
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochromism and electrochemical properties of complexes of transition metal ions with benzimidazole-based ligand†
Monika Wałęsa-Chorab *,
Radosław Banasz,
Damian Marcinkowski‡
,
Maciej Kubicki and
Violetta Patroniak
*,
Radosław Banasz,
Damian Marcinkowski‡
,
Maciej Kubicki and
Violetta Patroniak
Faculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89b, 61-614 Poznań, Poland. E-mail: mchorab@amu.edu.pl; Fax: +48 618291508; Tel: +48 618291772
First published on 1st November 2017
Abstract
Six new mononuclear complexes of transition metal ions (1–6) with (E)-2-(1-methyl-2-(1-(pyridin-2-yl)ethylidene)hydrazinyl)-1H-benzoimidazole L were synthesized and characterized by spectroscopic methods as well as X-ray diffraction analysis. Structures of these complexes can be divided into three groups depending on the coordination number and environment of the metallic center: the first one of general formula [ML2]X2, the second one - [MLX3] and the third one - [MLX2], where X is anion or solvent molecule. Electrochemical and electrochromic properties of complexes were investigated. Complexes Fe(II) 1, Cu(II) 2–4 and Co(II) 5 were found to be electroactive and their colors depend on the oxidation state of metal center and ligand molecule. Fe(II) complex 1 exhibits purple color in the neutral state and its color changes to yellow after Fe(II) → Fe(III) oxidation, followed by pale-yellow after further oxidation of ligand molecule and to green when the metal center is reduced to Fe(I). The original color can be restored after electrochemical oxidation Fe(I) to Fe(II). It was found that this complex exhibits high color stability in solution during multiple oxidation/reduction cycles. This complex can be therefore regarded as very interesting material for the construction of multielectrochromic devices.
Introduction
Electrochromism is defined as the capability of the material to change its optical properties in response to an external electric stimulus.1 New absorption bands at different parts of the visible or near infrared regions are generated by switching between different redox states. An ideal electrochromic material should exhibit high contrast ratio, good coloration efficiency and long-term stability.2 Very interesting are compounds exhibiting so-called multielectrochromism: various low redox states derived from distinct absorption bands at different wavelengths. Inorganic metal oxides,3–5 organic conjugated polymers6–8 or small molecules9–11 are typical materials for electrochromic uses.Well defined redox properties and intense coloration due to the metal-to-ligand charge transfer (MLCT), ligand-to-metal charge transfer (LMCT) or intervalence charge transfer (CT) of transition metal complexes make them promising materials for their use as electrochromic materials.12–14 The absorption wavelength of transition metal complexes significantly depends on the redox state of their building blocks and their properties can be modified by changing the ligands or metal ions.15 So far, mainly transition metal complexes with oligopyridines or phthalocyanines were examined as electrochromic materials, while electrochromic properties of transition metal complexes with hydrazones have not been widely investigated.
Herein we report the synthesis, crystal structures and electrochromic properties of Mn(II), Fe(II), Co(II) and Cu(II) complexes with the hydrazone-based ligand L ((E)-2-(1-methyl-2-(1-(pyridin-2-yl)ethylidene)hydrazinyl)-1H-enzo[d]imidazole) (Fig. 1).
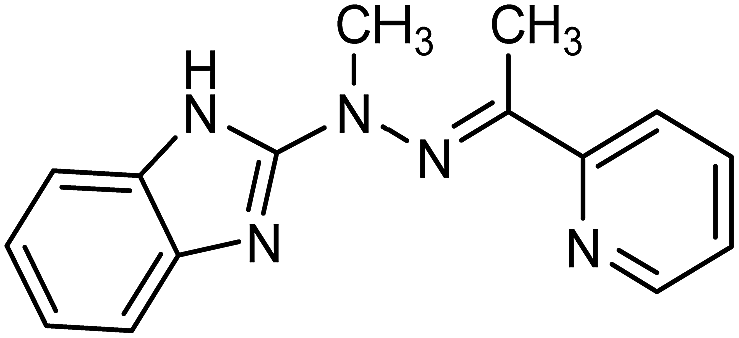 | ||
| Fig. 1 The ligand L. | ||
Ligand L is known to form polymeric complexes with silver(I) ions and mononuclear complexes of europium(III), terbium(III) and gadolinium(III) in which L acts as tridentate N3-donor ligand.16 The complexation reactions of L with different salts of transition metal ions (Fe(II), Cu(II), Co(II) and Mn(II) lead to formation of three types of supramolecular complexes. Obtained complexes have been characterized by spectroscopic methods as well as by X-ray crystallography. Electrochromic properties of complexes have been also investigated.
Result and discussion
The self-assembly of ligand L and appropriate salts of Mn(II), Fe(II), Co(II) and Cu(II) leads to formation of three types of complexes: the first type in which metal ion coordinates with two ligand molecules and has coordination number 6 ([FeL2](CF3SO3)2 (1)), the second one of stoichiometry M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L 1:1 and general formula [MLX3], where X is anion or solvent molecule ([CuL(CF3SO3)2(CH3OH) (2)) and the third one of general formula [MLX2] (M = Cu(II) (3 and 4), Co(II) (5) or Mn(II) (6); X – anion or solvent molecule) where central atom adopts coordination number 5 and its coordination sphere is filled by donor atoms from one ligand molecule and two anions and/or solvent molecules. Different transition metal salts were used to determine the influence of metallic center on electrochemistry and electrochromic properties of obtained compounds. Reactions were carried out in M:L 1:2 (complex 1) or 1:1 (complexes 2–6) molar ratios at room temperature for 48 h in acetonitrile/methanol/dichloromethane solutions (1:1:1 v/v). All complexes were obtained as colored solids by evaporation of the solvent and recrystallization of the residue from a minimum volume of acetonitrile by the gradual addition of diethyl ether. Obtained compounds were characterized by conventional characterization methods (elemental analysis, infrared spectroscopy and electrospray mass spectrometry) as well as by X-ray diffraction, which gave the unequivocal evidence of structures of compounds and allowed to establish coordination environments, coordination numbers and geometries of metallic centers. Electrospray mass spectra of compound 1 showed peaks at m/z = 585 and 294 clearly assigned respectively to [FeL2]2+ and [FeL(L − H)]+ molecular cations which confirm that stoichiometry M:L 1:2 in complex 1 remains unchanged in solution. For complexes 2–6 molecular cations of [MLX]+ formula were observed in the ESI-MS spectra, at e.g. m/z = 477, 359 or 399 assigned to [CuLCF3SO3]+, [CoLCl]+ and [MnLBr]+ respectively, which confirms the coordination of anions to metal centers in those complexes.
:L 1:1 and general formula [MLX3], where X is anion or solvent molecule ([CuL(CF3SO3)2(CH3OH) (2)) and the third one of general formula [MLX2] (M = Cu(II) (3 and 4), Co(II) (5) or Mn(II) (6); X – anion or solvent molecule) where central atom adopts coordination number 5 and its coordination sphere is filled by donor atoms from one ligand molecule and two anions and/or solvent molecules. Different transition metal salts were used to determine the influence of metallic center on electrochemistry and electrochromic properties of obtained compounds. Reactions were carried out in M:L 1:2 (complex 1) or 1:1 (complexes 2–6) molar ratios at room temperature for 48 h in acetonitrile/methanol/dichloromethane solutions (1:1:1 v/v). All complexes were obtained as colored solids by evaporation of the solvent and recrystallization of the residue from a minimum volume of acetonitrile by the gradual addition of diethyl ether. Obtained compounds were characterized by conventional characterization methods (elemental analysis, infrared spectroscopy and electrospray mass spectrometry) as well as by X-ray diffraction, which gave the unequivocal evidence of structures of compounds and allowed to establish coordination environments, coordination numbers and geometries of metallic centers. Electrospray mass spectra of compound 1 showed peaks at m/z = 585 and 294 clearly assigned respectively to [FeL2]2+ and [FeL(L − H)]+ molecular cations which confirm that stoichiometry M:L 1:2 in complex 1 remains unchanged in solution. For complexes 2–6 molecular cations of [MLX]+ formula were observed in the ESI-MS spectra, at e.g. m/z = 477, 359 or 399 assigned to [CuLCF3SO3]+, [CoLCl]+ and [MnLBr]+ respectively, which confirms the coordination of anions to metal centers in those complexes.
The single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether of diisopropyl ether into acetonitrile/methanol (1:1 v/v) (complexes 1–4 and 6) or acetonitrile/ethanol (1:1 v/v) (complex 5) solutions of complexes. Fig. 2–5 show perspective views of the complexes as observed in their crystal structures. Table 1 lists the relevant geometrical parameters.
| 1 (M = Fe) | 2 (M = Cu) | 3 (M = Cu) | 4 (M = Cu) | 5 (M = Co) | 6 (M = Mn) | |
|---|---|---|---|---|---|---|
| M–N1 | 1.973(2) | 1.9608(16) | 1.956(3) | 1.964(3) | 2.039(2) | 2.179(2) |
| 1.955(2) | ||||||
| M–N12 | 1.912(2) | 1.9745(15) | 1.993(3) | 2.032(3) | 2.199(2) | 2.292(2) |
| 1.909(2) | ||||||
| M–N16 | 1.955(2) | 1.9943(16) | 1.994(3) | 1.999(3) | 2.074(2) | 2.250(2) |
| 1.962(2) | ||||||
| M–Br | 2.3706(6) | 2.5196(5) | ||||
| 2.7063(6) | 2.5227(5) | |||||
| M–Cl | 2.2978(8) | |||||
| 2.3347(8) | ||||||
| M–O(MeOH) | 1.9873(13) | 2.212(3) | ||||
| M–O(nitrate) | 1.977(3) | |||||
| M–O(triflate) | 2.4036(12) | |||||
| 2.4645(13) | ||||||
| Angles (3) | 175.78(10) | 170.03(6) | 162.16(15) | 162.43(7) | 158.23(7) | 149.68(6) |
| 161.62(10) | 168.92(5) | 159.68(15) | 156.46(11) | 143.58(10) | 135.65(8) | |
| 161.27(10) | 160.73(6) | 109.549(16) | 103.98(2) | 107.66(3) | 105.07(2) | |
| A/B | 8.8(3) | 10.72(13) | 4.23(14) | 5.57(19) | 2.4(3) | 18.03(14) |
| 6.6(2) | ||||||
| B/C | 12.8(2) | 6.88(11) | 4.86(18) | 3.42(2) | 2.3(3) | 7.17(8) |
| 5.1(2) | ||||||
| A/C | 17.24(13) | 16.89(6) | 6.14(19) | 5.44(16) | 2.91(17) | 24.34(7) |
| 8.68(14) |
Complex 1, the only one of the 1:2 metal to ligand ratio, namely dicationic FeL22+ (Fig. 2), is accompanied in the crystal structure by two CF3SO3− anions and solvent – acetonitrile molecule. Fe ion is six coordinated in quite regular manner (cf. Table 1 – the distances and three largest angles are listed there). In the crystal structure, each cationic complex is connected by N–H⋯O hydrogen bonds (Table 2 lists hydrogen bond data), with two triflate anions. Such neutral molecular complex is in fact the structural unit – these units are only weakly bound with one another and with acetonitrile guests.
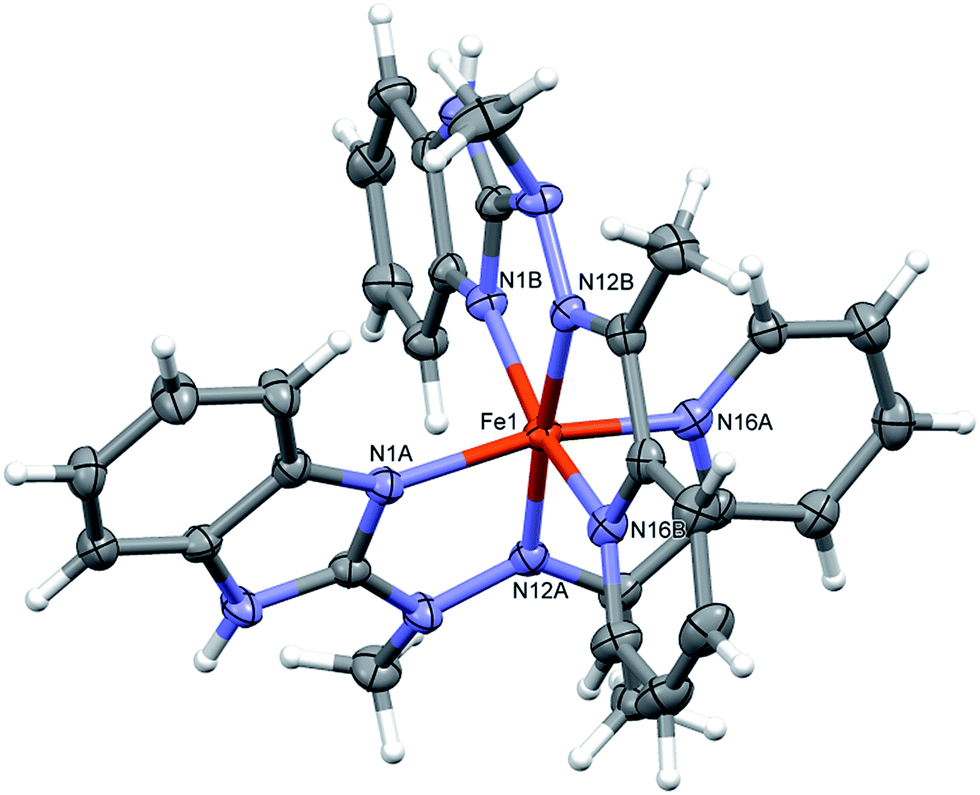 | ||
| Fig. 2 Perspective views of the cationic complex 1; ellipsoids are drawn at the 50% probability level; hydrogen atoms are represented by spheres of arbitrary radii. | ||
| D | H | A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|---|---|
| a 1 − x, 1 − y, 1 − z.b −1 + x, y, z.c 1/2 + x, 1/2 − y, 1/2 + z.d x, y, 1 + z.e 1 + x, 1 + y, z.f 1/2 + x, y, 3/2−z. | ||||||
| 1 | ||||||
| N3A | H3A | O2D | 0.86(4) | 2.06(4) | 2.880(5) | 158(4) |
| N3B | H3B | O3C | 0.77(4) | 2.13(4) | 2.871(4) | 162(4) |
|
||||||
| 2 | ||||||
| N3A | H3A | O3Ba | 0.76(32) | 2.16(2) | 2.871(2) | 157(2) |
| O1C | H1C | O2Bb | 0.75(2) | 2.01(2) | 2.7388(19) | 162(2) |
|
||||||
| 3 | ||||||
| N3A | H3A | O2Dc | 0.88 | 2.00 | 2.753(6) | 143 |
| O1C | H1C | O2Dd | 0.84 | 1.90 | 2.715(6) | 162 |
|
||||||
| 4 | ||||||
| N3A | H3A | O1D | 0.88 | 1.89 | 2.752(4) | 165 |
| O1D | H1D | Br1Ce | 0.84 | 2.44 | 3.227(2) | 157 |
|
||||||
| 5 | ||||||
| N3A | H3A | Cl1Ca | 0.88 | 2.30 | 3.125(3) | 157 |
| O1D | H1D | Cg1f | 0.84 | 2.43 | 3.254(4) | 166 |
|
||||||
| 6 | ||||||
| N3A | H3A | Br1Ca | 0.88 | 2.52 | 3.327(2) | 153 |
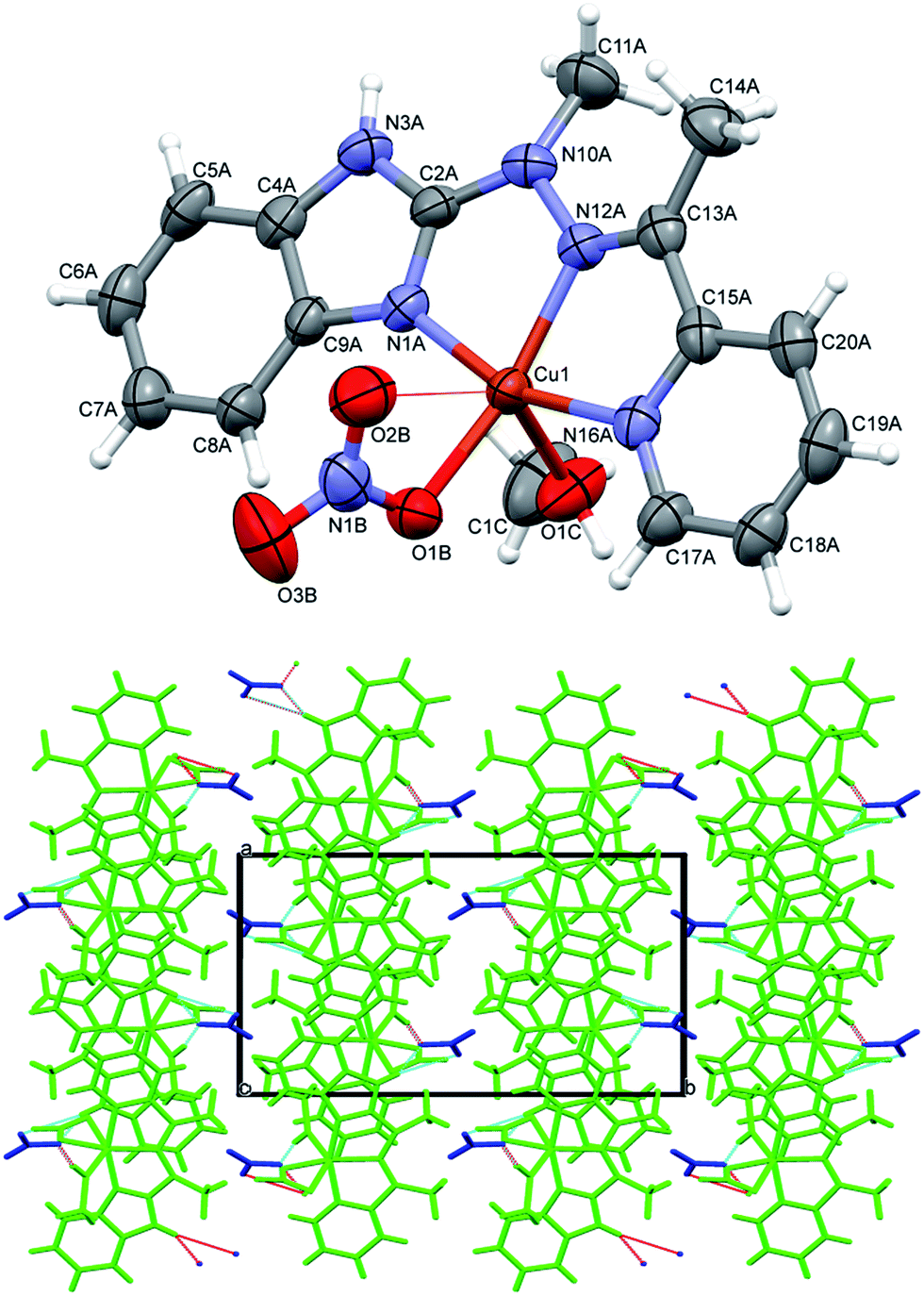
In the neutral complex 2, CuL(CH3OH)(CF3SO3)2, copper ion is six-coordinated, in square bipyramidal (rather than octahedral) fashion (Fig. 3 top). The basal plane is created by three nitrogen atoms from the ligand and one oxygen from methanol molecule – the bonds are almost equal and four coordination centers make quite regular square. The distances to two other oxygen atoms, from two triflate anions, are significantly longer (cf. Table 1). In the crystal structure two kinds of hydrogen bonds (N–H⋯O and O–H⋯O, both with triflate oxygen atoms as acceptors) create the regular three-dimensional network: O–H⋯O bonds connect molecules into infinite chains along x-direction (Table 2), and these chains are bonded by N–H⋯O bonds, which, in turn, connect molecules into centrosymmetric dimers (Fig. 3 bottom).
 | ||
| Fig. 3 Top: perspective view of the complex 2; ellipsoids are drawn at the 50% probability level, hydrogen atoms are represented by spheres of arbitrary radii; bottom: hydrogen-bond network in the crystal structure of 2. Hydrogen bonds are shown as dashed lines. | ||
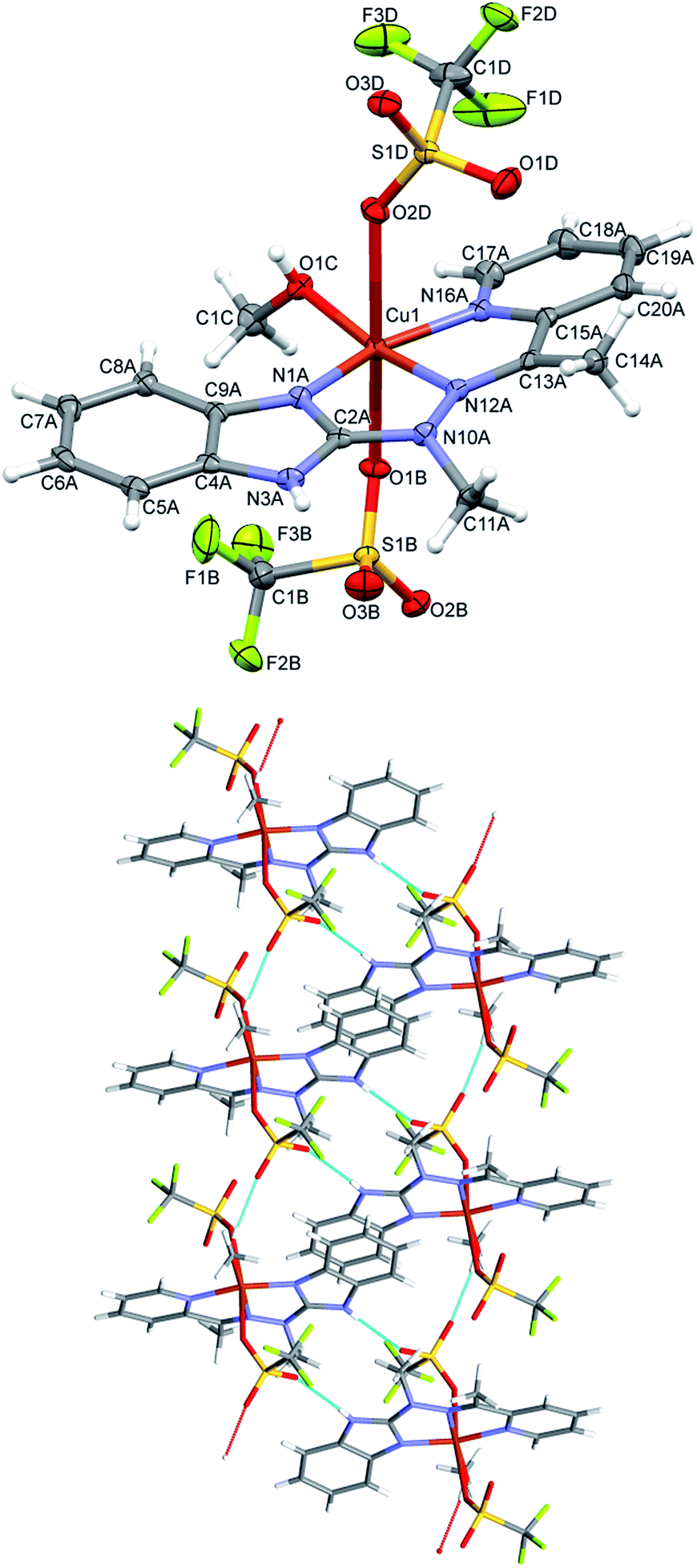
In the cationic complex 3, CuL(NO3)(CH3OH)+, which crystallizes with the nitro anions, the Cu cation is five coordinated (the sixth contact with the second oxygen atom from nitro group is much longer – by 0.6 Å - and should not be regarded as bonding interaction), and coordination geometry is close to tetragonal pyramid (Fig. 4 top). Three ligand nitrogen atom and closely bound oxygen from nitro group are approximately coplanar (within 0.115 Å), the fifth coordination center, oxygen from methanol molecule is at the apex of the polyhedron; Cu is also displaced from the base towards apex (by 0.209 Å). In the crystal, hydrogen bonds with oxygen atoms of the nitro-group as acceptors are main structure-creating factors. In this case however (unlike the structure 1) these hydrogen bonds do not create closed units, but expand in the crystal making three dimensional network (Fig. 4 bottom).
 | ||
| Fig. 4 Top: perspective views of the complex 3; ellipsoids are drawn at the 50% probability level, hydrogen atoms are represented by spheres of arbitrary radii. Thin line represents secondary Cu–O(nitro) contact. Bottom: hydrogen-bond network in the crystal structure of 3. Hydrogen bonds are shown as dashed lines. | ||
In complexes 4–6, of general formula MLX2 (M = Cu, Co, Mn, X = Cl, Br), in all cases, the metal is five coordinated, in more regular then in 3 tetragonal pyramid geometry (Fig. 5). Three nitrogen atoms from ligand and one halide create almost planar base, the second halide lies at the apex. The metal ion is duly displaced towards the apex. The maximum deviations of atoms creating the basal plane are 0.056 Å in 4, 0.0051 Å in 5 and 0.045 Å in 6, metal atoms are displaced by 0.3–0.58 Å from this plane, and the apex is displaced by almost 3 Å in the same direction as metal ion.
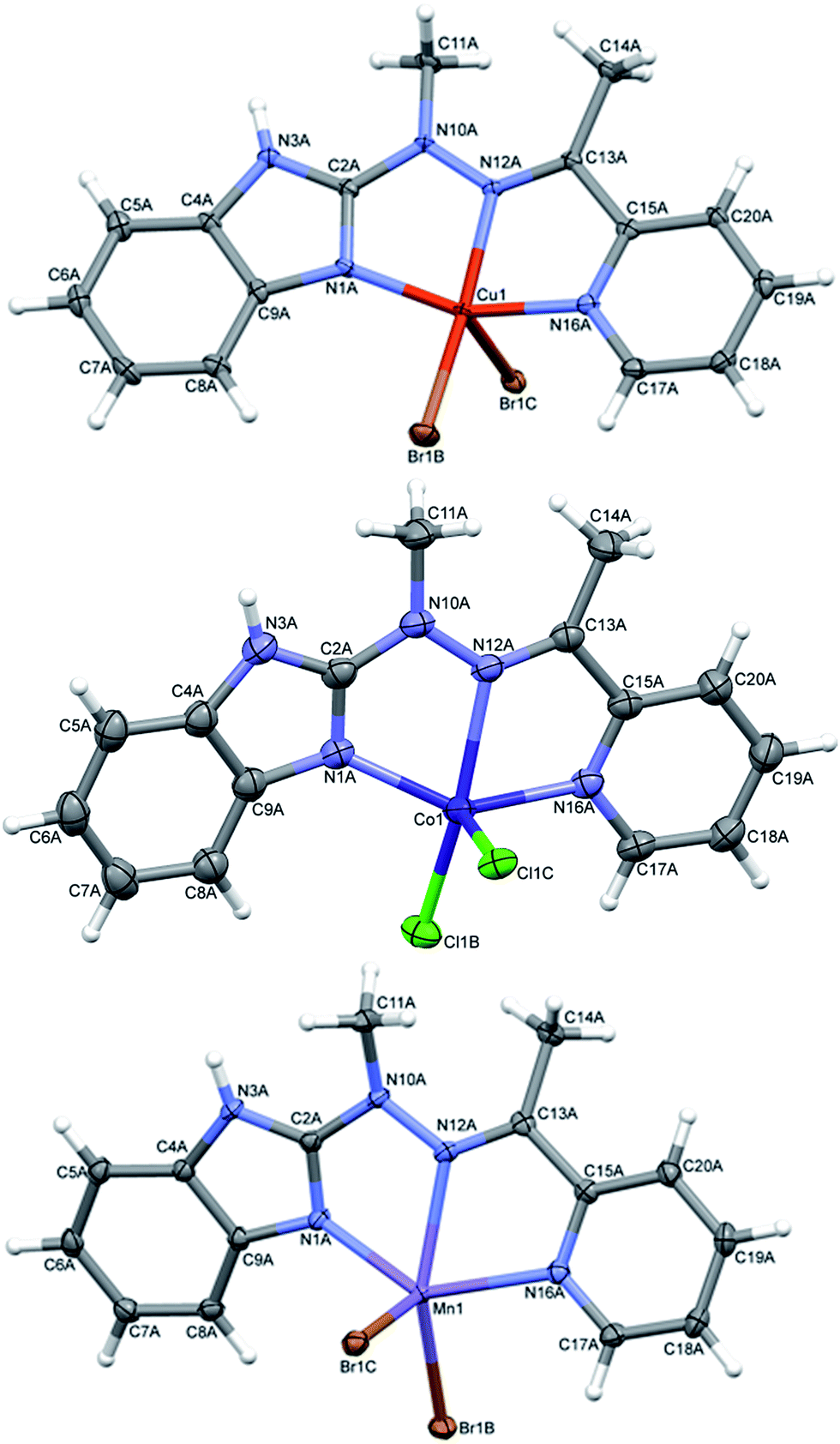 | ||
| Fig. 5 Perspective views of the complex 4–6; ellipsoids are drawn at the 50% probability level, hydrogen atoms are represented by spheres of arbitrary radii. | ||
Despite similar molecular structures, the crystal packing modes are different. Structures 4 and 5 contain additionally guests, solvent molecules: methanol in 4, ethanol in 5, which add the hydrogen bond donating/accepting potential. In the structure 4 the methanol molecules act as linkers between subsequent complex molecules (Fig. 6 top). Quite unusual situation is observed in 5: the N–H⋯Cl hydrogen bonds make typical centrosymmetric dimers but OH groups from ethanol molecule is involved in very short and directional O–H⋯π hydrogen bond (Fig. 6 bottom, Table 2). Complex 6 crystallizes without additional molecules and the building block is hydrogen bonded dimer, similar to that in 5.
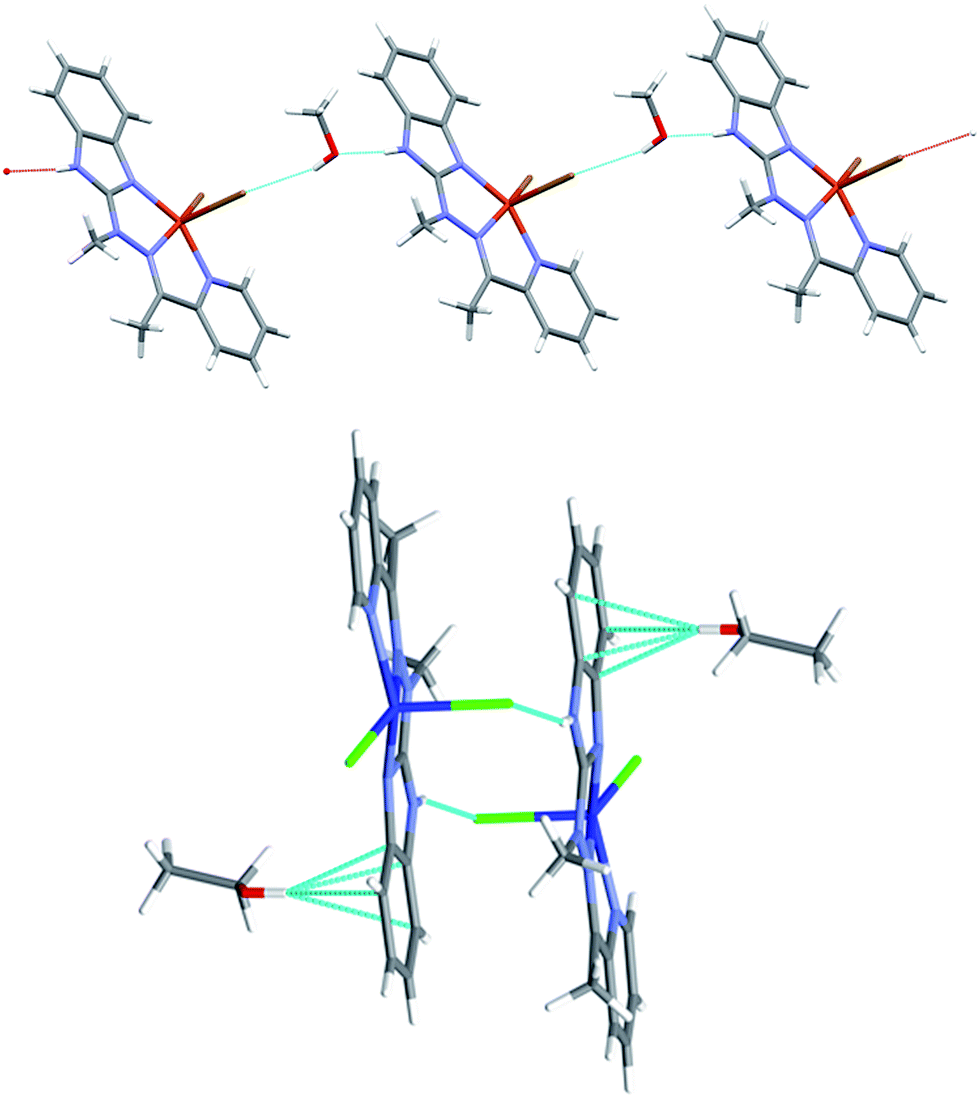 | ||
| Fig. 6 Top: hydrogen-bonded chain of subsequent complex/methanol molecules in 4 bottom: hydrogen bonded dimer in 5 and the strong O–H⋯π hydrogen bonds. | ||
The reversible electrochemistry and the distinct color changes of compounds are the most desired properties for using them as active materials in electrochromic devices. To determine whether investigated compounds could be reversibly oxidized, electrochemical measurements were done. To evaluate the usefulness of ligand L and its complexes 1–6 as electrochromes, their spectroelectrochemical properties were investigated. By gradually applying positive or negative potential to generate oxidized or reduces species, while examining the change in the absorption, the track of color changes is possible. Cyclic voltammetry of ligand L and its complexes 1–6 with transition metal ions were investigated in anhydrous and deaerated acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte. The cyclic voltammogram of ligand L obtained in the negative direction showed one irreversible anodic peak at +975 mV which is assigned to the oxidation of imine bond of ligand L17 (Fig. 7).
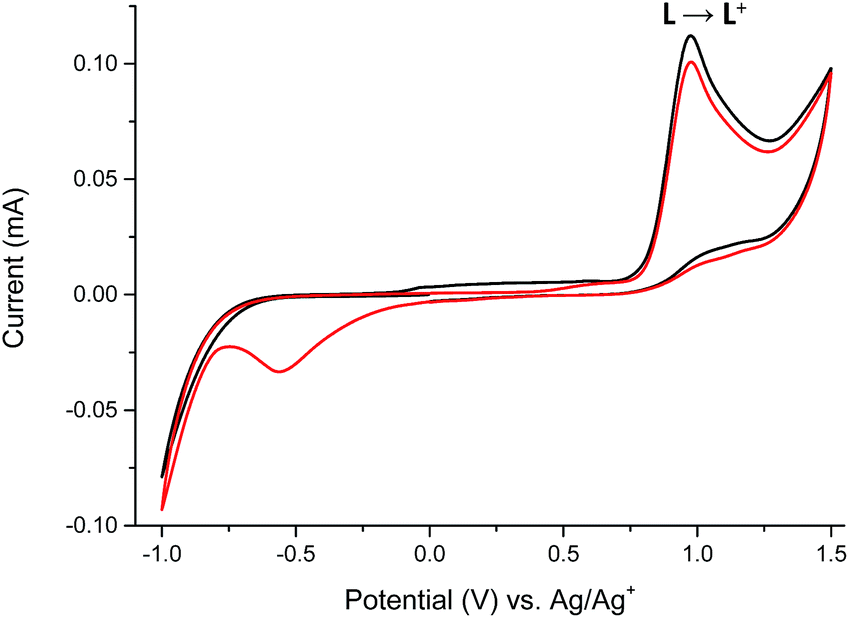 | ||
| Fig. 7 The cyclic voltammogram of ligand L: 1st cycle (black) and 2nd cycle (red) measured in anhydrous and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte at a scan rate 100 mV s−1 scanned in the negative direction. | ||
The presence of the cathodic peak at −555 mV is dependent on the former oxidation of the imine bond. It can be explained that the oxidized species of L undergo a chemical reaction and form some undefined species which can be reduced at −555 mV to restore the ligand L. This is why no cathodic signal is observed at the first cycle in the negative direction. In the UV-vis region ligand L showed one absorption band at 345 nm which can be attributed to the π–π* and n–π* electronic transitions originating from the pyridyl rings of ligand L16,18 (Fig. 8). By gradual applying of positive potential absorption band at 345 nm gradually decreases and shifts by ∼15 nm to lower absorption wavelengths forming absorption band at 330 nm. The color changes from slightly yellow to transparent were observed. The original UV-vis spectrum of L can be restored by applying negative potential (−500 mV).
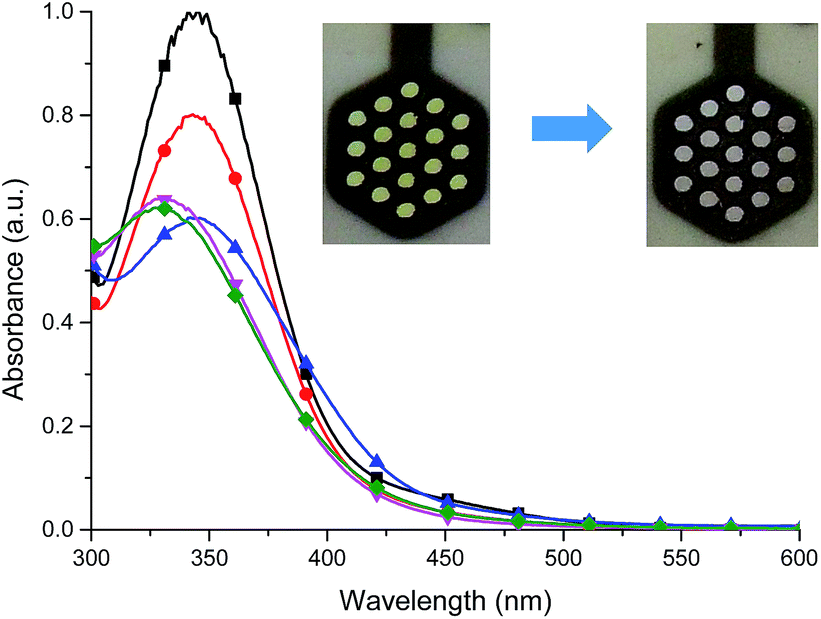 | ||
Fig. 8 Spectral changes of ligand L in dehydrated and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte by applying 0 ( ), +800 ( ), +800 ( ), +900 ( ), +900 ( ), +1000 ( ), +1000 ( ) and +1100 mV ( ) and +1100 mV ( ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically oxidized (right) L by applying a potential for 1 min. ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically oxidized (right) L by applying a potential for 1 min. | ||
The redox properties of Fe(II) complex 1 were studied in the potential range of −1000 to +1500 mV (Fig. 9).
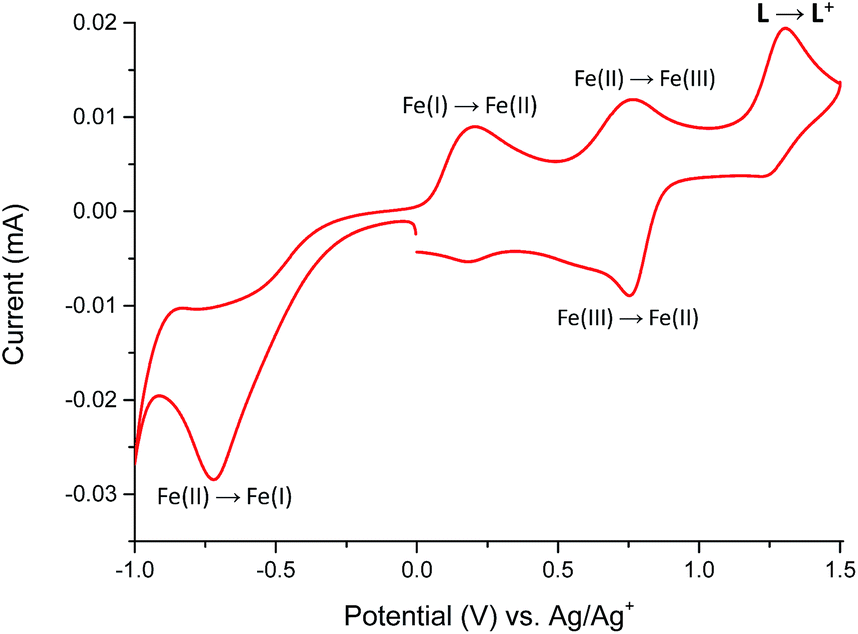 | ||
| Fig. 9 The cyclic voltammogram of Fe(II) complex 1 measured in anhydrous and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte at a scan rate 100 mV s−1 scanned in the negative direction. | ||
The Fe(II) complex is electroactive with respect to both metal center and ligand L molecule. The irreversible reduction peak at Epc = −720 mV is attributed to the Fe(II) → Fe(I) process, while the oxidation peak at Epa = +200 mV can be assigned to Fe(I) → Fe(II) process.19,20 It can indicate on the stability of formed Fe(I) complex in the solution. The second reversible oxidation at E1/2 = 755 mV is attributed to the Fe(II)/Fe(III) couple.21 The oxidation peak of ligand L in complex 1 was observed at Epa = +1300 mV and it is shifted to a more positive value by 325 mV in comparison to the free ligand molecule. The positive potential shift is a consequence of a stabilization of ligand L molecule in transition metal complexes.22 To support the assignment of the redox processes, spectroelectrochemical measurements were performed and the results summarized in Fig. 10.
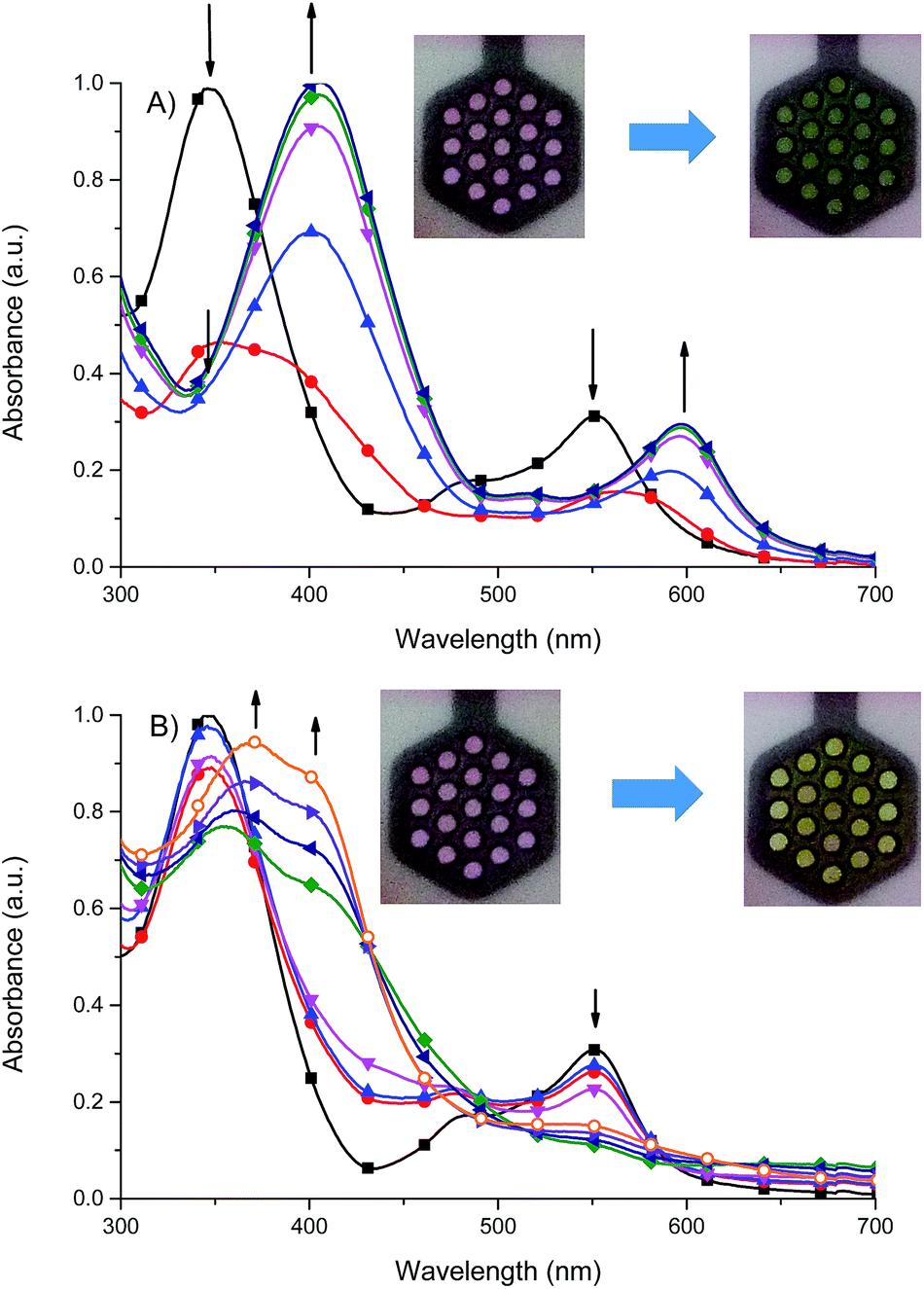 | ||
Fig. 10 Spectral changes of 1 in dehydrated and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte by applying A) 0 ( ), −100 ( ), −100 ( ), −200 ( ), −200 ( ), −300 ( ), −300 ( ), −400 ( ), −400 ( ) and −500 mV ( ) and −500 mV ( ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically reduced (right) 1 by applying a potential for 1 min. B) 0 ( ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically reduced (right) 1 by applying a potential for 1 min. B) 0 ( ), +500 ( ), +500 ( ), +600 ( ), +600 ( ), +700 ( ), +700 ( ), +800 ( ), +800 ( ), +900 mV ( ), +900 mV ( ), +1000 ( ), +1000 ( ) and +1100 mV ( ) and +1100 mV ( ) potentials versus Ag/Ag+ reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically oxidized (right) 1 by applying a potential for 1 min. ) potentials versus Ag/Ag+ reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically oxidized (right) 1 by applying a potential for 1 min. | ||
It was found that the oxidized state of both ligand molecule and metal center strongly affect the UV-vis absorption spectra and color of the complex. Complex Fe(II) ions 1 in its neutral state exhibits two absorption bands at 348 nm and 550 nm. The first one is the ligand-based absorption, while the second one can be attributed to the metal-to-ligand charge transfer (MLCT).23 By stepwise applying of negative potential the ligand-based absorption band at 350 nm decreases and shifts to 405 nm and it is concomitant with the decrease of the MLCT band at 550 nm and formation of new absorption band at 600 nm (Fig. 10a). In terms of colors discernible by a user, neutralism of 1 was purple and the reduced form was green. The original purple color was obtained after applying potential of +400 mV. While the ligand molecule is spectroelectrochemically inactive during reduction, the spectral changes must be due to the metal-centered redox process and support the Fe(II) ↔ Fe(I) process. During the oxidation of 1 (Fig. 10b), at 800 mV Fe(II) ions are oxidized to Fe(III) ions, the MLCT band of Fe(II) complex at 550 nm diminished and the color changed from purple to yellow what was also observed for Fe(II)/Fe(III) process in another Fe(II) complexes.24 The ligand-based absorption band decreases and shifts by ∼20 nm, the absorption band appears at 370 nm with a shoulder at around 400 nm which can be attributed to the ligand-to-metal charge transfer (LMCT) in Fe(III) complex.25 Further oxidation leads to oxidation of ligand molecule and spectral changes are similar to those observed for the free ligand (Fig. S1†), it was concomitant with the visible color change from yellow to slightly yellow. Applying of negative potential (−500 mV) leads to neutralization of ligand molecule and reduction of metal center from Fe(III) to Fe(I), what was visible as a color change from slightly yellow to green.
Fig. 11 shows the cyclic voltammograms of Cu(II) complexes 2–4.
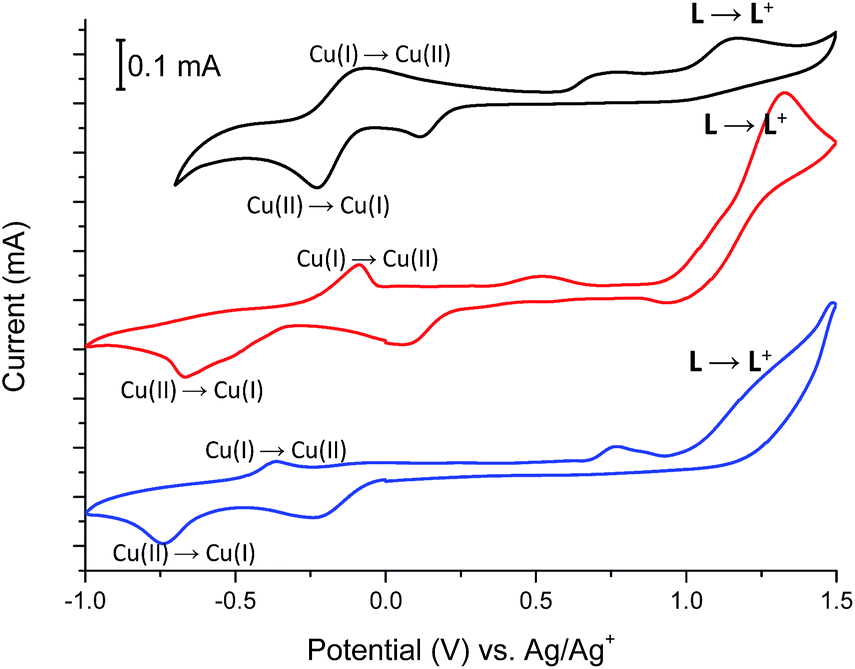 | ||
| Fig. 11 The cyclic voltammograms (2nd cycles) of Cu(II) complexes 2 (black), 3 (blue) and 4 (red) measured in anhydrous and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte at a scan rate 100 mV s−1 scanned in the negative direction. | ||
In all Cu(II) complexes, Cu(II) centers undergo reduction to Cu(I) ions at Epc –230 mV, −675 mV and −740 mV for 2, 3 and 4 respectively what is similar to the reduction potential of Cu(II)/Cu(I) couple observed for another Cu(II) complexes.26 More negative reduction potential of Cu(II) → Cu(I) for complexes 3 and 4 than for complex 2 may be because of different coordination numbers of metallic centers, which is 6 for complex 2 and 5 for complexes 3 and 4 or due to the coordination of metal centers by nitrate (3) and bromide (4) anions which are known to have higher coordination strength to transition metal ions than trifluoromethanesulfonate anions (2).27 The reduction peaks are associated with the reverse peaks at Epa −75 mV, −90 mV and −365 mV for 2, 3 and 4 respectively, attributed to the Cu(I) → Cu(II) process. The Cu(II) → Cu(I) reduction is probably associated with the change of coordination geometry of metal center what leads to disassembling/assembling process,28 as well as to the formation of a solvated copper(I) species with acetonitrile molecules, since this solvent forms stable Cu(I)-acetonitrile complexes in solution.29 The presence of solvated copper(I) ions in solution is supported by the broad peaks at about +750 mV, +525 mV and +765 mV for 2, 3 and 4 respectively.30 Spectral changes observed during reduction of Cu(II) complexes 2–4 are shown in Fig. 12, S2 and S3† and are similar for all investigated Cu(II) complexes.
For complex 2, when negative potential was applied concomitant decrease and shift to higher absorption wavelengths of the ligand-based absorption band at 347 nm occurred and after reduction of Cu(II) ions to Cu(I) ions the absorption band with maximum at 368 nm was observed (Fig. 12). The sharp isosbestic point at 368 nm confirms the presence of only two species in solution: the neutral and reduced ones and their independence. The original spectrum of the neutral complex 2 could be recovered when the potential was applied at +100 mV. The color change from yellow to red-brown was observed. Similar spectral and color changes were perceived for complexes 3 and 4 (Fig. S2 and S3†).
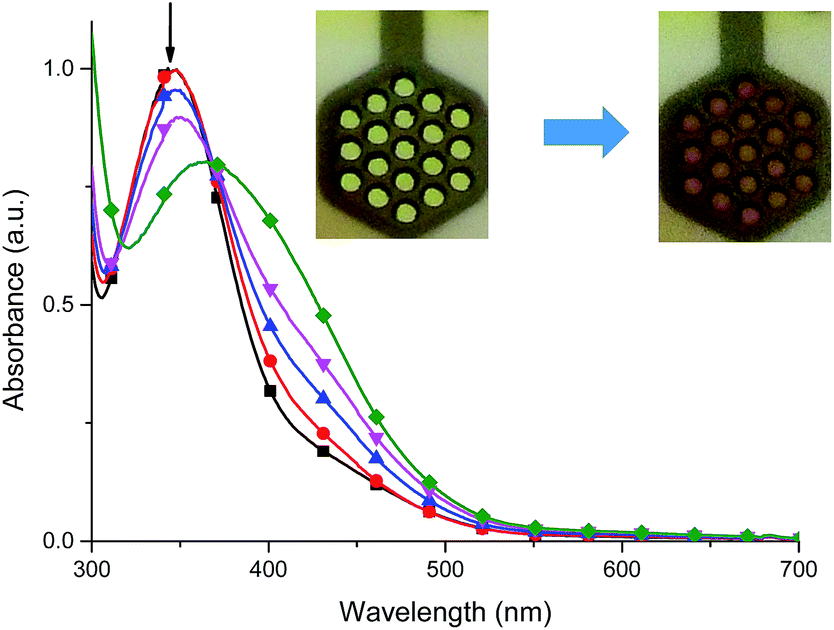 | ||
Fig. 12 Spectral changes of 2 in dehydrated and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte by applying 0 ( ), −100 ( ), −100 ( ), −200 ( ), −200 ( ), −300 ( ), −300 ( ) and −400 mV ( ) and −400 mV ( ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically reduced (right) 2 by applying a potential for 1 min. ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically reduced (right) 2 by applying a potential for 1 min. | ||
The cyclic voltammogram of Co(II) complex 5 (Fig. 13) reveals two oxidation peaks centered at +210 mV and +1380 mV. The first one is associated with the irreversible Co(II) → Co(III) oxidation,31 while the second one is attributed to the oxidation of ligand L coordinated to metal center. Neutralization of the electrogenerated oxidized species of L occurs at −85 mV, whereas the irreversible cathodic peak at −520 mV can be assigned to the Co(III) → Co(II) redox process.32,33 This assignment of oxidation and reduction potentials is consistent with spectroelectrochemical data (Fig. 14).
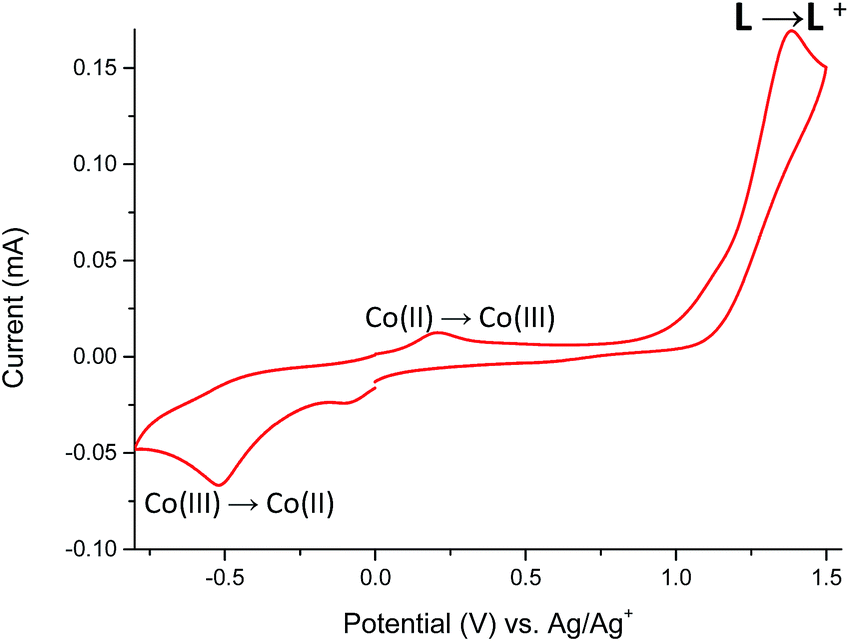 | ||
| Fig. 13 The cyclic voltammogram (2nd cycle) of Co(II) complex 5 measured in anhydrous and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte at a scan rate 100 mV s−1 scanned in the negative direction. | ||
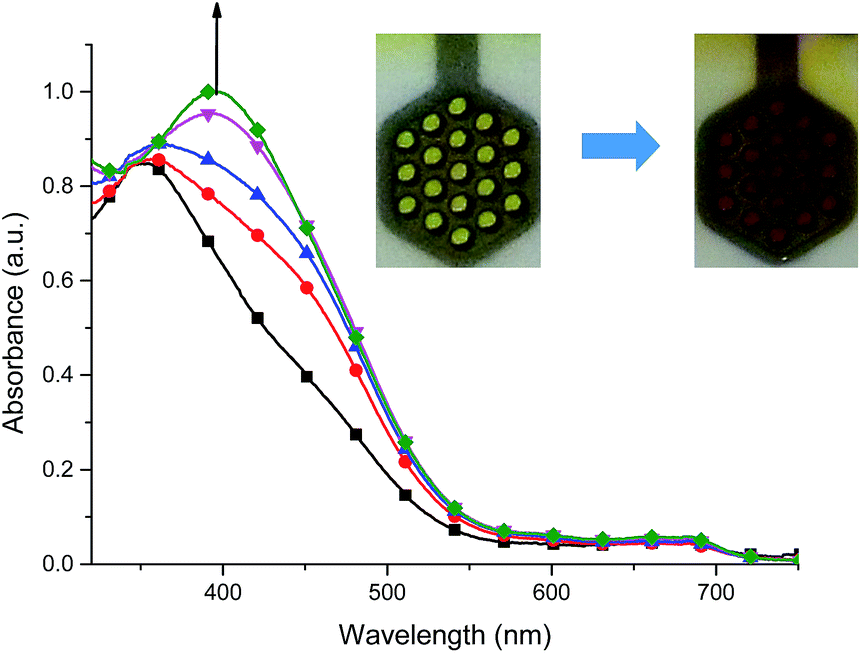 | ||
Fig. 14 Spectral changes of 5 in dehydrated and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte by applying 0 ( ), +100 ( ), +100 ( ), +200 ( ), +200 ( ), +300 ( ), +300 ( ) and +400 mV ( ) and +400 mV ( ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically oxidized (right) 5 by applying a potential for 1 min. ) potentials versus Ag/AgCl gel reference electrode held for 30 s per potential. Insert: photographs of the original (left) and electrochemically oxidized (right) 5 by applying a potential for 1 min. | ||
Co(II) complex 5 exhibited one, ligand-based absorption band at 353 nm. During stepwise oxidation, the red shift of this band was observed leading to formation of absorption band at 395 nm when applied potential value reached the first oxidation potential with the observed color change of the solution from yellow to red-brown. Further increasing of potential leads to the oxidation of ligand molecule and ligand-based color changes (Fig. S4†). Applying potential of −600 mV caused the neutralization of the oxidized state of ligand molecule and reduction of Co(III) to Co(II) leading to the restoration of the original spectra of 5.
The electrochemical oxidation of manganese(II) complex 6 leads to decomposition of Mn(II) complex and precipitation of dark-brown solid of MnO2. In the cyclic voltammogram, except ligand-based anodic and cathodic waves located at respectively Epa = +1165 mV and Epc = −280 mV, one irreversible anodic peak at +920 mV was observed (Fig. S5†). This peak is attributed to the Mn(II) → Mn(III) oxidation process, what can lead to formation of some undefined mixed-valent di-μ-oxo bridged binuclear complexes of Mn(III)/Mn(IV) formed due to decomposition of the instable corresponding Mn(III) complex. This process is known to occur during electrochemical oxidation of Mn(II) complexes with polypyridyl bidentate and tridentate ligands.34 Further decomposition of di-μ-oxo bridged binuclear complexes probably leads to precipitation of MnO2.
The color changes can be quantified by the CIE L*a*b* color space. In this convention, the a* and b* parameters respectively refer to the red/green and yellow/blue contributions to the perceived color and the L* parameter quantifies the color brightness. The colors of the neutral and charged states perceived by a standard user at 2° observer angle for the studied compounds are quantified with D65 illuminant which corresponds to the midday light. CIE L*a*b* coordinates are summarized in Table S1.† For ligand L only b* parameter slightly decreases what confirms lowering of contribution of yellow color during oxidation, meanwhile, the red/green contribution (a* parameter) to the perceived color did not vary between the neutral and oxidized state. In case of complex 1, the a* value confirmed that its red component was bleached during oxidation process and it was replaced with green component when negative potential was applied, whereas the b* parameter increases during oxidation what confirms that the blue component of the neutral state was replaced with a yellow one.
According CIE color parameters of neutral and reduced states of complexes 2–4, during electrochemical reduction, the contribution of red component increases, while for complex 5 the most significant changes were observed when complex was oxidized to its first oxidation state - both parameters a* and b* increase what indicate the higher contribution of yellow and red components in the final color of the first oxidized state of 5.
The stability of color change was measured by switching between two different potentials and monitoring of transmittance changes at appropriate wavelength. For Fe(II) complex 1 potential was switched at every 30 seconds between −300 mV and +900 mV (60 seconds per one cycle), what corresponds to green ↔ yellow color change, and changes in transmittance were monitored at 600 nm. Compounds exhibit high color stability and green ↔ yellow color change during multiple oxidation/reduction cycles are especially interesting in the view of their applications as active materials in adaptive military camouflage technologies35,36 and due to this switching between these two colors has been chosen for investigation of the color stability. As seen in Fig. 15 transmittance difference between reduced and oxidized states was ∼20% and it did not change after 65 minutes of measurement (65 cycles), whereas complexes Cu(II) 2–4 and Co(II) 5 decompose after few oxidation/reduction cycles (Fig. S6–S9†).
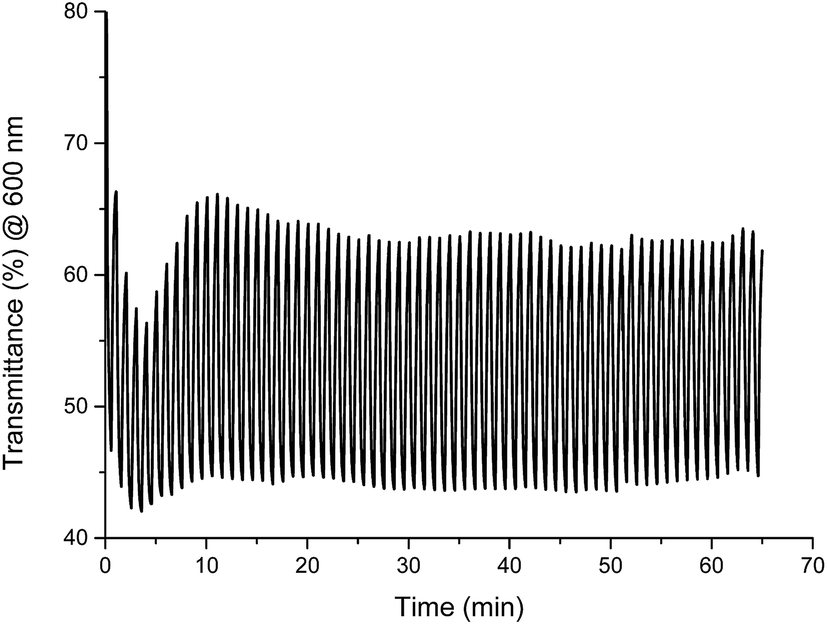 | ||
| Fig. 15 Changes in transmittance of Fe(II) complex 1 measured in anhydrous and deaerated acetonitrile with 0.1 M TBAPF6 as a supporting electrolyte and monitored at 600 nm when switching between −300 mV and +900 mV potential at 60 s cycles. | ||
Experimental section
General
Ligand L ((E)-2-(1-methyl-2-(1-(pyridin-2-yl)ethylidene)hydrazinyl)-1H-benzo[d]imidazole) was prepared according to literature methods.16 Solvents and metal salts, were used without further purification as supplied from Aldrich. ESI mass spectra of acetonitrile solutions ∼10−4 M were measured using a Waters Micromass ZQ spectrometer. Microanalyses were performed using a Perkin–Elmer 2400 CHN microanalyser. IR spectra were obtained with a Nicolet iS50 FT-IR spectrophotometer in ATR mode and peak positions are reported in cm−1. The electrochemical and spectroelectrochemical measurements were done with a VSP Bio-Logic multichannel potentiostat. Compounds were dissolved in anhydrous and deaerated acetonitrile at 10−4 M with 0.1 M tetrabutylammonium hexafluorophosphate as the electrolyte. The solutions were purged with nitrogen for 20 min. to remove the dissolved oxygen. A nitrogen blanket was then maintained over the solvent to prevent oxygen diffusion during the measurements. A platinum electrode was used as the working electrode and a platinum wire was used as the auxiliary electrode. The reference electrode was an Ag/Ag+ electrode. Spectroelectrochemical measurements were done using a commercially available platinum honeycomb working electrode on a ceramic support in a narrow optical path quartz cuvette using miniature Ag/AgCl gel electrode as a reference electrode. The potential was controlled and switched with the potentiostat. The resulting spectroscopic changes were measured with a Jasco V-770 UV-vis-NIR spectrometer.Crystallographic data
Diffraction data were collected by the ω-scan technique on Agilent Technologies four-circle diffractometers: 1 at 180(1) K, 2, 4, and 6 at 120(1) K and 3 at 200(1) on Xcalibur with Eos CCD detector and graphite-monochromated MoKα radiation (λ = 0.71069 Å), 5 at 130(1)K on SuperNova with Atlas CCD detector, equipped with Nova microfocus CuKα radiation source (λ = 1.54178 Å). The data were corrected for Lorentz-polarization as well as for absorption effects.37 Precise unit-cell parameters were determined by a least-squares fit of 6656 (1), 4277 (2), 581 (1B), 2571 (3), 1268 (4), 11197 (5), and 2571 (6) reflections of the highest intensity, chosen from the whole experiment. The structures were solved with SHELXT38 and refined with the full-matrix least-squares procedure on F2 by SHELXL-2013.38 All non-hydrogen atoms were refined anisotropically, NH and OH hydrogen atoms in 1 and 2 were found in the difference Fourier maps and isotropically refined, all other hydrogen atoms were placed in idealized positions and refined as ‘riding model’ with isotropic displacement parameters set at 1.2 (1.5 for methyl groups) times Ueq of appropriate carrier atoms. Table S2† lists the relevant experimental data and refinement details.
Synthesis
:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The purple solid was centrifuged, washed with diethyl ether and dried. Yield: 44%. ESI-MS: m/z (%) = 735 (10)[FeL2]CF3SO3+, 585 (100) [FeL(L − H)]+, 294 (50) [FeL2]2+. IR (ATR) ν = 3059 ν(C–H)ar, 2944 νas.(CH3), 2882 νs.(CH3), 1622 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)imin., 1560, 1552, 1498, 1461 ν(CC), 1437, 1389 ν(CN), 1322, 1265 ν(C–N), 1241, 1224, 1028 ν(CF3SO3), 1151 ν(N–N), 1045, 929, 900, 814 ρ(C–H), 742, 696, 634, 572, 515 γ(C–H) cm−1. Anal. calc. for [Fe(C15H15N5)2](CF3SO3)2(CH3CN) (925.13): C, 44.12; H, 3.59; N, 16.64; S, 6.93. Found: C, 44.28; H, 3.53; N, 16.58; S, 6.98%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 67%. ESI-MS: m/z (%) = 477 (70) [CuLCF3SO3]+, 327 (100) [Cu(L − H)]+. IR (ATR) ν = 3104, 3055, 3001 ν(C–H)ar, 2948 νas.(CH3), 2810 νs.(CH3), 1625 ν(CN)imin., 1582, 1566, 1506, 1468ν(CC), 1437, 1365ν(CN), 1288, 1268 ν(C–N), 1238, 1218, 1026 ν(CF3SO3), 1159 ν(N–N), 1055, 935, 903, 828, 783 ρ(C–H), 759, 744, 636, 572 γ(C–H) cm−1. Anal. calc. for [Cu(C15H15N5)(CF3SO3)2(CH3OH)] (657.99): C, 32.80; H, 2.91; N, 10.63; S, 9.73. Found: C, 32.87; H, 2.83; N, 10.70; S, 9.75%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 77%. ESI-MS: m/z (%) = 390 (60) [CuLNO3]+, 327 (100) [Cu(L − H)]+. IR (ATR) ν = 3084 ν(C–H)ar, 2943, 2888 νas.(CH3), 2804 νs.(CH3), 1622 ν(CN)imin., 1603, 1573, 1556, 1472 ν(CC), 1428νas.(ONO), 1409, 1334 ν(CN), 1377 v(NO3−ionic) 1278νs.(ONO), 1255ν(C–N), 1180ν(N–N), 1007ν(NO), 1059, 1030, 942, 902, 831, 812, 782 ρ(C–H), 758, 747, 726, 653, 621, 596, 565 γ(C–H) cm−1. Anal. calc. for [Cu(C15H15N5)(NO3)(CH3OH)]NO3(484.06): C, 39.63; H, 3.95; N, 20.22. Found C, 39.69; H, 3.88; N, 20.27%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 77%. ESI-MS: m/z (%) = 409 (100) [CuLBr]+, 327 (50) [Cu(L − H)]+. IR (ATR) ν = 3083, 2026ν(C–H)ar, 2911 νas.(CH3), 2840νs.(CH3), 1617ν(CN)imin., 1578, 1561, 1493, 1473, 1462 ν(CC), 1432, 1380ν(CN), 1305, 1286 ν(C–N), 1230, 1209 ν(C–C), 1052 ν(N–N), 1024, 1011, 936, 901, 815, ρ(C–H), 784, 759, 726, 611 γ(C–H) cm−1. Elemental analysis calc. for [Cu(C15H15N5)Br2]·CH3OH(520.71): C, 36.91; H, 3.68; N, 13.45. Found C, 36.97; H, 3.61; N, 13.40%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 77%. ESI-MS: m/z (%) = 359 (100) [CoLCl]+. IR (ATR) ν = 3033ν(C–H)ar, 2933νas.(CH3), 1628 ν(CN)imin., 1599, 1573, 1561, 1490, 1464 ν(CC), 1443, 1382 ν(CN), 1303, 1283 ν(C–N), 1229, 1207 ν(C–C), 1049 ν(N–N), 1008, 997, 801 ρ(C–H), 783, 762, 745, 664, 617γ(C–H) cm−1. Elemental analysis calc. for [Co(C15H15N5)Cl2]·CH3OH (440.05): C, 46.28; H, 4.80; N, 15.87. Found C, 46.19; H, 4.88; N, 15.79%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The yellow solid was centrifuged, washed with diethyl ether and dried. Yield: 75%. ESI-MS m/z (%) = 399 (20) [MnLBr]+, 266 (100) [L + H]+. IR (ATR) ν = 3222ν(C–H)ar, 1624 ν(CN)imin., 1595, 1568, 1557, 1489, 1466, ν(CC), 1439, 1428, 1372 ν(CN), 1316, 1282 ν(C–N), 1251, 1226, 1205 ν(C–C), 1046 ν(N–N), 1010, 992, 902, 825 ρ(C–H), 784, 743, 637, 595 γ(C–H) cm−1. Elemental analysis calc. for [Mn(C15H15N5)Br2] (480.06): C, 37.53; H, 3.15; N, 14.59. Found: C, 37.60; H, 3.03; N, 14.50%.
N)imin., 1560, 1552, 1498, 1461 ν(CC), 1437, 1389 ν(CN), 1322, 1265 ν(C–N), 1241, 1224, 1028 ν(CF3SO3), 1151 ν(N–N), 1045, 929, 900, 814 ρ(C–H), 742, 696, 634, 572, 515 γ(C–H) cm−1. Anal. calc. for [Fe(C15H15N5)2](CF3SO3)2(CH3CN) (925.13): C, 44.12; H, 3.59; N, 16.64; S, 6.93. Found: C, 44.28; H, 3.53; N, 16.58; S, 6.98%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 67%. ESI-MS: m/z (%) = 477 (70) [CuLCF3SO3]+, 327 (100) [Cu(L − H)]+. IR (ATR) ν = 3104, 3055, 3001 ν(C–H)ar, 2948 νas.(CH3), 2810 νs.(CH3), 1625 ν(CN)imin., 1582, 1566, 1506, 1468ν(CC), 1437, 1365ν(CN), 1288, 1268 ν(C–N), 1238, 1218, 1026 ν(CF3SO3), 1159 ν(N–N), 1055, 935, 903, 828, 783 ρ(C–H), 759, 744, 636, 572 γ(C–H) cm−1. Anal. calc. for [Cu(C15H15N5)(CF3SO3)2(CH3OH)] (657.99): C, 32.80; H, 2.91; N, 10.63; S, 9.73. Found: C, 32.87; H, 2.83; N, 10.70; S, 9.75%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 77%. ESI-MS: m/z (%) = 390 (60) [CuLNO3]+, 327 (100) [Cu(L − H)]+. IR (ATR) ν = 3084 ν(C–H)ar, 2943, 2888 νas.(CH3), 2804 νs.(CH3), 1622 ν(CN)imin., 1603, 1573, 1556, 1472 ν(CC), 1428νas.(ONO), 1409, 1334 ν(CN), 1377 v(NO3−ionic) 1278νs.(ONO), 1255ν(C–N), 1180ν(N–N), 1007ν(NO), 1059, 1030, 942, 902, 831, 812, 782 ρ(C–H), 758, 747, 726, 653, 621, 596, 565 γ(C–H) cm−1. Anal. calc. for [Cu(C15H15N5)(NO3)(CH3OH)]NO3(484.06): C, 39.63; H, 3.95; N, 20.22. Found C, 39.69; H, 3.88; N, 20.27%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 77%. ESI-MS: m/z (%) = 409 (100) [CuLBr]+, 327 (50) [Cu(L − H)]+. IR (ATR) ν = 3083, 2026ν(C–H)ar, 2911 νas.(CH3), 2840νs.(CH3), 1617ν(CN)imin., 1578, 1561, 1493, 1473, 1462 ν(CC), 1432, 1380ν(CN), 1305, 1286 ν(C–N), 1230, 1209 ν(C–C), 1052 ν(N–N), 1024, 1011, 936, 901, 815, ρ(C–H), 784, 759, 726, 611 γ(C–H) cm−1. Elemental analysis calc. for [Cu(C15H15N5)Br2]·CH3OH(520.71): C, 36.91; H, 3.68; N, 13.45. Found C, 36.97; H, 3.61; N, 13.40%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The green solid was centrifuged, washed with diethyl ether and dried. Yield: 77%. ESI-MS: m/z (%) = 359 (100) [CoLCl]+. IR (ATR) ν = 3033ν(C–H)ar, 2933νas.(CH3), 1628 ν(CN)imin., 1599, 1573, 1561, 1490, 1464 ν(CC), 1443, 1382 ν(CN), 1303, 1283 ν(C–N), 1229, 1207 ν(C–C), 1049 ν(N–N), 1008, 997, 801 ρ(C–H), 783, 762, 745, 664, 617γ(C–H) cm−1. Elemental analysis calc. for [Co(C15H15N5)Cl2]·CH3OH (440.05): C, 46.28; H, 4.80; N, 15.87. Found C, 46.19; H, 4.88; N, 15.79%.:1:1 v/v (15 mL) was stirred at room temperature for 48 h under the normal atmosphere. The solution was concentrated and diethyl ether was added to precipitate of the complex. The yellow solid was centrifuged, washed with diethyl ether and dried. Yield: 75%. ESI-MS m/z (%) = 399 (20) [MnLBr]+, 266 (100) [L + H]+. IR (ATR) ν = 3222ν(C–H)ar, 1624 ν(CN)imin., 1595, 1568, 1557, 1489, 1466, ν(CC), 1439, 1428, 1372 ν(CN), 1316, 1282 ν(C–N), 1251, 1226, 1205 ν(C–C), 1046 ν(N–N), 1010, 992, 902, 825 ρ(C–H), 784, 743, 637, 595 γ(C–H) cm−1. Elemental analysis calc. for [Mn(C15H15N5)Br2] (480.06): C, 37.53; H, 3.15; N, 14.59. Found: C, 37.60; H, 3.03; N, 14.50%.Conclusions
In the present study, we have synthesized six mononuclear complexes of transition metal ions (Fe(II), Cu(II), Co(II) and Mn(II)) with benzimidazole-based ligand L and their crystal structures as well as electrochemical and spectroelectrochemical properties were reported. All complexes were found to be electroactive in voltammetric studies. Complexes Fe(II), Cu(II) and Co(II) showed both metal-centered and ligand-based color changes, while Mn(II) complex decomposes during electrochemical oxidation. For Fe(II) complex four different colors were observed depending on the redox state of both metal ion and ligand molecule and it is very interesting compound for its use in multielectrochromic devices. Complex Fe(II) was found to be stable in solution during multiple oxidation/reduction cycles. Switching between Fe(I) form of complex (green color) and Fe(III) form (yellow color) can be done in solution more than 60 cycles with no decrease in difference in transmittance percent between these two states.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support received from the National Science Centre, Poland (Grant No. 2016/21/D/ST5/01631) is gratefully acknowledged.Notes and references
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- L. Sicard, D. Navarathne, T. Skalski and W. G. Skene, Adv. Funct. Mater., 2013, 23, 3549–3559 CrossRef CAS.
- R.-T. Wen, C. G. Granqvist and G. A. Niklasson, ChemElectroChem, 2016, 3, 266–275 CrossRef CAS.
- H. Kim, Y. Park, D. Choi, S.-H. Ahn and C. S. Lee, Appl. Surf. Sci., 2016, 377, 370–375 CrossRef CAS.
- Z. Tong, H. Yang, L. Na, H. Qu, X. Zhang, J. Zhao and Y. Li, J. Mater. Chem. C, 2015, 3, 3159–3166 RSC.
- M. Walesa-Chorab and W. G. Skene, RSC Adv., 2014, 4, 19053–19060 RSC.
- A. M. Österholm, D. E. Shen, J. A. Kerszulis, R. H. Bulloch, M. Kuepfert, A. L. Dyer and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2015, 7, 1413–1421 Search PubMed.
- T. Abidin, Q. Zhang, K.-L. Wang and D.-J. Liaw, Polymer, 2014, 55, 5293–5304 CrossRef CAS.
- M. Wałęsa-Chorab, M.-H. Tremblay, M. Ettaoussi and W. G. Skene, Pure Appl. Chem., 2015, 87, 649–661 CrossRef.
- M. Wałęsa-Chorab, M.-H. Tremblay and W. G. Skene, Chem.–Eur. J., 2016, 22, 11382–11393 CrossRef PubMed.
- F. Ito and T. Nagamura, J. Photochem. Photobiol., C, 2007, 8, 174–190 CrossRef CAS.
- F. Hasanain and Z. Y. Wang, Dyes Pigm., 2009, 83, 95–101 CrossRef CAS.
- D. Arıcan, A. Aktaş, H. Kantekin and A. Koca, Sol. Energy Mater. Sol. Cells, 2015, 132, 289–295 CrossRef.
- C.-W. Hu, T. Sato, J. Zhang, S. Moriyama and M. Higuchi, J. Mater. Chem. C, 2013, 1, 3408–3413 RSC.
- K. Cheng and B. Tieke, RSC Adv., 2014, 4, 25079–25088 RSC.
- D. Marcinkowski, M. Wałęsa-Chorab, A. Bocian, J. Mikołajczyk, M. Kubicki, Z. Hnatejko and V. Patroniak, Polyhedron, 2017, 123, 243–251 CrossRef CAS.
- R. Borthakur, A. Kumar, A. K. De and R. A. Lal, Arabian J. Chem., 2015 DOI:10.1016/j.arabjc.2014.12.040.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953–1963 RSC.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- M. Najafi and M. Sadeghi, J. Braz. Chem. Soc., 2013, 24, 1679–1684 CAS.
- A. K. Mengel, C. Forster, A. Breivogel, K. Mack, J. R. Ochsmann, F. Laquai, V. Ksenofontov and K. Heinze, Chem.–Eur. J., 2015, 21, 704–714 CrossRef CAS PubMed.
- A. Mishra, E. Mena-Osteritz and P. Bäuerle, Beilstein J. Org. Chem., 2013, 9, 866–876 CrossRef CAS PubMed.
- N. K. Shee, M. G. B. Drew and D. Datta, New J. Chem., 2017, 41, 452–456 RSC.
- K. Takada, R. Sakamoto, S.-T. Yi, S. Katagiri, T. Kambe and H. Nishihara, J. Am. Chem. Soc., 2015, 137, 4681–4689 CrossRef CAS PubMed.
- P. Chábera, Y. Liu, O. Prakash, E. Thyrhaug, A. E. Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. B. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695–699 CrossRef PubMed.
- M. M. Kimani, D. Watts, L. A. Graham, D. Rabinovich, G. P. Yap and J. L. Brumaghim, Dalton Trans., 2015, 44, 16313–16324 RSC.
- M. O. Awaleh, A. Badia, F. Brisse and X.-H. Bu, Inorg. Chem., 2006, 45, 1560–1574 CrossRef CAS PubMed.
- A. Adamski, M. Osińska, M. Kubicki, Z. Hnatejko, G. Consiglio and V. Patroniak, Eur. J. Inorg. Chem., 2017, 2017, 859–872 CrossRef CAS.
- A. M. Sosa, V. M. Ugalde-Saldívar, I. González and L. Gasque, J. Electroanal. Chem., 2005, 579, 103–111 CrossRef CAS.
- G. Arena, R. P. Bonomo, A. Contino, C. Sgarlata, G. Spoto and G. Tabbi, Dalton Trans., 2004, 3205–3211, 10.1039/B408724K.
- T. T. Tasso, T. Furuyama and N. Kobayashi, Inorg. Chem., 2013, 52, 9206–9215 CrossRef CAS PubMed.
- R. S. Gaddie, C. B. Moss and C. M. Elliott, Langmuir, 2013, 29, 825–831 CrossRef CAS PubMed.
- D. K. Sau, N. Saha, R. J. Butcher and S. Chaudhuri, Transition Met. Chem., 2003, 28, 229–234 CrossRef CAS.
- S. Romain, C. Baffert, C. Duboc, J. C. Lepretre, A. Deronzier and M. N. Collomb, Inorg. Chem., 2009, 48, 3125–3131 CrossRef CAS PubMed.
- L. M. N. Assis, L. Ponez, A. Januszko, K. Grudzinski and A. Pawlicka, Electrochim. Acta, 2013, 111, 299–304 CrossRef CAS.
- E. Kavak, C. N. Us, E. Yavuz, A. Kivrak and M. İçli Özkut, Electrochim. Acta, 2015, 182, 537–543 CrossRef CAS.
- Agilent Technologies, CrysAlis PRO (Version 1.171.33.36d), Agilent Technologies Ltd, 2011 Search PubMed.
- G. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Selected spectral changes of investigated compounds, CIE parameters of L and compounds 2–5, crystal data, data collection and structure refinement parameters. CCDC 1570187 (1), 1468757 (2), 1468756 (3), 1468758 (4), 1570188 (5), and 1570189 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10451k |
| ‡ Current address: Institute of Technology and Life Sciences, Biskupińska 67, 60-463 Poznań, Poland. |
| This journal is © The Royal Society of Chemistry 2017 |