
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphine oxide based polyimides: structure–property relationships
Irina Butnaruab,
Maria Brumaa and
Sabyasachi Gaan *b
*b
a“Petru Poni” Institute of Macromolecular Chemistry, Aleea Grigore Ghica Voda, 41A, Iasi, 700487, Romania. E-mail: ibacosca@icmpp.ro
bLaboratory for Advanced Fibers, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, St. Gallen 9014, Switzerland. E-mail: sabyasachi.Gaan@empa.ch
First published on 30th October 2017
Abstract
Several aromatic polyimides containing phosphine oxide moiety and different flexible linkages were synthesized and evaluated for their properties. The influence of flexible units on solubility, optical, wetting, static contact angles, thermal, and dielectric properties, as well as flame retardant behaviour were investigated. The results evidenced an improvement of the physical properties of the synthesized polyimides as compared to the classical aromatic polyimides. Thus, high values of transmittance of 80–88% at 450 nm were obtained, while the static contact angles of water and ethylene glycol were in the range of 74–92° and 52–62°, respectively. The thermogravimetric analysis confirmed that the initial decomposition temperature of these polyimides was in the range of 473–487 °C; they exhibited a single-stage decomposition process in the domain of 492–511 °C and had a residual weight in the range of 49–62% at 800 °C in nitrogen. Glass transition temperatures of the polyimides were in the domain of 216–271 °C strongly depending on the rigidity of the dianhydride segment. The dielectric constant values of thin free-standing films made from these polyimides were in the range of 3.36–3.64, close to those registered for commercially available polyimide (Kapton®). Flame retardant properties of the polyimides showed reduced values for peak heat release rate which were in the range of 27–429 W g−1 and for total heat release in the domain of 1.4–8.3 kJ g−1.
1. Introduction
Synthetic polymeric materials find wide scale applications and, over the last decades, many conventional materials have been replaced by polymers, due to their versatility, low density and improved properties.1,2 The most important drawback of most organic polymeric materials is their combustibility. During the burning process, the decomposition of a polymer results not only in the destruction of its properties, but also in the possible release of heat, toxic gases and smoke. This has led to the introduction of stricter legislation and safety standards concerning flammability of materials and as a result led to extensive research on development of flame retardant polymers.3–5A successful strategy to reduce the flammability of a polymeric material involves interrupting the complex combustion process (in solid or in vapour phase) by reducing the rate and/or by changing the burning mechanism. A flame retardant interferes with one or more steps of the fire cycle, during heating of the polymeric material, its subsequent degradation and burning of the resulting volatiles. Although very successful, the use of halogenated compounds usually results in an increase in formation of smoke and toxic decomposition products during polymer combustion and thus poses health hazard. As a result of these drawbacks, there has been increasing research to develop new, environmentally friendly, halogen-free flame retardants. Phosphorus-based flame retardants are both non-toxic,6 very effective and act in solid phase during combustion of polymers containing amino and hydroxyl functional groups.7,8 In case when the phosphorus based flame retardants are applied as non-reactive additives in polymers, they may leach out and thus the materials lose their flame retardant efficacy. In some case it may even create environmental issues. One way to avoid such problems is to either use reactive flame retardant additives or use polymers which are inherently flame retardant. Some examples of such polymers are polyetheretherketon (PEEK), polyethersulfon (PES), fully aromatic polyesters and polyimides. Polyimides represent an important class of organic polymers with outstanding properties; they have thermal and thermo-oxidative stability, are inherently flame retardant, exhibit high tensile strength and high modulus, excellent electrical properties and superior chemical resistance. Therefore, polyimides are used among others as matrix resins, adhesives, coatings and films for printed circuit boards, insulators for high performance applications in the aerospace, automotive, electrical, electronics and packaging industries.9,10 Despite their many attractive and desirable properties, the use of aromatic polyimides is limited because they are usually insoluble in common organic solvents and infusible under conventional processing conditions. The rigid structural components of polyimide determine its high glass transition temperatures or high crystalline melting points and thus are responsible for their poor solubility and processability. Strong interactions arise from the intra- and interchain charge transfer complex formation and electronic polarization, which also determine the brown coloration of the polyimides. To overcome these limitations continuous research on modifying their structure so as to improve flow and moldability, while maintaining the inherent attractive properties, has been performed over the last decades. It has been demonstrated that by reducing the stiffness of the polymer backbone or by lowering the interchain interactions melt flow and solubility can be improved.11 The main approaches to achieve these results include the incorporation of thermally stable and flexible or asymmetrical linkages, introduction of large polar or non-polar pendant substituents and/or interruption of the chain symmetry and of molecular iteration by copolymerization reactions.9,12–14
One approach which is worth to investigate is the incorporation of phosphorus based moieties in the polyimide backbone not only to improve the physical properties and its processability but also to simultaneously enhance its flame resistance. Moreover, chemically incorporated reactive phosphorus-containing monomers into the polymer backbone can avoid several problems, such as leaching over time. Reactive phosphorus moieties tend to stay in the condensed phase during the burning process and increase the char formation which acts as a thermal insulation for underlying layers.
In this research we have chosen phosphine oxide based reactive moiety to construct the polyimides. Compared to other phosphorus chemistries, the choice of phosphine oxide moiety has obvious advantages. P–C bond (compared to P–O–C)15 and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (compared to phosphine) offer hydrolytic and oxidative stability, respectively. Polymers containing phosphine oxide moieties typically display good flame resistance, high thermal oxidative stability, enhanced solubility, and improved miscibility and adhesion.16,17 On the other hand, the incorporation of short bridges such as –CO–, –CH2–, –C(CF3)2– into the polyimide chains has proven particularly beneficial to improve the solubility. In general, such a structural modification results in decrease in glass transition temperature and crystalline melting temperature as well as significant improvement in solubility and other processing characteristics of the polymers without greatly compromising other useful properties.18–20 The incorporation of hexafluoroisopropylidene (–C(CF3)2–) groups into polymer backbone also increases the glass transition temperature and flame resistance with a concomitant decrease of water absorption. The bulky –C(CF3)2– groups also serve to increase the free volume of the polymers, thus improving their electrical insulating characteristics.21,22
O bond (compared to phosphine) offer hydrolytic and oxidative stability, respectively. Polymers containing phosphine oxide moieties typically display good flame resistance, high thermal oxidative stability, enhanced solubility, and improved miscibility and adhesion.16,17 On the other hand, the incorporation of short bridges such as –CO–, –CH2–, –C(CF3)2– into the polyimide chains has proven particularly beneficial to improve the solubility. In general, such a structural modification results in decrease in glass transition temperature and crystalline melting temperature as well as significant improvement in solubility and other processing characteristics of the polymers without greatly compromising other useful properties.18–20 The incorporation of hexafluoroisopropylidene (–C(CF3)2–) groups into polymer backbone also increases the glass transition temperature and flame resistance with a concomitant decrease of water absorption. The bulky –C(CF3)2– groups also serve to increase the free volume of the polymers, thus improving their electrical insulating characteristics.21,22
Studies on polymers synthesized from bis(3-aminophenyl) methyl phosphine oxide and two dianhydrides namely, benzophenone-3,3′,4,4′-tetracarboxylic dianhydride and 4,4′-(4,4′-isopropylidenediphenoxy) bis(phthalic anhydride) involve basic characterization and thermal stability analyses.23,24 We have reported a characterization of polyimide fibers obtained by the polycondensation reaction of bis(3-aminophenyl) methyl phosphine oxide and 4,4′-(4,4′-isopropylidenediphenoxy) bis(phthalic anhydride).25
The present study aims to provide further detailed insight on the influence of various flexible linkages on the properties of four structurally analogues phosphine oxide containing aromatic polyimides. Thus, the influence of ether, carbonyl, hexafluoroisopropylidene and isopropylidene units on solubility, optical, surface morphology, wettability, dielectric constant, thermal and flammability properties of polyimides were measured and correlated to their chemical structures. It was expected that the combination of phosphine oxide moiety with aromatic polyimide ring and flexible linkages should offer polymers with enhanced solubility, film forming ability, good dielectric properties and thermal stability together with an increased flame retardant behaviour.
2. Experimental
2.1 Materials
N-Methyl pyrrolidone (NMP) (HPLC, 99.9% purity), N,N-dimethylformamide (DMF) (anhydrous, 99.8%), N,N-dimethylacetamide (DMAc) (anhydrous, 99.8%), dimethyl sulfoxide (DMSO) (anhydrous, ≥99.9%), 4,4′-oxydiphthalic anhydride (ODPA), benzophenone-3,3′,4,4′-tetracarboxylic dianhydride (BTDA) 4,4′-(hexafluoroisopropylidene) diphthalic anhydride (6FDA) and 4,4′-(4,4′-isopropylidenediphenoxy) bis(phthalic anhydride) (6HDA), were purchased from Aldrich and used without further purification.2.2 Monomers and polymers
Bis(3-aminophenyl) methyl phosphine oxide (BAMPO) was prepared by the two-step reaction according to published procedures.24,26 Analysis of BAMPO: m.p. = 150.4 °C (DSC). 1H NMR (DMSO-d6, 400 MHz, ppm): 1.76 (3H, d), 5.29 (4H, s), 6.65 (2H, d), 6.75 (2H, t), 6.83 (2H, d), 7.08 (2H, t). 31P NMR (DMSO-d6, 400 MHz, ppm): 28.7 (s).Phosphorus containing polyimides PI1–PI4 were prepared by a two-step polycondensation reaction of equimolar amounts of aromatic diamine BAMPO with different aromatic dianhydrides, such as ODPA, BTDA, 6FDA and 6HDA, in NMP as solvent (Scheme 1). The concentration of solids in the resulting solutions was of 15%. Thus, the dianhydride was added to a solution of diamine in NMP at room temperature and the reaction was run for 24 h under nitrogen, giving the corresponding poly (amidic acid)s. The cyclodehydration of the intermediary compound was performed by heating the poly (amidic acid) solution at 195–200 °C for 10 h, under a slow stream of nitrogen. The final polyimide was isolated by precipitation in ethanol. The solid product was filtered, washed with ethanol and dried at 120 °C for 12 h.
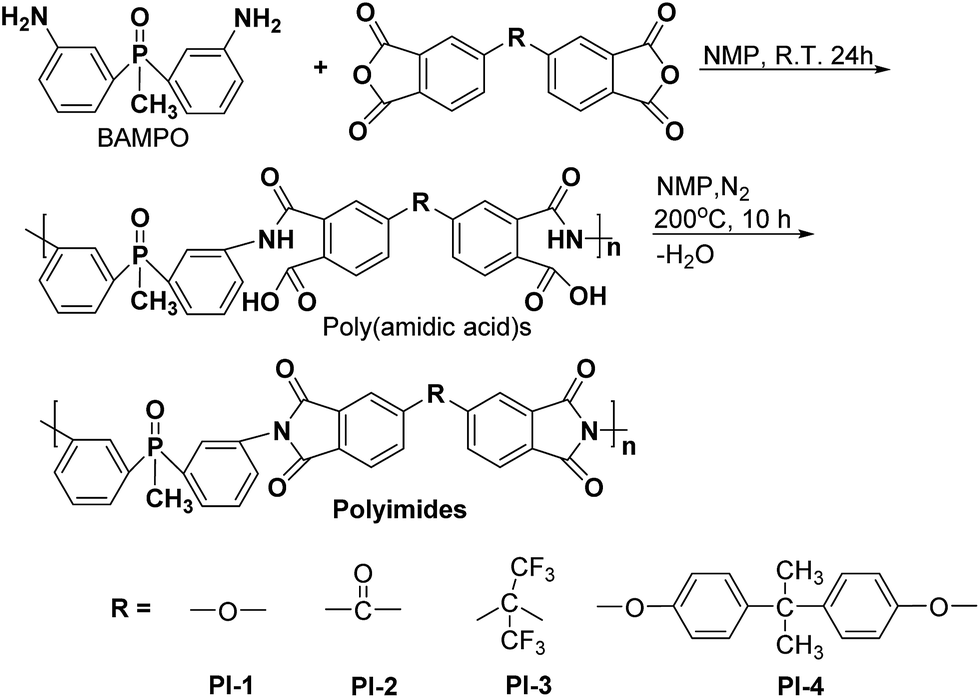 | ||
| Scheme 1 Synthesis of polyimides PI1–PI4. | ||
O stretching), 1720 (symmetric CO stretching), 1425 (P–Ar stretching), 1368 (C–N stretching), 1237 (Ar–O–Ar stretching), 1177 (PO stretching), 743 (imide ring deformation). 1H NMR (DMSO-d6, 400 MHz, ppm): 2.08 (3H, d), 7.55 (4H, d), 7.60 (4H, m), 7.75 (2H, t), 7.90 (2H, d), 7.97 (2H, d). 31P NMR (DMSO-d6, 400 MHz, ppm): 27.42 (s). N content: 5.08 ± 0.01 wt%, P content: 4.71 ± 0.12 wt%.O stretching), 1719 (symmetric CO stretching), 1668 (ketone CO stretching), 1424 (P–Ar stretching), 1370 (C–N stretching), 1164 (PO stretching), 714 (imide ring deformation). 1H NMR (DMSO-d6, 400 MHz, ppm): 2.10 (3H, d), 7.67 (4H, m), 7.87 (2H, t), 7.95 (2H, d), 8.11 (4H, m), 8.20 (2H, d). 31P NMR (DMSO-d6, 400 MHz, ppm): 27.41 (s). N content: 5.01 ± 0.03 wt%, P content: 4.63 ± 0.06 wt%.O stretching), 1723 (symmetric CO stretching), 1426 (P–Ar stretching), 1371 (C–N stretching), 1209 and 1191 (C–F stretching), 1157 (PO stretching), 720 (imide ring deformation). 1H NMR (DMSO-d6, 400 MHz, ppm): 2.07 (3H, d), 7.63 (6H, m), 7.80 (2H, t), 7.92 (4H, d), 8.14 (2H, d). 31P NMR (DMSO-d6, 400 MHz, ppm): 27.40 (s). N content: 4.13 ± 0.04 wt%, P content: 3.88 ± 0.12 wt%.O stretching), 1722 (symmetric CO stretching), 1424 (P–Ar stretching), 1366 (C–N stretching), 1239 (Ar–O–Ar stretching), 1173 (PO stretching), 742 (imide ring deformation). 1H NMR (DMSO-d6, 400 MHz, ppm): 1.67 (6H, s), 2.05 (3H, d), 7.06 (4H, d), 7.28 (8H, m), 7.58 (4H, t), 7.76 (2H, t), 7.88 (4H, d). 31P NMR (DMSO-d6, 400 MHz, ppm): 27.35 (s). N content: 3.79 ± 0.02 wt%, P content: 3.72 ± 0.10 wt%.2.3 Preparation of polyimide films
Free-standing thin films of polyimides PI1–PI4 were prepared from polymer solutions of 10% concentration in NMP by casting onto glass plates, followed by gradual heating from room temperature up to 225 °C. The resulting films showed a strong adhesion to the glass support and were stripped off the plates by immersion in water, followed by drying in oven at 110 °C. The obtained films had the thickness in the range of tens of micrometers and were used afterwards for the study of dielectric properties.Very thin films having the thickness in the range of nanometers were prepared from polymer solutions in NMP with concentrations of 1% by spin-coating onto silicon wafers at a speed of 500 rpm. These films, as-deposited, were gradually heated up to 225 °C in the same way as described earlier to remove the solvent and they were used afterwards for atomic force microscopy, optical and wetting measurements.
2.4 Measurements
Infrared spectra were recorded on FT-IR Bruker Vertex 70 analyzer, by using KBr pellets.1H and 31P were recorded on Bruker Avance400 NMR spectrometer (frequencies of 400.13 and 100.61 MHz, respectively) in DMSO-d6 at 25 °C. Chemical shifts are reported in δ units (ppm) relative to the remaining resonances of the solvent at 2.49 ppm for 1H-NMR. The 31P-NMR spectra are externally referenced to a sample of neat H3PO4 at 0.0 ppm.
The phosphorus and nitrogen content of the polymers were estimated using the inductively coupled plasma optical emission spectrometry method (ICP-OES), on Perkin Elmer Optima 3000 apparatus. Sample preparation for ICP-OES consists of mixing 300 mg of polymer with 1 mL H2O2 and 3 mL HNO3, followed by etching in the microwave.
The molecular weight and their distribution were determined by GPC with a PLGPC 220 instrument with a 2× PL-Gel Mix-B LS column set equipped with RI (refractive index), viscosity and LS (light scattering with 15° and 90° angle) detectors (DMF + 1 g L−1 LiBr as eluent at 80 °C). Universal calibration was done using polystyrene standards of known molecular weight.
The quality and morphology of very thin films as-deposited on silicon wafers were investigated by using AFM with a Scanning Probe Microscopy Solver PRO-M, NT-MDT equipment (Russia), in semi-contact mode, semi-contact topography technique using a SEM with Quanta 200 ESEM equipment.
Wetting properties of polymer films on glass substrates were studied by measuring the static contact angles of water and ethylene glycol droplets deposited onto the polymer film surface. Deionized water and ethylene glycol microdrops with an average size of 1–2 μL were deposited on the sample surface with a microliter syringe. Images of the drops on polymer films were recorded with a video camera. The experiments were carried out using a CAM 101 Optical Video Contact Angle System from KSV Instruments Ltd, Finland, in open-air atmosphere and at room temperature. Contact angles were calculated as average values over a large number of measurements (typically 10). The measurements were repeated three times on different parts of the film samples. Temperature and moisture were constant during the experiment (23 °C and 68%, respectively). Ethylene glycol and deionized water were used as solvents for calculation of the surface free energy of the polymer films. The surface free energy and its components were calculated using the following equations:
Young equation:
γLV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ = γSV − γSL, cosθ = γSV − γSL,
| (1) |
Dupré equation:
| Wa = γLV + γSV − γSL, | (2) |
Young–Dupré equation:
|
Wa = γLV (1 + cosθ),
| (3) |
Fowkes equation:
| γSV = γpSV + γdSV, | (4) |
Owen–Wendt equation:
|
γLV (1 + cosθ) = 2(γdSVγdLV)0.5 + 2(γpSVγpLV)0.5,
| (5) |
Wenzel equation:
|
cosθr = rcosθ,
| (6) |
The dispersion (γdLV) and the polar (γpLV) force components of the surface free energy of water are 21.8 and 51 mJ m−2, respectively, and those of ethylene glycol are 29 and 19 mJ m−2, respectively.29 By introducing these values in the Owens–Wendt equation, the following equations were obtained:
| 72.8(1 + cosθw) = 2(21.8γdLV)0.5 + 2(51γpLV)0.5 |
| 48(1 + cosθeg) = 2(29γdLV)0.5 + 2(19γpLV)0.5 |
After the contact angles of the liquid drops on polymer films were measured (θw = water contact angle and θeg = ethylene glycol contact angle), two equations with two unknowns (γdSV)0.5 and (γdLV)0.5 were obtained.
UV-visible absorption spectra of the polymers were recorded with Specord M42 apparatus using very dilute polymer solutions (cca 10−5 mol L−1).
Thermogravimetric analysis (TGA) of the polyimides was carried out by using NETZSCH TG209 F1 Iris instrument. The sample, weighing approx. 3 mg, was heated from 25 to 800 °C at a heating rate of 10 °C min−1. The measurements were performed two times under nitrogen atmosphere with a total gas flow of 50 mL min−1.
Differential scanning calorimetry (DSC) measurements were carried out with Mettler T28E calorimeter. Samples (3–5 mg) were heated from 25 to 250C with a heating rate of 20 °C min−1 under nitrogen atmosphere.
Broad dielectric spectroscopy (BDS) measurements of the polymer films at various temperatures in the range of −150 to 270 °C and in the frequency range of 10−1 to 106 Hz were performed by using Novocontrol Dielectric Spectrometer (GmbH Germany), CONCEPT 40. Polymer films were placed in a flat parallel plate capacitor arrangement having gold plated electrodes with 20 mm diameter. The thickness of the films was in the range of 0.04–0.07 mm. The amplitude of AC applied voltage was 1 V.
The combustibility of polyimides was determined by pyrolysis combustion flow calorimetry (PCFC) by using FTT PCFC instrument. The samples weighing 1–3 mg were pyrolysed by heating up to 750 °C, at a heating rate of 1 °C s−1. The combustor of the PCFC instrument was maintained at 900 °C. The specific principle of the PCFC analysis is well described in the literature.30
3. Results and discussion
To make a correlation between chemical structure and properties of phosphorous containing polyimides (PIs) a series of polymers resulting from the reaction of an aromatic diamine containing phosphine oxide moiety with different aromatic dianhydrides incorporating various flexible linkages were synthesized.3.1 Basic characterization of polyimides
The confirmation of the chemical structure of polyimides was made by elemental analysis (measuring N and P content), FTIR, 1H and 31P NMR spectroscopy. Infrared spectra of the polymers evidenced the formation of imide ring, the incorporation of phosphine oxide unit and of flexible linkages: ether, carbonyl, hexafluoroisopropylidene or isopropylidene. Thus, strong bands of absorption were observed in the FTIR spectra at about 1780 and 1720 cm−1 which were assigned to the stretching of asymmetric and symmetric CO unit, at about 1370 cm−1 due to the vibration of C–N stretching and at approximately 740 cm−1 due to the imide ring deformation. The absorption band for aromatic C–H linkage appeared at about 3070 cm−1, while the band characteristic for aliphatic C–H unit was evidenced at about 2916 cm−1. The successful incorporation of phosphine oxide moiety was evidenced by the absorption bands which appeared at about 1170 cm−1 which were ascribed to PO unit stretching and at 1420 cm−1 characteristic for P–Ar linkage. Also, sharp absorption bands characteristic of aromatic ether linkage were evidenced for polymers PI-1 and PI-4 at approximately 1240 cm−1, the ketone unit appeared at 1668 cm−1 in the spectrum of polyimide PI-2, the hexafluoroisopropylidene group appeared at 1209 and 1191 cm−1 in the FTIR spectrum of polyimide PI-3, while the isopropylidene group was evidenced at 2967 and 2870 cm−1 in polymer PI-4.
1H NMR spectra of polyimides evidenced the complete imidization process, no traces of the intermediary polyamidic acids were detected. The aromatic protons appeared in the 7.0–8.20 ppm region in all polyimide NMR spectra. The presence of methyl protons from phosphine oxide unit were identified at about 2.1 ppm, while the protons from isopropylidene group in polyimide PI-4 appeared at 1.67 ppm. 31P NMR spectra evidenced a single peak at about 27.4 ppm which demonstrates the existence of a single P species and the successful incorporation of phosphine oxide unit in the PIs backbone.
3.2 Solubility and film forming ability
All polyimides were soluble at room temperature in polar organic solvents such as NMP, DMF, DMAc, DMSO, and PI-4 was soluble in less polar solvents such as chloroform. The good solubility is also due to the structural modifications made by the incorporation of flexible linkages such as ether, carbonyl, hexafluoroisopropylidene or isopropylidene units in the main chain which disturb the packing of PIs. As a consequence, the shape of the respective macromolecules is not a linear rigid rod like the one characteristic to conventional fully aromatic polyimides. Thus, the interchain interactions are diminished and the packing of the macromolecules is prevented facilitating the diffusion of small molecules of solvents between the polymer chains leading to a better solubility. Due to this good solubility the present polyimides were processed into thin free-standing films and into very thin coatings on silicon wafers.The GPC data of these polyimides showed relatively high molecular weight values similar to those reported in the literature.20 In summary, the number-average molecular weight (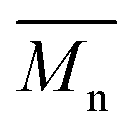 ) values are in the range of 8700–24000 Da and the weight-average molecular weight (
) values are in the range of 8700–24000 Da and the weight-average molecular weight (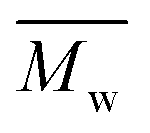 ) values are in the domain of 18000–52000 Da. At the same time, the polymers show a relatively narrow distribution of molecular weight as indicated by the value of
) values are in the domain of 18000–52000 Da. At the same time, the polymers show a relatively narrow distribution of molecular weight as indicated by the value of 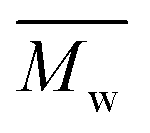 /
/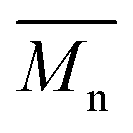 , ranging between 1.67 and 2.18 (Table 1), being similar to those reported previously by other authors.31 Such values of molecular weight enabled the formation of flexible thin films which were prepared afterwards from these polymers.
, ranging between 1.67 and 2.18 (Table 1), being similar to those reported previously by other authors.31 Such values of molecular weight enabled the formation of flexible thin films which were prepared afterwards from these polymers.
| Sample | (
|
(
|
(
|
|---|---|---|---|
| PI-1 | 9600 | 21000 |
2.18 |
| PI-2 | 8700 | 18000 |
2.07 |
| PI-3 | 15600 |
26000 |
1.67 |
| PI-4 | 24000 |
52000 |
2.16 |
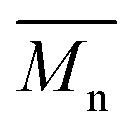
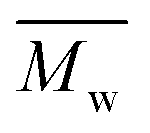
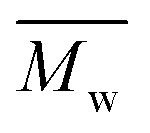
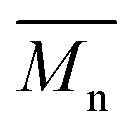
A high quality of thin film requires smooth surfaces, free of pinholes in order to avoid leakage current of a device which could cause short circuits. Therefore, atomic force microscopy was used to analyse the surface of polyimide films. The results obtained from the scanning over an area of 5 × 5 μm2 evidenced a low value for average roughness in all PI films, in the range of 0.35–0.74 nm (Table 2). The measurement showed that the surface of the thin films was smooth, without cracks or pinholes. Fig. 1 presents typical AFM images of polyimide PI-3 containing phosphine oxide and hexafluoroisopropylidene groups.
| Sample | Ra [nm] | T450 [%] | λcut-off [nm] |
|---|---|---|---|
| a Ra-average roughness, measured by AFM; T450-transmittance at 450 nm; λcut-off-cut-off wavelength. | |||
| PI-1 | 0.40 | 86.0 | 317 |
| PI-2 | 0.63 | 77.3 | 319 |
| PI-3 | 0.35 | 88.2 | 299 |
| PI-4 | 0.74 | 88.3 | 300 |
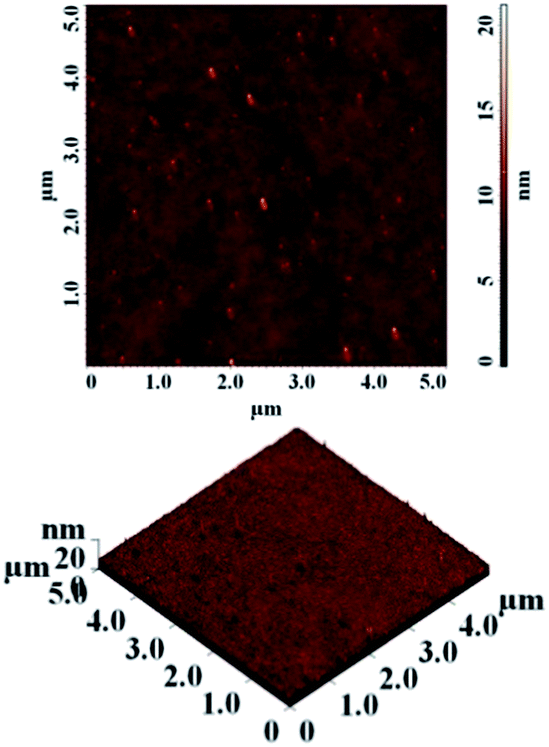 | ||
| Fig. 1 AFM images: 2D (top) and 3D (bottom) of polyimide PI-3. | ||
The relationship between chemical structure and optical properties in phosphine oxide containing polyimides was investigated. The PI films showed good optical transparency in the ultraviolet-visible light region with cut-off wavelengths lower than 319 nm and transmittances close or higher than 80% at 450 nm (Table 2). The higher optical transparency registered in the case of polyimides PI-3 and PI-4 (about 88%) may be attributed to flexible hexafluoroisopropylidene and isopropylidene groups, respectively, present in the dianhydride unit. This data may be explained by the inhibition of intra- and/or intermolecular charge–transfer interactions. It has been established that the charge transfer complex formation (CTC) between the alternating electron-donor (diamine) and electron-acceptor (dianhydride) units in all-aromatic PI chains is the main reason for the coloration of the films.32 The introduction of flexible ether, hexafluoroisopropylidene or isopropylidene groups reduced the formation of CTC, thus improving the transparency of the polyimides. The lower value of T450 obtained for PI-2 film may be attributed to the incorporation of more polar carbonyl unit (>CO). The cut-off wavelengths registered the lowest value for PI-3 (299 nm) and the highest value for PI-2 (319 nm), being comparable with the ones measured for classical aromatic polyimides.33
3.3 Wetting properties of polyimide films
In order to investigate the hydrophobic properties of these polyimides static contact angles of water and ethylene glycol on thin polymer film surface were measured. The obtained values were in the range of 74.3–92.2° for water and 52.2–62.3° for ethylene glycol (Table 3). As can be observed, polyimides PI-1, PI-2 and PI-4 do not display significant variations in the values of contact angle. The small differences are due to the structural feature of the dianhydride segment in the repeating units of the macromolecular chains and to polymer film topography. It is known that the wettability behaviour of surfaces depends on several parameters and, in particular, on the surface chemical composition and topography (geometry and roughness).33 Polymer PI-1 has the lowest contact angle value (74.3°) due to the presence in its structure of ether bridges which interact with water or ethylene glycol molecules forming hydrogen bonds. As expected PI-3, containing hexafluoroisopropylidene groups shows the highest contact angle (92.2°), confirming the idea that the incorporation of fluorine into polymers increases its hydrophobicity. Polyimide PI-4 has a higher contact angle (81.8°) than polymer PI-1 although it contains more ether bridges in the molecule. This could be attributed to the surface topography differences of the two polymer films. Polyimide film PI-4 has rougher surface (Ra = 0.74 nm) compared to PI-1 (Ra = 0.40 nm), probably due to a higher degree of packing in the solid state. The influence of the packing degree on the contact angle value has a greater impact than the structural differences between these polymers PI-1 and PI-4.| Polymer | θw (°) | θeg (°) | γpSV (mJ m−2) | γdSV (mJ m−2) | γSV (mJ m−2) | γSL (mJ m−2) |
|---|---|---|---|---|---|---|
| a θw – water contact angle, θeg – ethylene glycol contact angle, γpSV – polar part of surface free energy, γdSV – dispersive part of surface free energy, γSV – total interfacial tension at solid/vapor interface (total surface free energy), γSL – interfacial tension at solid/liquid interfaces. | ||||||
| PI-1 | 74.28 ± 0.53 | 52.23 ± 0.87 | 14.28 | 17.03 | 31.31 | 11.60 |
| PI-2 | 79.52 ± 0.51 | 55.12 ± 0.75 | 9.41 | 20.44 | 29.86 | 16.16 |
| PI-3 | 92.16 ± 1.13 | 62.33 ± 0.20 | 1.84 | 29.47 | 31.31 | 34.05 |
| PI-4 | 81.77 ± 0.55 | 62.31 ± 0.51 | 10.97 | 14.80 | 25.77 | 15.34 |
The surface free energy and its components were calculated using the Young, Dupré, Young–Dupré, Fowkes, Owen–Wendt and Wenzel equations. By solving the system of these equations the values of the dispersive and polar parts of the surface free energy were obtained.
The polar part of the surface free energy ranges from 1.84 to 14.28 mJ m−2. The lowest value was obtained for the highest hydrophobic polymer film surface PI-3 incorporating non-polar hexafluoroisopropylidene group, while the highest value of γpSV was calculated for PI-1 which contains polar ether groups. The values achieved for the dispersive component of the surface free energy were in the range of 14.80–29.47 mJ m−2, close to those reported by other authors (γdSV ≈ 30 mJ m−2).34,35 The increased values of the dispersive components of PI-2 and PI-3 compared with PI-1 and PI-4 can be explained by the increased degree of molecular packing of the polyimides chains.
3.4 Thermal properties
The thermal stability of polyimides is a very important aspect when the material has to resist high temperatures. The thermal properties of the polyimides were evaluated using the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). A series of parameters are crucial factors when a material is analysed as possible thermally stable candidate for high temperature applications: the initial decomposition temperature (T5) taken in air, the value of maximum speed of decomposition temperature (Tmax), the residue at 800 °C (W800) under inert condition and the glass transition temperature (Tg).According to the TGA measurements presented in Table 4, all polyimides showed good thermal stability. The initial decomposition temperature (taken as T5) measured in nitrogen was in the range of 473–487 °C. The polymers exhibited single-step decomposition process with a maximum rate of decomposition in the domain of 492–511 °C which corresponds to the scission of large segments of macromolecular chains. The decomposition processes in air environment, as expected, was different compared to in the nitrogen atmosphere. The T5 value in air was slightly lower (467–487 °C) compared to those registered under inert atmosphere. Also, the decomposition behaviour in air evidenced a multi-step degradation process with maxima which started in the range of 485–506 °C. The data showed that the polyimides retained most of their weights up to 467 °C and exhibited rapid weight loss after 485 °C in both environments. The polymers registered a residual weight in the range of 49–62% at 800 °C in nitrogen (Fig. 2) and of about 1.36–33% in air environment. Their high thermal stability could be attributed to the introduction of phosphine oxide moiety between the imide rings. The difference in the residue contents at 800 °C for various polymers may be correlated to the relative weight percentage of P in polyimides. Thus, for polyimides PI-1 (4.71% P) and PI-2 (4.63% P) with identical phosphorus content the residue is approximately the same (62%); for PI-3 with 3.88% P it registered the lowest value for W800 of about 49%, while the high value of char yield (about 61%) obtained in the case of PI-4 with 3.72% P may be due to the increased aromatic carbon content from the main chain. Compared to a related polyimide structure based on 6HDA and 4,4′-oxydianiline the thermal stability has a similar behaviour as PI-4 except the value of residual weight at 800 °C in nitrogen: the first has a value of 48% while in the case of polyimide PI-4 W800 is higher (61%).36 Thus, the introduction of phosphine oxide moieties leads to an increase in char yield of the corresponding polyimides while maintaining the high thermal stability. Also, the introduction of flexible ether, carbonyl, hexafluoroisopropylidene and isopropylidene units in the main chain did not affect the thermal stability of the present polyimides while the solubility and film forming ability were improved.
| Sample | In N2 | In air | Tg (°C) | ||||
|---|---|---|---|---|---|---|---|
| T5 (°C) | Tmax (°C) | W800 (%) | T5 (°C) | Tmax (°C) | W800 (%) | ||
| a T5-initial decomposition temperature taken as the 5% weight loss; Tmax-temperature of the maximum mass loss rate; W800-char yield at 800 °C; Tg-glass transition temperature. | |||||||
| PI-1 | 473 ± 0.0 | 492 ± 0.1 | 62.3 ± 0.0 | 472 ± 1.4 | 489 ± 0.0/541 ± 3.5/775 ± 4.9 | 21 ± 1.8 | 269 |
| PI-2 | 479 ± 0.18 | 503 ± 2.0 | 62.9 ± 1.7 | 467 ± 0.5 | 485 ± 0.0/526 ± 2.5/688 ± 3.0 | 1.36 ± 0.6 | 270 |
| PI-3 | 483 ± 1.7 | 511 ± 1.4 | 49.5 ± 0.1 | 480 ± 1.3 | 506 ± 1.4/584 ± 1.7 | 3 ± 0.4 | 271 |
| PI-4 | 487 ± 1.8 | 497 ± 0.0 | 61.2 ± 0.1 | 486 ± 1.4 | 495 ± 2.1/572 ± 7.0/790 ± 8.0 | 33.3 ± 6.0 | 216 |
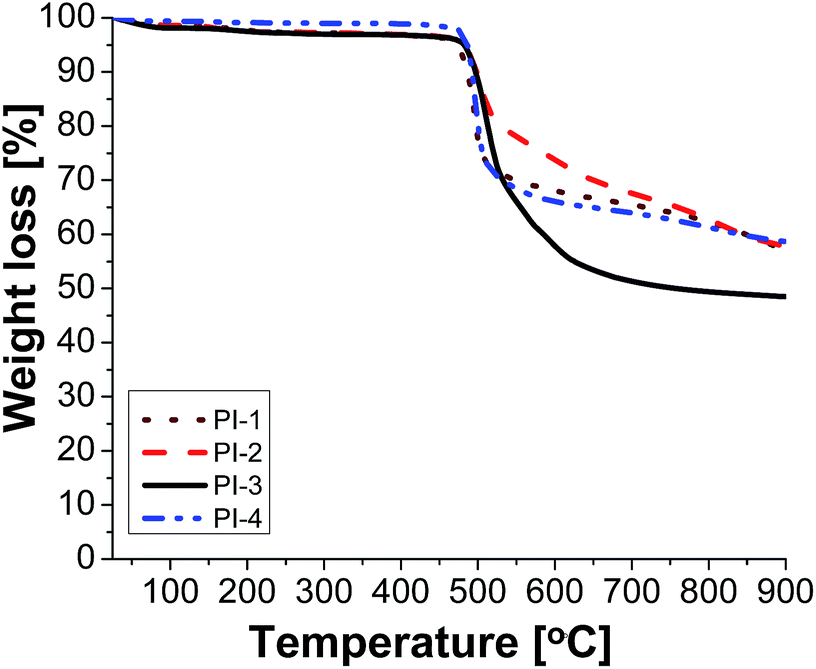 | ||
| Fig. 2 TGA curves of polyimides PI1–PI4 in nitrogen. | ||
The DSC study of polyimides confirmed the glass transition temperatures of the polymers in the range of 216–271 °C (Table 4). The Tg of the polymers strongly depends on the rigidity of the dianhydride segment. Thus, the lowest value corresponds to the polyimide PI-4 which incorporates a more flexible isopropylidene linkage, while PI-2 containing rigid carbonyl unit exhibited the highest Tg.
3.5 Dielectric properties
Electrical insulating properties of polymer films PI1–PI4 were evaluated on the basis of dielectric constant and dielectric loss, and their variation with frequency and temperature. The dielectric permittivity of a material is a complex quantity, when measured in the frequency domain. It is composed of two parts: the real part (ε′), called the “dielectric constant” decreases with the increase of frequency with characteristic steps. Its imaginary part (ε′′), usually called the “dielectric loss” may show two or more maxima on the diagrams versus frequency, or versus temperature. The “steps” on dielectric constant diagrams and the maxima on dielectric loss curves correspond to different molecular relaxation phenomena. The dielectric constant is frequency dependent and it is composed of electronic, atomic, and dipole orientational contributions. The dielectric constant is associated to the polarizability of a material and is therefore strongly dependent on its chemical structure. The dielectric loss is a measure of the energy required for molecular motion, which is the energy dissipated in this motion in the presence of an electric field. It consists of two contributions: energy losses due to the orientation of molecular dipoles, and energy losses due to the conduction of ionic species.37–39 Therefore, the dielectric permittivity of polyimide films was analysed from 1 to 106 Hz and in the domain of −150 to 250 °C.Fig. 3 presents the dependence of dielectric constant on frequency at low, moderate and high temperatures for polyimides PI1–PI4. It can be observed that the dielectric constant of polyimides increases with the increase of temperature and it decreases with the raise of frequency, taking low values at high frequencies for all analysed temperatures. This behaviour may be attributed to the response of the electronic, atomic and dipolar polarizable units which varies with frequency in the specific domain. It is known that the magnitude of the dielectric constant is dependent upon the ability of the polarizable units to orient fast enough to keep up with the oscillation of the alternating electric field. When frequency increases, the orientational polarization decreases since the orientation of dipole moments needs a longer time than electronic and ionic polarizations.36,40
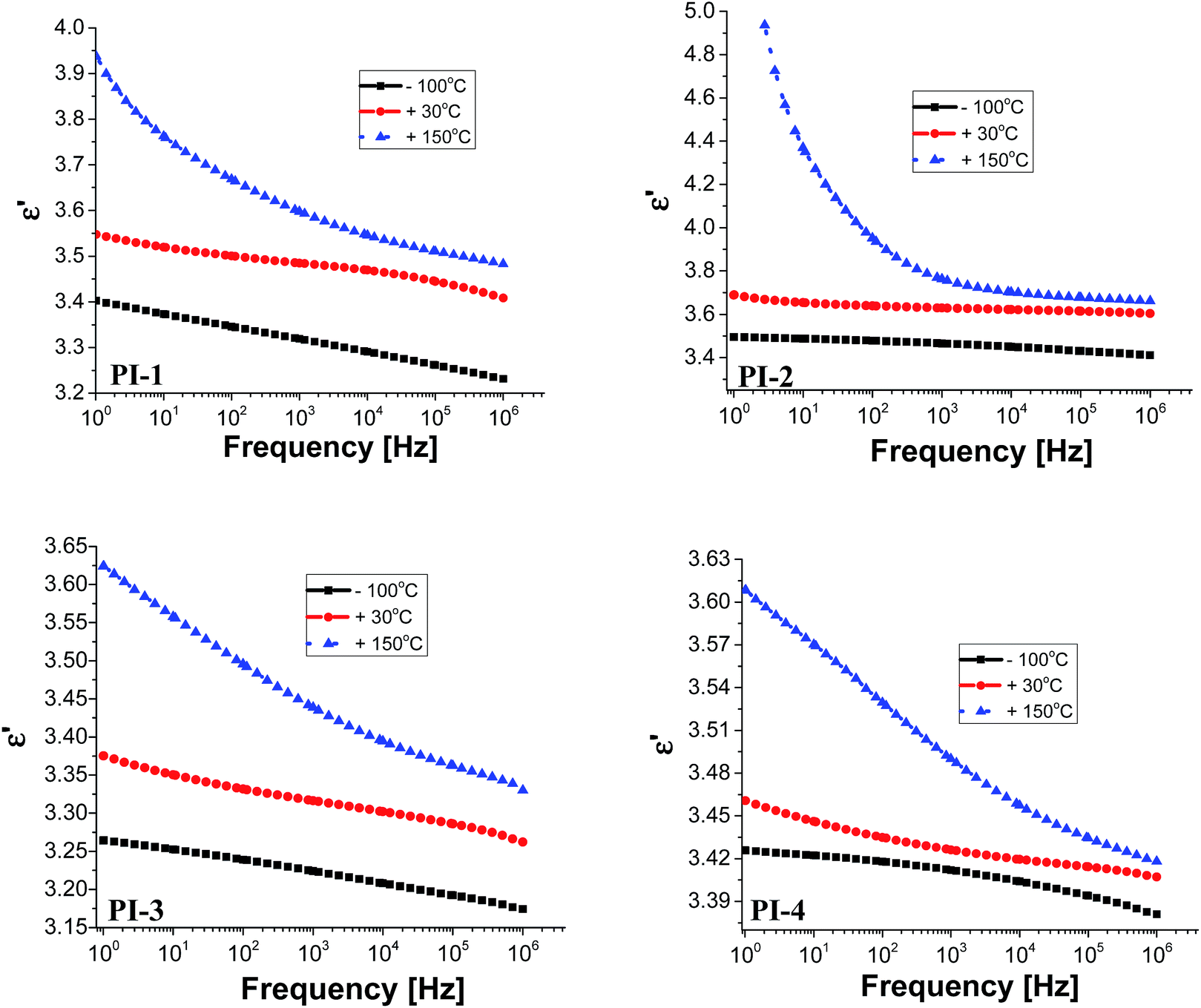 | ||
| Fig. 3 Dielectric constant at different temperatures for polyimides PI1–PI4. | ||
Fig. 3 shows that the increasing rate of the dielectric constant with decreasing frequency for polyimides at low, moderate and high temperatures is comparable, demonstrating a similar polarization capability of the polymers. A strong increase of the dielectric constant can be observed at high temperature (150 °C) due to the mobility of the charge carriers. The dielectric constants of the PI films at 1 Hz, 1 kHz and 100 kHz, at 20 °C, taken from the second heating cycle are presented in Table 5. The dielectric constant values of these polyimides are in the range 3.36–3.64, being comparable or slightly higher than those of commercially available Kapton HN polyimide film.41 The relatively low dielectric constant value (3.36) registered for PI-3 is due to the presence of flexible hexafluoroisopropylidene groups in the polymer chains, which decreases the chain packing and increases the free volume of the polymer.42 A similar situation can be observed in PI-4 that showed a lower value of dielectric constant (3.43) as compared to PI-1 and PI-2, which can be attributed to the flexible isopropylidene units from the main chain. The incorporation of flexible units determines a reduction of polarization by decreasing the number of polarizable groups per volume unit and thus lowering the dielectric constant.43 Higher dielectric constant were obtained for polyimides PI-1 (3.53) and PI-2 (3.64), which contain more polar ether and carbonyl units in the structure. This may increase the number of polar groups per structural unit thus can explain the higher dielectric constant obtained for PI-2 and PI-1.
| Polymer | Dielectric constant | Dielectric loss | ||||
|---|---|---|---|---|---|---|
| 1 Hz | 1 kHz | 100 kHz | 1 Hz | 1 kHz | 100 kHz | |
| PI-1 | 3.53 | 3.47 | 3.43 | 2.11 × 10−2 | 1.05 × 10−2 | 2.01 × 10−2 |
| PI-2 | 3.64 | 3.61 | 3.58 | 3.33 × 10−2 | 9.84 × 10−3 | 8.14 × 10−3 |
| PI-3 | 3.36 | 3.30 | 3.27 | 1.71 × 10−2 | 9.72 × 10−3 | 1.28 × 10−2 |
| PI-4 | 3.43 | 3.41 | 3.36 | 1.35 × 10−2 | 1.18 × 10−2 | 1.08 × 10−2 |
Generally, polymers show relaxation processes that influence the physical properties of materials made from them.44 Typically, in polyimide films, three relaxation processes are observed with increasing temperature, designated as γ, β and α, respectively, the last corresponding to the glass–rubber relaxation.45,46
Fig. 4 displays the dielectric loss versus temperature at various frequencies, in the first heating and second heating cycle. As can be observed in the first scan, in the negative temperature region a relaxation peak appears in the ε′′ curve, which corresponds to γ local relaxations. It seems that the introduction of different flexible units in the dianhydride segment does not have a significant influence on the temperature at which this relaxation is detected, at about −100 °C, in all polyimide films. γ transition can be associated with phenyl ring oscillations and it is influenced by moisture absorption content, aging history and morphology.47,48 On the medium temperature region another sub-glass relaxation process β, at about 60 °C, was detected. β relaxation is related to the rotation of rigid segments of phenylene or imide groups around flexible hinges.45
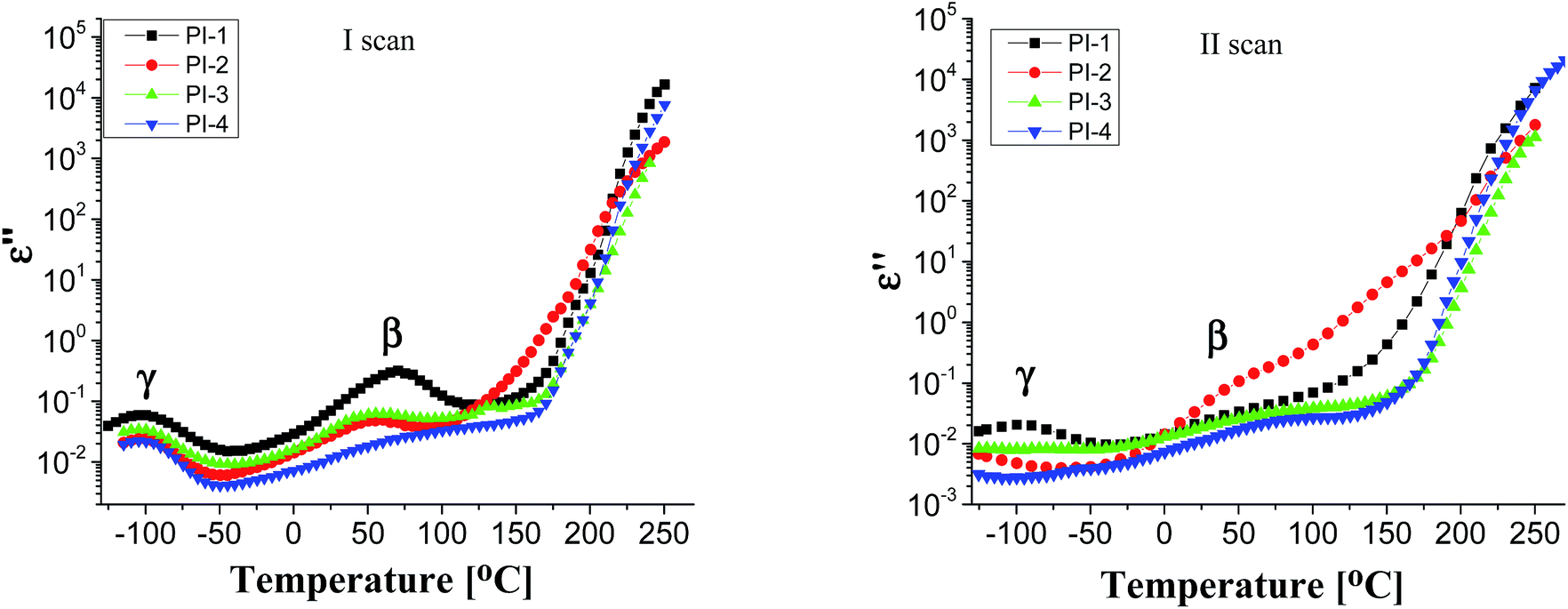 | ||
| Fig. 4 Dielectric loss at 1 Hz at various temperatures for polyimides PI1–PI4. | ||
In the second heating cycle, it can be observed that the γ relaxation disappears completely (for PI-3 and PI-4) or the intensity is reduced (for PI-1 and PI-2). It is possible that some polar molecules (e.g. water) adsorbed during storage are removed from the polyimide films on heating. Similar processes have been observed in other aromatic polyimides.48 The curves representing the dielectric loss in the frequency domain show conductive contributions which hide the low-frequency side of the β relaxation. The broadening of β process may be determined by the intensity of the intramolecular charge transfer between diamine and diimide segments.45 The local or segmental chain mobility allows for the molecular dipoles to respond to changes in frequency. With increasing temperature there is an increase of permittivity that causes the material to become less insulating.49 The values of dielectric loss registered at 1 Hz for polyimides PI1–PI4 are in the range of 1.35 × 10−2 to 3.33 × 10−2 being lower when compared with that of Kapton HN (ε′′ = 6 × 10−2 at 1 Hz).42 Low values of dielectric loss are indicative of minimal conversion of electrical energy into heat in the dielectric material. It is advantageous to have low values for both dielectric constant and dielectric loss because the electrical signals loss will be lower in the dielectric medium.
3.6 Flame retardancy of polyimides
PCFC is a small-scale flammability testing technique which enables fast screening of material at milligram scale. This technique provides useful data regarding fire behaviour of a material e.g. peak of heat release rate (pHRR), the temperature at the maximum heat release rate (TM) and total heat release (THR) that reflects the combustion properties of materials. The HRR plots for polyimides PI1–PI4 are shown in Fig. 5 and the corresponding combustion data are presented in Table 6.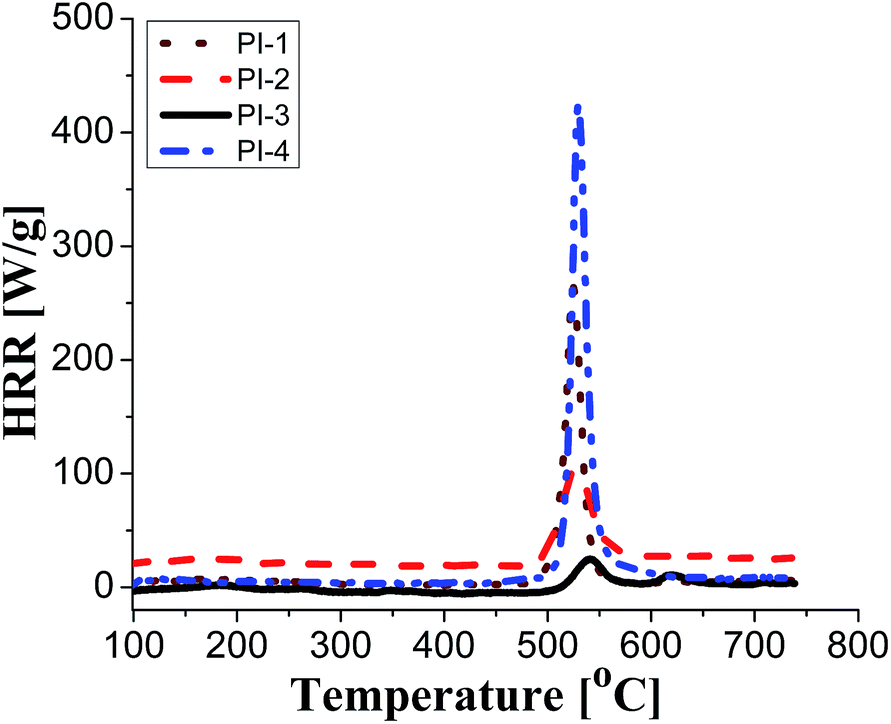 | ||
| Fig. 5 HRR curves of PI1–PI4 from PCFC data. | ||
| Polymer | R | Ψi (kJ mol−1 K−1) | TM (°C) | pHRR (W g−1) | THR (kJ g−1) | W800 (%) |
|---|---|---|---|---|---|---|
| a Ψi-molar contribution of components to heat release capacity; TM-the temperature at the maximum heat release rate; pHRR-peak of heat release rate; THR-total heat release; W800-char residue at 800 °C. | ||||||
| PI-1 |  |
11.1 | 523.6 ± 3.5 | 260.9 ± 19.9 | 5.22 ± 0.2 | 63.7 ± 6.1 |
| PI-2 | 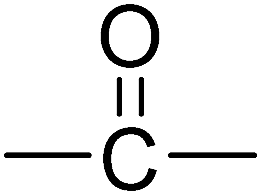 |
−12.6 | 526.5 ± 2.8 | 80.42 ± 8.8 | 2.97 ± 0.3 | 67.7 ± 3.2 |
| PI-3 | 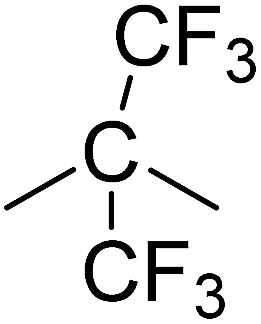 |
−13.3 | 539.9 ± 1.5 | 27.38 ± 1.8 | 1.4 ± 0.2 | 58.4 ± 12.9 |
| 619 ± 2.0 | 9.55 ± 1.7 | |||||
| PI-4 | 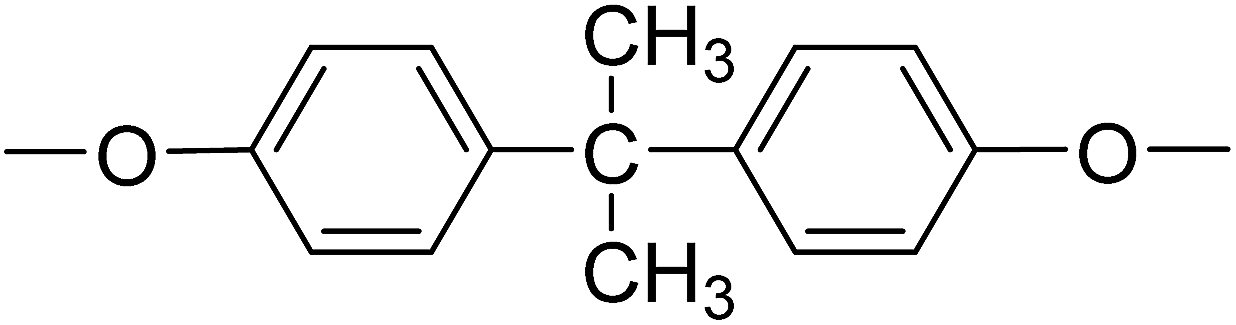 |
85.5 | 529.6 ± 0.8 | 428.6 ± 4.9 | 8.3 ± 0.3 | 65.4 ± 10 |
It can be observed that the peak HRR (pHRR) values of PI-3 (∼27 W g−1) and PI-2 (∼81 W g−1) are much lower compared to PI-4 (∼429 W g−1) and PI-1 (∼261 W g−1). The pHRR values correlate well with the calculated molar group contributions to the heat capacity (Ψi) of different flexible segments (Table 6) from each polyimide structure. The data for the molar group contributions of each segment (R in Table 6) from the polyimide repeating unit were obtained by summing the additive molar group contributions of the comprising units, as previously reported by other authors.50 Knowing that heat release capacity is calculated by dividing the maximum specific heat release rate (HRR) by the actual heating rate used in the experiment, the chemical structure of R segment has a significant influence on the data obtained from PCFC. Therefore, the lowest value of pHRR obtained for PI-2 may be correlated with a low value for Ψi (−12.6 kJ mol−1 K−1), while in the case of PI-3 the negative contribution to the heat release capacity (Ψi = −13.3 kJ mol−1 K−1) may be due to the strong synergistic interactions between phosphine oxide unit and hexafluoroisopropylidene heteroatom group. Interestingly, in the case of PI-3 there is another heat release peak of about 9.5 W g−1, which may indicate a secondary decomposition for char residue at high temperature (∼619 °C). This behaviour was also observed by other authors in the case of fluorinated polymers.51 Polyimide PI-4 has the highest value for pHRR which may be correlated with the high value of molar group contribution to the heat of capacity of isopropylidene group (Ψi = 85.5 kJ mol−1 K−1). These results suggest that polyimides PI-1 and PI-3 can provide higher flame retardancy compared to many traditional polymers. Similar trend was observed in the case of total heat release: the lowest value was obtained for polymer PI-3 (THR = ∼1.4 kJ g−1), followed by PI-2 (THR = ∼2.97 kJ g−1) and PI-1 (THR = ∼5.22 kJ g−1), while the highest value was registered for polyimide PI-4 (THR = ∼8.3 kJ g−1). Therefore, the flame retardance has been greatly influenced by the variation of R segments in the polyimide chain, leading to a beneficial reduction in the value of THR in the case of PI-3 and PI-2. The temperature for maximum heat release rate are in the range of 527–619 °C which are in the same domain as the ones registered for other polyimide structures.52,53 The char residue values (Table 6) obtained for the polyimides in the PCFC experiments are comparable to the values obtained in TGA in nitrogen (Table 4). Thus, the W800 values are high, in the domain of 58–68%, corresponding to the structural modification induced by the flexible segments (R in Table 6).
These results indicate that the polyimides, depending on the chemical structure, exhibit varying levels of flame retardancy. PCFC being a small scale technique to investigate the combustion behaviour, a more detailed fire evaluation in future should involve larger scale fire tests, such as the cone calorimeter and UL94 tests.
4. Conclusions
Aromatic polyimides containing phosphine oxide moieties and various flexible linkages were synthesized and the influence of such flexible groups on the physical properties of the polymers was investigated. The presence of ether, carbonyl, hexafluoroisopropylidene and isopropylidene groups in the main chain of the corresponding polyimides improved the solubility, optical, wetting, thermal and dielectric properties, and produced a higher flame retardant behaviour at the same time. Thus, the highest values for optical transmittances (88% at 450 nm), the largest static contact angles (92.16° and 81.77°) and the lowest dielectric constants (3.36 and 3.43) were obtained for polymers containing hexafluoroisopropylidene and isopropylidene units, respectively. The thermal stability maintained high values for all polymers up to 467 °C; the polymers lost 5% of their initial weight rapidly after 485 °C in air and nitrogen environment. The polyimides registered a residual weight in the range of 49–62% at 800 °C in nitrogen and of about 1.36–33% in air environment. The highest values of char residue were obtained, as expected, for the polymer containing isopropylidene unit. Glass transition temperatures, strongly dependent on the rigidity of the flexible unit from the dianhydride segment, were in the range of 216–271 °C. The introduction of different flexible units has not a significant influence on the temperature of sub-glass relaxation processes at which γ and β relaxations appear in dielectric studies. Pyrolysis combustion flow calorimetry showed that the peak heat release rate values correlate well with the calculated molar group contributions to the heat capacity of different flexible segment from each polyimide structure. The lowest values were obtained for polyimide containing hexafluoroisopropylidene (∼27 W g−1) and carbonyl units (∼81 W g−1). The reduction of total heat release values indicate a higher flame retardant behaviour for these polymers, which correlates well with the data obtained from thermal analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support provided by SCIEX project no 12.379/2013 and by the Executive Agency for Higher Education, Research, Development and Innovation Funding (UEFISCDI) – Romania, under project code PN-III-P4-ID-PCE-2016-0708, contract no. 66/2017 is acknowledged. We thank Dr Thomas Schweizer from ETH Zurich for GPC measurements, to Dr Valentina Musteata and Dr Gabriela Hitruc from “Petru Poni” Institute of Macromolecular Chemistry, Iaşi, Romania for BDS and AFM analyses.References
- K. Singh, in Chemistry in Daily Life, ed. A. K. Gosh, PHI Learning Private Limited, New Delhi, 2012 Search PubMed.
- Handbook of Research on Functional Materials: Principles, Capabilities and Limitations, ed. C. A. Wilkie, G. Geuskens and V. M. de Matos Lobo, CRC Press, Boca Raton, 2014 Search PubMed.
- Hazardous chemicals: Safety management and global regulations, ed. T. S. S. Dikshith, CRC Press, Boca Raton, 2013 Search PubMed.
- R. Renner, U.S., flame retardants will be voluntarily phased out, Environ. Sci. Technol., 2004, 38, 14A–15A Search PubMed.
- L. S. Birnbaum and D. F. Staskal, Environ. Health Perspect., 2004, 112, 9–17 CrossRef CAS PubMed.
- C. Hirsch, B. Striegl, S. Mathes, C. Adlhart, M. Edelmann, E. Bono, S. Gaan, K. A. Salmeia, L. Hoelting, A. Krebs, J. Nyffeler, R. Pape, A. Bürkle, M. Leist, P. Wick and S. Schildknecht, Arch. Toxicol., 2017, 91, 407–425 CrossRef CAS PubMed.
- I. Butnaru, M. Fernandez-Ronco, J. Czech-Polak, M. Heneczkowski, M. Bruma and S. Gaan, Polymers, 2015, 7, 1541–1563 CrossRef CAS.
- K. A. Salmeia, S. Gaan and G. Malucelli, Polymers, 2016, 8, 319 CrossRef.
- D. J. Liaw, K. L. Wang, Y. C. Huang, K. R. Lee, J. Y. Lai and C. S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- H. Banda, D. Damien, K. Nagarajan, M. Hariharan and M. M. Shaijumon, J. Mater. Chem. A, 2015, 3, 10453–10458 CAS.
- C. H. Lin, T. I. Wong, M. W. Wang, H. C. Chang and T. Y. Juang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 513–520 CrossRef CAS.
- J. R. Wiegand, Z. P. Smith, Q. Liu, C. T. Patterson, B. D. Freeman and R. Guo, J. Mater. Chem. A, 2014, 2, 13309–13320 CAS.
- H. Y. Yao, Y. H. Zhang, Y. Liu, K. Y. You, N. N. Song, B. J. Liu and S. W. Guan, J. Membr. Sci., 2015, 480, 83–92 CrossRef CAS.
- L. Ma, Y. Yang, C. Y. He, Z. X. Jia, X. Y. Liu and J. Q. Qin, Colloid Polym. Sci., 2015, 293, 1281–1287 CAS.
- S. Wendels, T. Chavez, M. Bonnet, K. A. Salmeia and S. Gaan, Materials, 2017, 10, 784 CrossRef PubMed.
- C. W. Chang, C. H. Lin, P. W. Cheng, H. J. Hwang and S. A. Dai, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2486–2499 CrossRef CAS.
- Y. Wang, W. Wang, M. Zhu and D. Yu, RSC Adv., 2017, 7, 42891–42899 RSC.
- J. de Abajo and J. G. de la Campa, Adv. Polym. Sci., 1999, 140, 23–59 CrossRef CAS.
- I. Bacosca, E. Hamciuc, M. Cristea, G. Lisa and M. Bruma, J. Appl. Polym. Sci., 2012, 124, 1956–1966 CrossRef CAS.
- J. U. Wieneke and C. Staudt, Polym. Degrad. Stab., 2010, 95, 684–693 CrossRef CAS.
- G. L. Tullos, P. E. Cassidy and A. K. S. Clair, Macromolecules, 1991, 24, 6059–6064 CrossRef CAS.
- C. Hamciuc, E. Hamciuc, I. Bacosca and M. Olariu, Mater. Plast., 2010, 47, 11–15 CAS.
- I. K. Varma and B. S. Rao, J. Appl. Polym. Sci., 1983, 28, 2805–2812 CrossRef CAS.
- B. Tan, C. N. Tchatchoua, L. Dong and J. E. McGrath, Polym. Adv. Technol., 1998, 9, 84–93 CrossRef CAS.
- D. Serbezeanu, I. Butnaru, C.-D. Varganici, M. Bruma, G. Fortunato and S. Gaan, RSC Adv., 2016, 6, 38371–38379 RSC.
- W. K. Chin, M. Shau and W. C. Tsai, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 373–379 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- F. M. Fowkes, J. Phys. Chem., 1963, 67, 2538–2541 CrossRef CAS.
- H. Y. Erbil, Surface Chemistry of Solid and Liquid Interfaces, Blackwell Publishing, Oxford, 2006, pp. 308–337 Search PubMed.
- ASTM, Standard Test Method for Determining Flammability Characteristics of Plastics and Other Solid Materials Using Microscale Combustion Calorimetry, ASTM D7309-11, 2011 Search PubMed.
- D. T. Padavana and W. K. Wan, Mater. Chem. Phys., 2010, 124, 427–433 CrossRef.
- M. Hasegawa and K. Horie, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef.
- M. Messori, M. Toselli, F. Pilati and C. Tonelli, Polymer, 2001, 42, 9877–9885 CrossRef CAS.
- M. D. Damaceanu, C. P. Constantin, A. Nicolescu, M. Bruma, N. Belomoina and R. S. Begunov, Eur. Polym. J., 2014, 50, 200–213 CrossRef CAS.
- I. Sava and S. Chisca, Mater. Chem. Phys., 2012, 134, 116–121 CrossRef CAS.
- I. Bacosca, M. Bruma, T. Koepnick and B. Schulz, J. Polym. Res., 2013, 20, 1–14 CAS.
- D. W. Van Krevelen, Properties of Polymers, Elsevier Science, Amsterdam, 1990 Search PubMed.
- M. Ree, K. J. Chen, D. P. Kirby, N. Katzenellenbogen and D. Grischkowsky, J. Appl. Phys., 1992, 72, 2014–2022 CrossRef CAS.
- G. Hougham, G. Tesoro, A. Viehbeck and J. D. Chapple-Soko, Macromolecules, 1994, 27, 5964–5971 CrossRef CAS.
- H. Deligoz, T. Yalcinyuva, S. Ozgumus and S. Yildirim, J. Appl. Polym. Sci., 2006, 100, 810–818 CrossRef CAS.
- M. D. Damaceanu, V. E. Musteata, M. Cristea and M. Bruma, Eur. Polym. J., 2010, 46, 1049–1062 CrossRef CAS.
- G. Hougham, in Fluoropolymers 2: Properties, ed. G. Haugham, P. E. Cassidy, K. Johns and T. Davidson, Plenum Press, New York, 1999, pp. 233–276 Search PubMed.
- K. Xie, J. G. Liu, H. W. Zhou, S. Y. Zhang, M. H. He and S. Y. Yang, Polymer, 2001, 42, 7267–7274 CrossRef CAS.
- S. Sen and R. H. Boyd, Eur. Polym. J., 2008, 44, 3280–3287 CrossRef CAS.
- C. Bas, C. Tamagna, T. Pascal and N. D. Alberola, Polym. Eng. Sci., 2003, 43, 344–355 CAS.
- A. C. Comer, D. S. Kalika, B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2009, 50, 891–897 CrossRef CAS.
- W. Qu, T. M. Ko, R. H. Vora and T. S. Chung, Polymer, 2001, 42, 6393–6401 CrossRef CAS.
- S. Chisca, V. E. Musteata, I. Sava and M. Bruma, Eur. Polym. J., 2011, 47, 1186–1197 CrossRef CAS.
- M. D. Damaceanu, R. D. Rusu, V. E. Musteata and M. Bruma, Polym. Int., 2012, 61, 1582–1591 CrossRef CAS.
- R. E. Lyon, M. T. Takemori, N. Safronava, S. I. Stoliarov and R. N. Walters, Polymer, 2009, 50, 2608–2617 CrossRef CAS.
- A. Ghosh, D. Bera, D. Y. Wang, H. Komber, A. K. Mohanty, S. Banerjee and B. Voit, Macromol. Mater. Eng., 2012, 297, 145–154 CrossRef CAS.
- R. E. Lyon, R. N. Walters and S. I. Stoliarov, Polym. Eng. Sci., 2007, 47, 1501–1510 CAS.
- A. Ghosh, S. Banerjee, D. Y. Wang, H. Komber and B. Voit, J. Appl. Polym. Sci., 2012, 123, 2959–2967 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |