
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis and structural revision of a mangrove alkaloid†
Michael T. Green‡
 a,
Gary R. Peczkowski‡b,
Aneesa J. Al-Ania,
Sophie L. Benjaminc,
Nigel S. Simpkinsb and
Alan M. Jones*d
a,
Gary R. Peczkowski‡b,
Aneesa J. Al-Ania,
Sophie L. Benjaminc,
Nigel S. Simpkinsb and
Alan M. Jones*d
aDivision of Chemistry and Environmental Science, Manchester Metropolitan University, M1 5GD, UK
bSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK
cSchool of Science and Technology, Nottingham Trent University, NG11 8NS, UK
dSchool of Pharmacy, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: a.m.jones.2@bham.ac.uk; Tel: +44 (0)1214-147288
First published on 17th October 2017
Abstract
We report the total synthesis of an alkaloid isolated from the mangrove fungi Hypocrea virens, based on the originally claimed structure, via a photochemical sequence. Inconsistencies between data sets led to a revision of the proposed structure followed by a concise synthetic sequence to deliver the revised natural product.
Natural products have served as the source or architectural inspiration for countless drug discovery efforts. In the field of oncology for instance, around half of all final drug entities were either natural products or directly derived from them during the past 70 years.1 This could be due to their pre-validated biological relevance and coverage of different regions of chemical space.2 More recently, natural products from the marine world have been subject to extensive exploration and yielded compounds that have become clinically approved therapeutics such as ziconotide and trabectedin.3 Marine derived fungal metabolites have become an important new avenue of research in identifying novel structures with potential bioactivity from spores and cultures.
Connected to our other efforts in medicinal chemistry we became interested in exploring novel nitrogen based heterocycles as new scaffolds.4 Recently, the isolation and spectral characterisation of the first naturally occurring example of an imidazo[1,5-b]isoquinoline-1,3,5(2H)-trione alkaloid (1, Fig. 1) from the mangrove fungus, Hypocrea virens has been reported by Hua and co-workers.5 Subsequently, Davies and co-workers have reported a single example of the related, 7-phenoxyimidazo[1,5-b]isoquinoline-1,3,5(2H)-trione skeleton (2, Fig. 1) that exhibits sub-micromolar potency for inhibition of the proto-oncogene serine/threonine-protein kinase, PIM1 (IC50 = 804 nM ± 37 nM).6
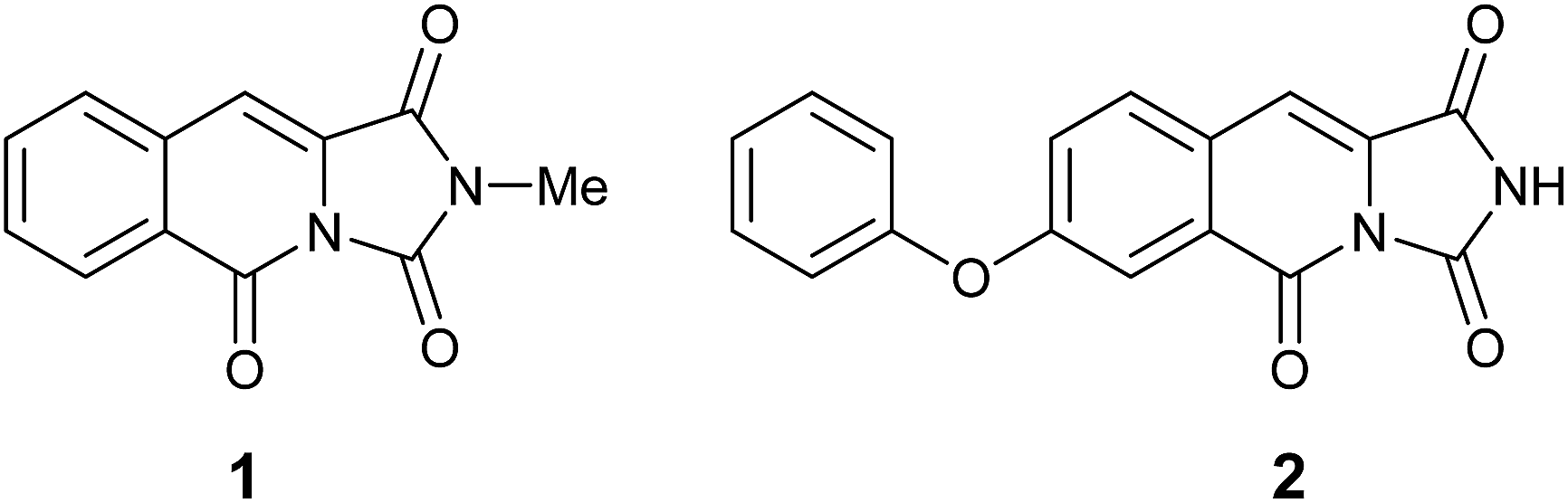 | ||
| Fig. 1 Reported structure of natural product 1 and a related bioactive analogue 2. | ||
Despite the apparent simplicity of 1, there have been no confirmatory syntheses reported to date of this mangrove alkaloid. Intrigued by both the structure of 1 and the recently reported bioactivity of a related analogue, we pursued a synthetic campaign to alkaloid 1.
Our initial approach to 1 envisioned a concise route; a double Knoevenagel and amide bond disconnection to 2-carboxybenzaldehyde 3 and 3-methylimidazolidine-2,4-dione 6 (Scheme 1). 6 in turn, could be prepared via a regioselective N3-alkylation of hydantoin with dimethylacetal dimethylacetamide (DMADMA) in 53% isolated yield.7 The reaction of 2-carboxybenzaldehyde 3 and a related hydantoin analogue, thiohydantoin 4a has previously been reported to afford the related heterocyclic scaffold 5a (Scheme 1).8
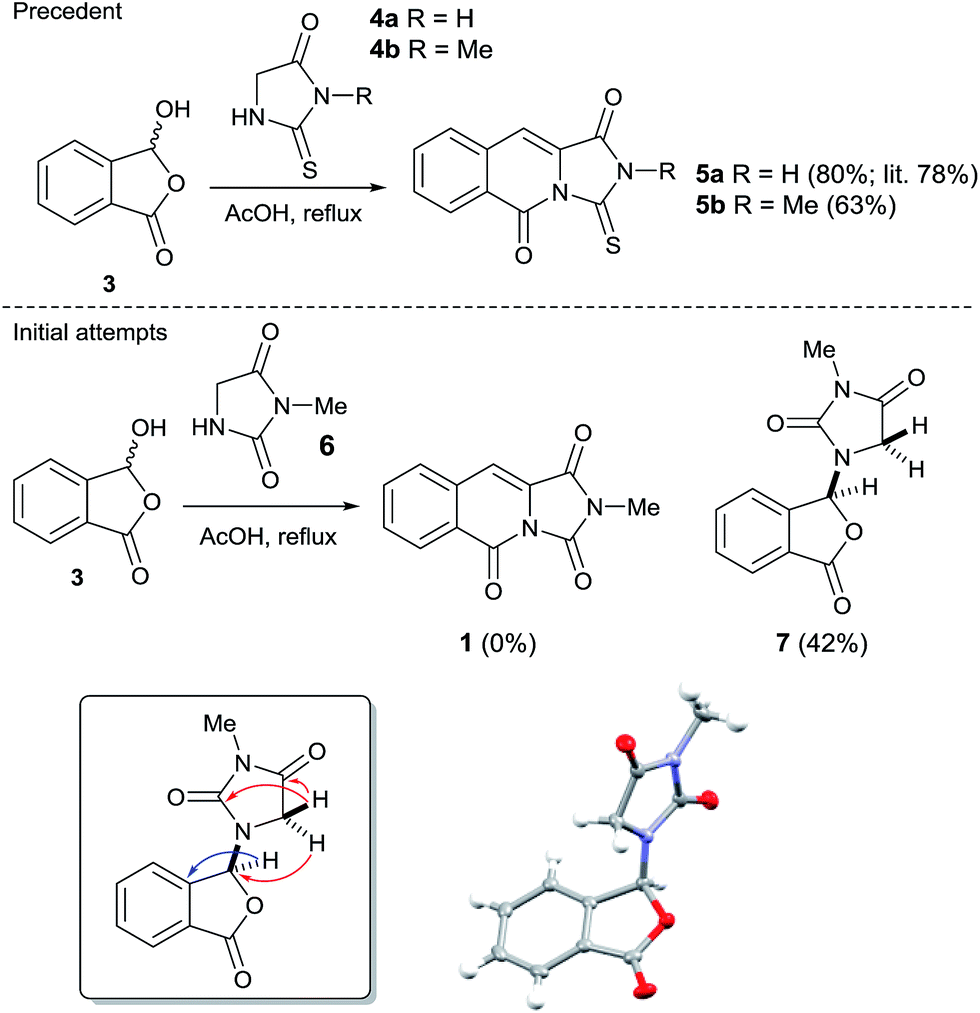 | ||
| Scheme 1 Literature precedent for the reaction of a thiohydantoin with 2-carboxybenzaldehyde to afford the tricyclic scaffold; result of the analogous hydantoin and 2-carboxybenzaldehyde experiment and inset selected instructive 1H–13C HMBC correlations and small molecule X-ray crystal structure of 7. | ||
However, treatment of 6 with 2-carboxybenzaldehyde 3 failed to deliver 1, instead an isobenzofuranone derivative 7 was afforded in 42% isolated yield. The connectivity of 7 was unambiguously assigned using a 1H–13C HMBC experiment (Scheme 1, inset).9 Other instructive spectral measurements included the splitting of the methylene signal in the 1H NMR spectrum, indicating diastereotopic protons which was confirmed by 1H–13C HSQC NMR.9 Conclusive evidence for the structure of 7 was obtained by small molecule X-ray crystallography (Scheme 1, inset).10 There are limited reports for the preparation of this isobenzofuranone derivative.11 In summary, unusual reactivity of hydantoin gave an unexpected reaction product compared to thiohydantoin with 2-carboxybenzaldehyde (Scheme 1).12 Attempts to intercept the reaction pathway via a protecting group strategy or sequentially performing the Knoevenagel step and the amide formation were also not successful.
We therefore sought to use the tricyclic manifold of 5b as a rapid entry to 1 via a desulfurisation approach. Stoichiometric strategies to convert thioamides and thioureas to amides or ureas, respectively are well known.13 On simple monofunctionalised thioamides a desulfurisation route using a photo-redox catalyst has been reported.14 Employing a photosynthetic route using Eosin-Y and green LEDs enabled the modest preparation of 1 but to the best of our knowledge this is the first report in a more complex heterocyclic setting (solvent and UV/vis studies are shown in the ESI).9 Alternatively, a two-step methylation and hydrolysis route also delivered 1 in an improved yield (Scheme 2).
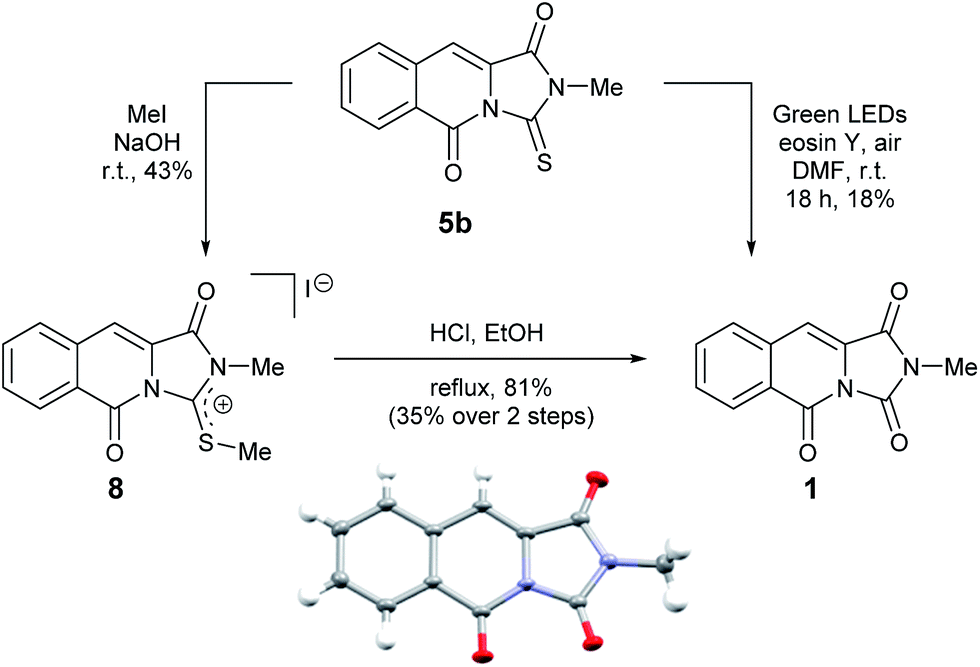 | ||
| Scheme 2 Two alternative desulfurisation routes to the reported mangrove alkaloid 1 and single crystal X-ray structure of 1 (inset). | ||
To our delight, compound 1 crystallised upon standing in CDCl3. Small molecule X-ray crystallography (Scheme 2) confirmed the preparation of structure 1. However, comparison of our NMR spectroscopic data with the originally reported5 revealed important disparities (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Isolated material 1 (CDCl3) | Synthesised material 1 (CDCl3) | |||||
| No. | δH (J in Hz) | δC | HMBC | δH (J in Hz) | δC | HMBC |
| a These δC values were reported to be interchangeable.5 | ||||||
| 1 | 156.5a | 159.3 | ||||
| 3 | 156.4a | 149.4 | ||||
| 5 | 156.5a | 156.9 | ||||
| 5a | 136.3 | 128.7 | ||||
| 6 | 8.49, d, 1H (8.2) | 117.2 | C8, C9a | 8.56, d, 1H (7.5) | 129.3 | C5, C8, C9a |
| 7 | 7.63, dd, 1H (8.2, 7.4) | 130.2 | C9 | 7.69–7.75, m, 2H | 130.4 | C9 |
| 8 | 7.48, dd, 1H (7.6, 7.4) | 126.9 | C6, C9a | 7.81, ddd, 1H (7.5, 1.5) | 134.1 | C6, C9a |
| 9 | 7.78, d, 1H (7.6) | 124.0 | C5a, C7, C10 | 7.69–7.74, m, 2H | 129.1 | C7 |
| 9a | 128.7 | 133.7 | ||||
| 10 | 7.71, s, 1H | 119.6 | C5a, C9a, C10a | 7.36, s, 1H | 109.1 | C1, C5a, C10a |
| 10a | 126.4 | 126.8 | ||||
| NCH3 | 3.47, s, 3H | 27.6 | C1, C3 | 3.27, s, 3H | 24.7 | C1, C3 |
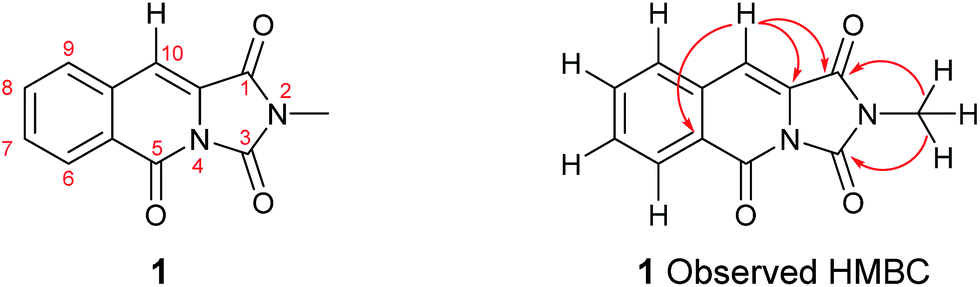
It became obvious by comparison of the NMR spectral data that the isolated example of 1 differed from that of synthesised 1 (Table 1). In particular, in the 1H NMR spectrum the location of the C10–H (+0.35 ppm) and N2-methyl group (+0.20 ppm) in isolated 1 vs. synthesised 1, respectively. Intriguingly, in the original report5 the amide, urea and arylamide 13C NMR spectral carbonyl shifts were all reported to be within ±0.1 ppm, which would be highly unlikely in the claimed structure.15 Furthermore, our synthesised example of 1 more closely replicates the known ethyl derivative reported by Niopas and Smail, in particular the location of the C10–H.16 It was therefore shown by both NMR spectroscopy and confirmation of the small molecule X-ray crystal structure that the previously isolated natural product had different connectivity. Therefore, taking account of all of the above it was proposed that the isolated structure claimed as 1, was in actual fact structural isomer 9 a metabolite of the gliotoxin family (Fig. 2).
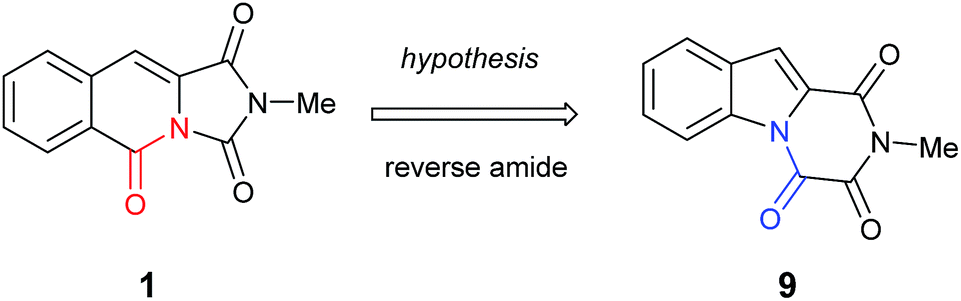 | ||
| Fig. 2 The connectivity pattern of the reported isolated material 1 (shown in black) and the most likely structural isomer 9. | ||
Furthermore, other metabolites isolated from Hypocrea virens and related mangrove fungi include gliotoxin derivatives demonstrating a potential common biosynthetic pathway.17 Compound 9 is a natural product recently isolated from the marine fungus Neosartorya pseudofischeri with NMR spectral data reported in d6-DMSO.18 In order to compare the spectra of 9 with the isolation data associated with the claimed structure of 1, a total synthesis and NMR spectra measurement in CDCl3 was required (Scheme 3), 9 having previously been prepared only via degradation studies of gliotoxin with selenium.19 Furthermore, the proposed structure 9 held promise, as it has also been reported to be found in isolates from Penicillium terlikowskii.20
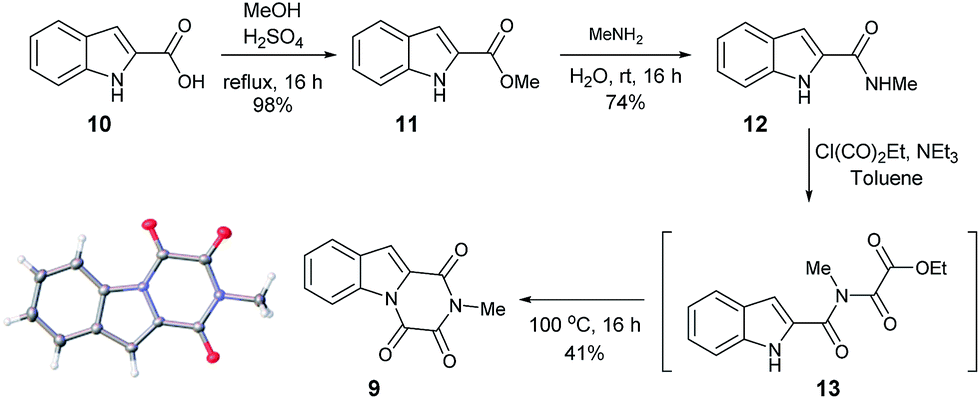 | ||
| Scheme 3 Synthetic route to the proposed natural product 9 and small molecule X-ray crystal structure of 9 (inset). | ||
Esterification of indole 2-carboxylic acid 10 followed by conversion to the N-methyl amide 12 in water proceeded smoothly.21 The conversion of 12 to 9 proved more challenging, with closure of the amine and amide motifs onto a range of reported oxalyl derivatives giving mixed results.22 The use of ethyl chlorooxoacetate proved effective via initial formation of 13 (observed in the crude 1H NMR after 1 h at reflux) to afford 9 in 41% isolated yield. Comparison of the 1H and 13C NMR spectra in CDCl3 revealed that 9 matched the isolation NMR details for the claimed structure of 1 (Table 2) with some subtle differences. The complete structural assignment began from the C6–H due to the effect of being forced into the deshielding cone of the peri-carbonyl group (C4).23 The 1H NMR spectral data was in good accordance between synthesised 9 and isolated 1 with <0.02 ppm differences for the C6–H and C10–H. Due to the pulse sequence used further splitting of the aromatic coupling pattern was observed at the 4J level as expected in a planar W-coupling system. The reported and observed 1H–13C HMBC correlations for 9 matched with the isolation data for 1 plus due to the strength of our sample further weak, but in agreement, correlations were observed (Table 2). Conclusive evidence for the structure of 9 was obtained by the small molecule X-ray crystal structure (Scheme 3, inset).10
|
||||||
|---|---|---|---|---|---|---|
| Isolated material 1 (CDCl3) | Revised material 9 (CDCl3) | |||||
| No. | δH (J in Hz) | δC | HMBC | δH (J in Hz) | δC | HMBC |
| 1 | 156.5 | 156.37 | ||||
| 3 | 156.4 | 156.45 | ||||
| 5 | 156.5 | 148.8 | ||||
| 5a | 136.3 | 136.3 | ||||
| 6 | 8.49, d, 1H (8.2) | 117.2 | C8, C9a | 8.47, dq, 1H (8.3, 0.9) | 117.2 | C8, C9a |
| 7 | 7.63, dd, 1H (8.2, 7.4) | 130.2 | C9 | 7.62, ddd, 1H (8.4, 7.3, 1.2) | 130.2 | C5a, C9 |
| 8 | 7.48, dd, 1H (7.6, 7.4) | 126.9 | C6, C9a | 7.47, ddd, 1H (8.2, 7.3, 1.0) | 126.9 | C6, C9a |
| 9 | 7.78, d, 1H (7.6) | 124.0 | C5a, C7, C10 | 7.78, dt, 1H (7.9, 1.0) | 124.0 | C5a, C7, C10 |
| 9a | 128.7 | 128.7 | ||||
| 10 | 7.71, s, 1H | 119.6 | C5a, C9a, C10a | 7.70, d, 1H (0.8) | 119.6 | C1, C5a, C9a, C10a |
| 10a | 126.4 | 126.4 | ||||
| NCH3 | 3.47, s, 3H | 27.6 | C1, C3 | 3.47, s, 3H | 27.6 | C1, C3 |
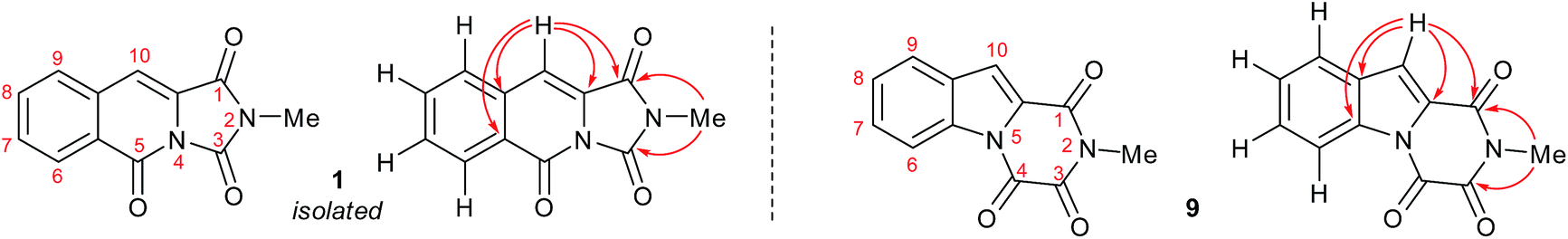
In summary, we have shown that the originally isolated natural product 1 through synthesis was gliotoxin derivative 9. We have reported the first synthesis, combining a photochemical step of the claimed structure of the mangrove alkaloid 1 and through structural revision, completed the first total synthesis of the revised natural product 9. The reassignment of the natural product isolated from Hypocrea virens opens the door to elucidating the biosynthesis or shunt metabolism of gliotoxin derivatives and provides a new scaffold to investigate the synthesis of gliotoxin family members. The biological evaluation of the reassigned natural product and related family members will be investigated and reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the EPSRC and AstraZeneca for iCASE support of G. R. P. The NMR instruments used in this research were obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). The authors thank Dr Phil Craven and Dr Stuart Langley for helpful discussions, MMU for a vacation scholarship to M. T. G. and Dr Louise Male and the Centre for Chemical and Materials Analysis in the School of Chemistry at the University of Birmingham for analytical support. We are indebted to Prof. Hui-Ming Hua (Shenyang Pharmaceutical University, China) for providing the scanned NMR spectra of 1 for comparison to our synthesised example and helpful correspondence.Notes and references
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461 CrossRef CAS PubMed; (b) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311 CrossRef CAS PubMed.
- S. Wetzel, A. Schuffenhauer, S. Roggo, P. Ertl and H. Waldmann, Chimia, 2007, 61, 355 CrossRef CAS.
- (a) T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2009, 8, 69 CrossRef CAS PubMed; (b) R. Montaser and H. Luesch, Future Med. Chem., 2011, 12, 1475 CrossRef PubMed; (c) H. Malve, J. Pharm. BioAllied Sci., 2016, 8, 83 CrossRef PubMed.
- A. W. Hensbergen, V. R. Mills, I. Collins and A. M. Jones, Tetrahedron Lett., 2015, 56, 6478 CrossRef CAS.
- T. Liu, Z.-L. Li, Y. Wang, L. Tian, Y. H. Pei and H.-M. Hua, Nat. Prod. Res., 2011, 25, 1596 CrossRef CAS PubMed.
- C. J. R. Bataille, M. B. Brennan, S. Byrne, S. G. Davies, M. Durbin, O. Fedorov, K. V. M. Huber, A. M. Jones, S. Knapp, G. Liu, A. Nadali, C. E. Quevedo, A. J. Russell, R. G. Walker, R. Westwood and G. M. Wynne, Bioorg. Med. Chem., 2017, 25, 2657 CrossRef CAS PubMed.
- (a) M. J. Porter, N. J. White, G. E. Howells and D. D. P. Laffan, Tetrahedron Lett., 2004, 45, 6541 CrossRef CAS; (b) Y. L. Janin, C. Huel, G. Flad and S. Thirot, Eur. J. Org. Chem., 2002, 1763 CrossRef CAS.
- A. I. Khodair, J.-P. Gesson and E. H. El-Ashry, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 2653 CrossRef CAS.
- See ESI.†.
- Crystallographic data for compounds 1, 7 and 9 have been deposited with the accession numbers CCDC 1569814, 1569815 and 1569797 contains the supplementary crystallographic data for this paper.†.
- (a) M. L. Baron, I. D. Rae and P. M. Simmonds, Aust. J. Chem., 1982, 35, 2567 CrossRef CAS; (b) D. R. Brittain and R. Wood, Imperial Chemical Industries PLC Patent, US4386100 A1, 1983.
- The differential reactivity of hydantoin vs. thiohydantoin with 2-carboxybenzaldehyde was postulated to be due to the greater nucleophilicity of the N1-nitrogen in hydantoin due to increased charge transfer from nitrogen to sulphur in the thiohydantoin structure.
- (a) A. Corsaro and V. Pistarà, Tetrahedron, 1998, 54, 15027 CrossRef CAS; (b) S. Sahu, P. R. Sahoo, S. Patel and B. K. J. Mishra, J. Sulfur Chem., 2011, 32, 171 CrossRef CAS.
- A. Yadav, V. Srivastava and L. Yadav, New J. Chem., 2013, 37, 4119 RSC.
- NMR spectroscopic data for 1 was supplied courtesy of Prof. Hui-Ming Hua (Shenyang Pharmaceutical University, China).
- I. Niopas and G. A. Smail, J. Chem. Soc., Perkin Trans. 1, 1991, 113 RSC.
- (a) A. Marion, M. Groll, D. Scharf, K. Scherlach, M. Glaser, H. Sievers, M. Schuster, C. Hertweck, A. A. Brakhage, I. Antes and E. M. Huber, ACS Chem. Biol., 2017, 12, 1874 CrossRef CAS PubMed; (b) T. Amatov and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 3312 CrossRef CAS PubMed.
- W.-L. Liang, X. Le, H.-J. Li, X.-L. Yang, J.-X. Chen, J. Xu, H.-L. Liu, L.-Y. Wang, K.-T. Wang, K.-C. Hu, D.-P. Yang and W.-J. Lan, Mar. Drugs, 2014, 12, 5657 CrossRef CAS PubMed.
- (a) J. D. Dutcher, J. R. Johnson and W. F. Bruce, J. Am. Chem. Soc., 1944, 66, 617 CrossRef CAS; (b) J. R. Johnson, R. B. Hasbrouck, J. D. Dutcher and W. F. Bruce, J. Am. Chem. Soc., 1945, 67, 423 CrossRef CAS; (c) M. I. P. Boente, G. W. Kirby, G. L. Patrick and D. J. Robins, J. Chem. Soc., Perkin Trans. 1, 1991, 1283 RSC.
- M. S. Ali, J. S. Shannon and A. Taylor, J. Chem. Soc. C, 1968, 16, 2044 RSC.
- S. N. Mistry, J. Shonberg, C. J. Draper-Joyce, C. K. Herenbrink, M. Michino, L. Shi, A. Cristopoulos, B. Capuano, P. J. Scammells and J. R. Lane, J. Med. Chem., 2015, 58, 6819 CrossRef CAS PubMed.
- (a) R. W. Foster, E. N. Lenz, N. S. Simpkins and D. Stead, Chem. –Eur. J., 2017, 23, 8810 CrossRef CAS PubMed; (b) M. Rees, N. S. Simpkins and L. Male, Org. Lett., 2017, 19, 1338 CrossRef CAS PubMed; (c) A. Cabanillas, C. D. Davies, L. Male and N. S. Simpkins, Chem. Sci., 2015, 6, 1350 RSC.
- A. M. Jones, T. Lebl, S. Patterson, T. van Mourik, H. A. Früchtl, D. Philp, A. M. Z. Slawin and N. J. Westwood, Tetrahedron, 2009, 65, 563 CrossRef CAS.
- The provided NMR spectra15 for isolated material 1 was found to be inconsistent with the structure of synthesised 1 and consistent with the spectroscopic data for 9. The only difference between synthesised 9 and isolated 1 was the 13C NMR shifts for the three carbonyl groups. In the original publication5 the three carbonyl groups were reported as being interchangeable at 156.4–156.5 ppm in CDCl3. As expected due to the different environments for the three carbonyl groups, three 13C NMR signals are seen in 9 at 156.45, 156.37 and 148.8 ppm. Resonances at 156.5, 156.4 and 148.8 ppm are also seen in the provided scanned 13C NMR spectra of isolated 1 including the previously unassigned carbonyl peak at 148.8 ppm.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1569814, 1569815, 1569797. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10483a |
| ‡ M. T. G and G. R. P contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |