrations) relationship obviously constituted the effect of the modified nanosilica content on the corrosion inhibition of the composite coatings having the hydrophobic TMDMS-grafted EP44 matrix, whose mechanism will be discussed in detail in the next subsection.
A tentative dual critical concentrations mechanism of the nanosilica content effect
As revealed from the water θ data of the composite coatings presented in Table 3, addition of the modified nanosilica by 0.5–5 wt% changed little their hydrophobicity. This suggests that the found double-extrema behaviour in corrosion inhibition, respectively, at the dual critical nanosilica concentrations originated essentially from variations in the physical barrier ability of the nanosilica instead of in the hydrophobic barrier capability of the composite coatings. As mentioned in the Introduction, incorporating IC such as silica nanoparticles into an EPR coating has generally been shown to improve its corrosion inhibition by filling the defective pores (free volumes, voids, cracks, etc.) present that are susceptible to corrosives;24–28 however, too many IC nanoparticles beyond their critical concentration may deteriorate the corrosion inhibition due to an increase in the composite coating porosity from size exclusion of significantly aggregated IC particles.24,29 As such, only the maximum (i.e. a single extremum) in the corrosion inhibition has been reported of EPR/IC composite coatings at a critical concentration (cc) of the IC over the 0–10 wt% IC loading levels investigated. Nevertheless in this work, we seemed to uniquely locate a second, higher cc (cc2) of the modified nanosilica, successively after its (first) cc (cc1) causing a maximum in the corrosion inhibition, that gave rise to a minimum in the corrosion inhibition of the composite coatings of the hydrophobic TMDMS-grafted EP44 matrix.This nanosilica content effect, mechanistically referred to as a dual critical concentrations behaviour, is therefore interpreted tentatively with reference to Scheme 5. First, be advised that a rise in the modified nanosilica concentration, due to its increased tendency to aggregate especially thermally during the vacuum drying at 80 °C for ≥8 h (cf. Scheme 3), led to an increase in its (mean) size. When a small concentration of them were added to the TMDMS-grafted EP44 coating full of increasing sizes of defective pores that were accessible to the corrosives, the nanosilica particles, at their small size, partially filled the pores whose sizes were larger, mostly at their plural form (Scheme 5a); this filling acted as a degree of physical barrier to the infiltration of the corrosives into the pores, reducing the effective cathodic area (i.e. pore volume) for the electrode reactions and hence improving the corrosion inhibition of the coating. As their concentration was raised to the cc1, the silica nanoparticles of an increased size filled, to the most extent, all the pores of larger sizes present (Scheme 5b), maximising the physical barrier protection and thus the corrosion inhibition of the coating. However, while their concentration further was increased above the cc1, the silica nanoparticles were large in size enough to be size-excluded from (i.e. vacate) such many pores of smaller sizes that the ability of physical barrier to the corrosives began to weaken, which deteriorated the corrosion inhibition of the coating; concomitant with this was a gradual transition of the particles filling mode from plural to singular (Scheme 5c). Typically, the moment their concentration was enhanced up to the cc2, the nanosilica particles grew too large in size to fill any of all the increasing sizes of pores due to their entire size exclusion (Scheme 5d), which maximised the effective cathodic area and thereby minimised the corrosion inhibition of the coating. And, ultimately, once their concentration was increased beyond the cc2, the silica particles, all size-excluded from the pores, became increasingly larger in size and denser in number, thus subject to formation of at least locally continuous, outside-of-pore physical barriers (Scheme 5e); this behaviour, cutting off to an extent the percolated diffusion of the corrosives, obviously turned to enhance the corrosion inhibition of the coating again.
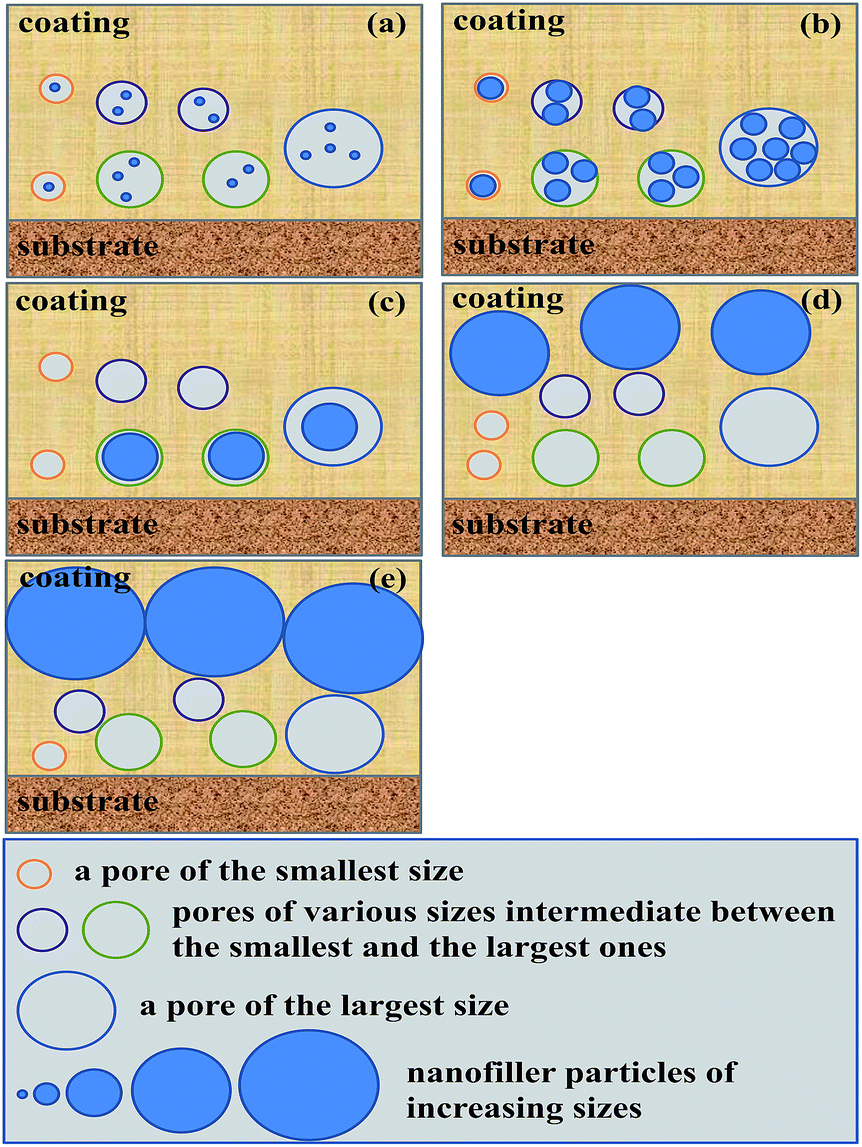 | ||
| Scheme 5 Schematic of a tentative, dual critical concentrations mechanism for the interpretation of the effect of nanofiller content on the corrosion inhibition, by filling the defective pores (free volumes, voids, cracks, etc.) for the diffusion detour of corrosives, of hydrophobic polymer-matrix composite coatings, where, with a steady enhancement of its concentration from (a) to (d), the nanofiller becomes increasingly larger in (mean) size due to its aggregation: (a) at their small concentrations and sizes, the nanofiller particles fill partially and largely plurally the pores of different sizes present in the coatings; (b) as their concentration is increased to a critical value, cc1, the nanoparticles fully occupy (i.e. jam) all the different-sized pores, mostly at their plural form; (c) when their concentration further is enhanced beyond the cc1, the nanoparticles are large enough to increasingly be size-excluded from part of the pores of smaller sizes; (d) while their concentration is up to a second, higher critical value, cc2, the nanofiller particles are so large (i.e. larger than the largest size pores) as to fill none of all the sizes of pores; and (e) as their concentration ultimately is raised beyond the cc2, the aggregated particles become extremely larger and denser, acting as an increasingly solid, outside-of-pore barrier to the diffusion of corrosives. | ||
As analysed in the last subsection, the cc1 and cc2, a couple of indices of the dual critical concentrations mechanism, were quantified as 0–0.5 and ∼3 wt%, respectively, for the composite coatings of the TMDMS-grafted EP44 filled with the HMDS-modified nanosilica. This is in sharp contrast to a much higher cc (i.e. cc1), ∼4 wt%, of the single critical concentration mechanism reported for the composite coatings of a bisphenol-A based EPR (Epikote 828) filled with a GPTMS-treated nanosilica.30 The rationale behind this contrast may lie in the following. Most importantly, owing to the significantly more hydrophobic nature of the TMDMS-grafted EP44 matrix than the Epikote 828 matrix, there were much fewer defective pores (or channels made of them), present in the composite coatings investigated in this work, that were accessible to water and the corrosive solutes (molecules, ions, etc.) therein. Secondly, by reducing the nanosilica hydroxyls more upon their higher-conversion reaction with HMDS than with GPTMS, the finer silica nanoparticles from more decreased aggregation prepared in this work filled more effectively the coating pores especially those small-sized ones. Thirdly, compared with the direct (single-step) cure reported of Epikote 828 at 120 °C for 30 min, the much-milder, step cure of the TMDMS-grafted EP44 (cf. step 6 to 7 of Scheme 3: cure in fume hood at RT for 7 days, followed by postcure in vacuo by a ramp to 70 °C in 1 h and then a hold there for 3 h) caused considerably fewer and smaller defective pores, from greatly slowed solvent evaporation, that intrinsically were developed in the composite coatings. These three factors contributed, sequentially decreasingly while all significantly, to the sharp decrease in the cc1, e.g. from ∼4 wt%, to only 0–0.5 wt%, which, further, interestingly was followed by the occurrence of the higher cc2 at ∼3 wt% that would not have otherwise arisen over the 0–10 wt% nanofiller contents investigated.
Conclusions
TMDMS has successfully been grafted onto EP44 molecules to substitute trifluoropropyls for hydroxyls via significant transesterification (i.e. alcoholysis) of the TMDMS methoxyls with the EP44 hydroxyls, as corroborated by FTIR and 1H NMR spectroscopies, under anhydrous DBTDL-catalyst conditions at 95 °C for 8 h. Meanwhile, a nanosilica powder of 30 nm in mean diameter has successfully been modified with HMDS to convert its surface hydroxyls into methyls through electrophilic substitution of the nanosilica hydroxyls by the HMDS trimethylsilyls at 50 °C for 20 h, as confirmed by FTIR spectroscopy and TGA. For comparative investigations, coatings of uniform thicknesses (52 ± 4 μm), onto an NaOH solution-treated tinplate substrate, of EP44, of the TMDMS-grafted EP44, and of the composites of the TMDMS-grafted EP44 filled with increasing contents (0.5, 1, 3 and 5 wt%) of the HMDS-modified nanosilica have been prepared from an n-butanol/xylene solution by the same mild, step cure with an amino-terminated polyamide-based curative. It is revealed from contact angle (θ) measurements of the six coatings that the θ of water against, and thus the hydrophobicity of, the EP44 coating considerably is increased from 83.8° to 105.5° upon its grafting with TMDMS, which then changes little with further addition of the increasing concentrations of the modified nanosilica. Likely, this is due predominantly to the introduction of superhydrophobic trifluoropropyl groups, as well as secondarily to a reduction in the hydrophilic hydroxyl concentration present, both upon the graft modification.The anticorrosion processes of the six coating specimens, upon immersion into an electrolyte (i.e. NaCl) aqueous solution, have therefore been characterised by EIS Bode plots, i.e. plots of the logarithm of impedance magnitude (log|Z|) vs. that of frequency (logf), and Nyquist plots, i.e. plots of the opposite of capacitive reactance (−Z′′) vs. resistance (Z′). These experimental data have further been simulated satisfactorily by appropriate electrochemical equivalent-circuit models, from which the coating resistance (Rc), a measure of the anticorrosive performance of the coating itself without respect to its interfacing with the electrolyte solution as well as with the substrate, has been evaluated for each of the coatings upon each of the immersion times (1, 24, 240, 480, 720 and 960 h) investigated. Generally, the capacitive arc(s) sizes observed from the Nyquist plots, the |Z|'s at 1 × 10−2 Hz read from the Bode plots, and/or the Rc's extrapolated from the model fits are decreased with increasing the immersion time until 240–720 h, while then vary abnormally (e.g. decrease little, increase slightly, or both (i.e. fluctuate)) as the immersion time is increased further, for all the six coatings under investigation. These indicate that the anticorrosive performance of the coatings deteriorates steadily in an early stage of the corrosion process due to progress in (i.e. intensification of) the corrosives (i.e. water, electrolyte (NaCl), oxygen, other molecules and ions) infiltration into their pores, whereas changes little in the later stage presumably owing to a significance of the cathodic deposition of the corrosion products (basically Fe(OH)3 and Fe2O3) into the coatings, which competes against the corrosive diffusion by occluding the coating pores or channels to act as a physical barrier to the corrosives.
More importantly, it has been found that the capacitive arc(s) size, the |Z| @1 × 10−2 Hz, and the Rc of the TMDMS-grafted EP44 coating, respectively, all are larger than those of the EP44 coating upon each of the immersion times, revealing that the anticorrosive performance of the former evidently is improved compared with the latter, as a consequence probably of the significantly increased hydrophobic barrier to the corrosives mediated by water, as well as possibly of the water digestion (i.e. “chemical barrier”) from hydrolysis of the residual, unreacted TMDMS methoxyls, both present in the coating upon the graft modification. Further, when the TMDMS-grafted EP44 coating is filled with the increasing contents (0–5 wt%) of the modified nanosilica, the little changes in its hydrophobicity dictate that the anticorrosive performance of the composite coatings has to be dominated by a corrosion inhibition mechanism by the nanosilica particles filling the defective pores (free volumes, voids, cracks, etc.), present in the coatings, that are accessible to the corrosives. In other words, the silica nanoparticles may act as physical barriers in the pores to the diffusion of water, the corrosive molecules and ions therein, making them take a significant detour to the substrate. Still from the associated, reconciled EIS data (capacitive arc(s) sizes and |Z|'s at 1 × 10−2 Hz) and simulations (Rc's), it has interestingly been found that, as the nanosilica particles concentration steadily is raised from 0 to 5 wt%, the corrosion inhibition of the composite coatings first intensifies probably thanks to an enhancement of the filling rate of the pores, subsequently weakens presumably due to an increase in the porosity from increased size-exclusion of aggregated nanoparticles from the pores, and finally improves again possibly owing to a densification of the outside-of-pore barriers of greatly aggregated, size excluded particles to the corrosives. This constitutes dual critical concentrations of the nanosilica at 0–0.5 and ∼3 wt%, respectively, that give rise to two extrema (a maximum followed by a minimum) in the corrosion inhibition of the composite coatings.
Particularly worth noting is that the dual critical concentrations behaviour discovered above contrasts with a single critical concentration of a GPTMS-modified nanosilica at ∼4 wt%, much higher than the 0–0.5 wt%, that causes the maximum in the corrosion inhibition of composite coatings of Epikote 828 matrix.30 Presumably, the rationale behind this contrast lies primarily in the much more hydrophobic nature of the TMDMS-grafted EP44 matrix that leads to fewer coating pores or channels accessible to the water-mediated corrosives, secondarily in the higher pore-filling efficiency of the less aggregated, finer HMDS-modified silica nanoparticles from their more reduced surface hydroxyls, and thirdly in the fewer and smaller total pores or channels developed within the coatings studied in this work from their milder, step cure process, all of which contribute to the minimisation of the (first) critical concentration to 0–0.5 wt% and hence the occurrence of the second, higher critical concentration at ∼3 wt% across the 0–10 wt% nanosilica contents investigated. To our knowledge, this dual critical concentrations behaviour has been the first work reported in the literature to rule the corrosion inhibition–nanosilica content relationship of composite coatings of an EPR matrix, which may universally be extended to other composite coatings of an organic polymer matrix filled with different contents of an IC nanofiller for corrosion prevention of a metallic substrate.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge with gratitude that this work was financially supported by the Natural Science Foundation of the Hubei Province of China (Contract no. 2016CFB205 and 2014CFA094), as well as by the National Natural Science Foundation of China (Contract no. 51601133) and the Select Overseas Chinese Scholars Science and Technology Activities Foundation of the Ministry of Human Resources and Social Security of China (Contract no. [2013]277).References
- X. Lu, Y. Liu, C. L. Zhou, W. F. Zhang and Z. Xin, RSC Adv., 2016, 6, 5805 RSC.
- T. H. Tran, A. Vimalanandan, G. Genchev, J. Fickert, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2015, 27, 3825 CrossRef CAS PubMed.
- M. Behzadnasab, S. M. Mirabedini, K. Kabiri and S. Jamali, Corros. Sci., 2011, 53, 89 CrossRef CAS.
- Z. Li, B. Y. Qin, X. Y. Zhang, K. Wang, Y. Wei and Y. Ji, RSC Adv., 2015, 5, 104451 RSC.
- L. B. Feng, Y. H. Che, Y. H. Liu, X. H. Qiang and Y. P. Wang, Appl. Surf. Sci., 2013, 283, 367 CrossRef CAS.
- F. Luo, Q. Li, X. K. Zhong, H. Gao, Y. Dai and F. N. Chen, Mater. Corros., 2012, 63, 148 CrossRef CAS.
- M. Qian, A. M. Soutar, X. H. Tan, X. T. Zeng and S. L. Wijesinghe, Thin Solid Films, 2009, 517, 5237 CrossRef CAS.
- B. Ramezanzadeh, E. Raeisi and M. Mahdavian, Int. J. Adhes. Adhes., 2015, 63, 166 CrossRef CAS.
- R. Suleiman, H. Dafalla and B. E. Ali, RSC Adv., 2015, 5, 39155 RSC.
- S. V. Harb, F. C. dos Santos, B. L. Caetano, S. H. Pulcinelli, C. V. Santilli and P. Hammer, RSC Adv., 2015, 5, 15414 RSC.
- A. M. Beccaria, G. Padeletti, G. Montesperelli and L. Chiaruttini, Surf. Coat. Technol., 1999, 111, 240 CrossRef CAS.
- T. O. Siyanbola, K. Sasidhar, B. Anjaneyulu, K. P. Kumar, B. V. S. K. Rao, R. Narayan, O. Olaofe, E. T. Akintayo and K. V. S. N. Raju, J. Mater. Sci., 2013, 48, 8215 CrossRef CAS.
- A. M. Mikhailova, M. Tamboura and M. Q. Jia, Silicon, 2012, 4, 197 CrossRef CAS.
- W. G. Ji, J. M. Hu, J. Q. Zhang and C. N. Cao, Corros. Sci., 2006, 48, 3731 CrossRef CAS.
- X. Yuan, Z. F. Yue, X. Chen, S. F. Wen, L. Li and T. Feng, Prog. Org. Coat., 2015, 86, 41 CrossRef CAS.
- Z. Y. Wang, F. C. Liu, E. H. Han and W. Ke, Mater. Corros., 2013, 64, 54 Search PubMed.
- L. Mathivanan and S. Radhakrishna, Anti-Corros. Methods Mater., 1998, 45, 301 CrossRef.
- X. Y. Lu, Y. Zuo, X. H. Zhao and Y. M. Tang, Corros. Sci., 2012, 60, 165 CrossRef CAS.
- S. H. Zaferani, D. Zaarei, I. Danaee and N. Mehrabian, J. Adhes. Sci. Technol., 2014, 28, 151 CrossRef CAS.
- S. A. Kumar, T. Balakrishnan, M. Alagar and Z. Denchev, Prog. Org. Coat., 2006, 55, 207 CrossRef.
- M.-Y. Jiang, L.-K. Wu, J.-M. Hu and J.-Q. Zhang, Corros. Sci., 2015, 92, 127 CrossRef CAS.
- P. Gupta and M. Bajpai, Adv. Chem. Eng. Sci., 2011, 1, 133 CrossRef CAS.
- W. G. Ji, J. M. Hu, L. Liu, J. Q. Zhang and C. N. Cao, Surf. Coat. Technol., 2007, 201, 4789 CrossRef CAS.
- F. Ansari, R. Naderi and C. Dehghanian, RSC Adv., 2015, 5, 706 RSC.
- C. Motte, M. Poelman, A. Roobroeck, M. Fedel, F. Deflorian and M.-G. Olivier, Prog. Org. Coat., 2012, 74, 326 CrossRef CAS.
- X. M. Shi, T. A. Nguyen, Z. Y. Suo, Y. J. Liu and R. Avci, Surf. Coat. Technol., 2009, 204, 237 CrossRef CAS.
- L. Allie, J. Thorn and H. Aglan, Corros. Sci., 2008, 50, 2189 CrossRef CAS.
- M. C. Lai, K. C. Chang, J. M. Yeh, S. J. Liou, M. F. Hsieh and H. S. Chang, Eur. Polym. J., 2007, 43, 4219 CrossRef CAS.
- V. Palanivel, D. Q. Zhu and W. J. van Ooij, Prog. Org. Coat., 2003, 47, 384 CrossRef CAS.
- A. Ghanbari and M. M. Attar, J. Ind. Eng. Chem., 2015, 23, 145 CrossRef CAS.
- Y. F. Li and J. Cheng, Adv. Mater. Res., 2006, 11, 379 CrossRef.
- V. R. Shayapov, M. N. Chagin and Y. M. Rumyantsev, Inorg. Mater., 2016, 52, 630 CrossRef CAS.
- E. Vyhmeister, H. V. González Lorenzo, R. Bozo, R. R. Maecker, A. Muscat, L. A. Estévez and D. Suleiman, Chem. Eng. Commun., 2016, 203, 908 CrossRef CAS.
- B. Xie and A. J. Muscat, Microelectron. Eng., 2004, 76, 52 CrossRef CAS.
- J. He, X. L. Li, D. Su, H. M. Ji, X. Zhang and W. S. Zhang, J. Mater. Chem. A, 2016, 4, 5632 CAS.
- M. J. Palimi, M. Rostami, M. Mahdavian and B. Ramezanzadeh, J. Coat. Technol. Res., 2015, 12, 277 CrossRef CAS.
- X. Y. Lu, Y. Zuo, X. H. Zhao, Y. M. Tang and X. G. Feng, Corros. Sci., 2011, 53, 153 CrossRef CAS.
- Y. J. Qiang, S. T. Zhang, L. Guo, X. W. Zheng, B. Xiang and S. J. Chen, Corros. Sci., 2017, 119, 68 CrossRef CAS.
- X. Yuan, Z. F. Yue, X. Chen, S. F. Wen and L. Li, Prog. Org. Coat., 2015, 78, 1 CrossRef.
- X. Yuan, Z. F. Yue, X. Chen, S. F. Wen, L. Li and T. Feng, J. Coat. Technol. Res., 2015, 12, 1 CrossRef.
- R. C. Zeng, L. J. Liu, T. T. Pang, F. Zhang, W. W. Zhang, S. Q. Li, H. Z. Cui and E. H. Han, Acta Metall. Sin., 2015, 28, 373 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |