
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkaloids from the flower of Erythrina arborescens†
Jing Wuab,
Bing-Jie Zhanga,
Wen-Na Xiaoa,
Mei-Fen Baoa and
Xiang-Hai Cai *a
*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, People's Republic of China. E-mail: xhcai@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 3rd November 2017
Abstract
Phytochemical investigations on the flower of Erythrina arborescens resulted in the isolation of eight new Erythrina alkaloid, erytharborines A–H (1–8), together with 17 known alkaloids. Erytharborines A/B (1–2) and C (3) possessed an 2H-imidazole ring and a unique oxime moiety, respectively. The structures were elucidated on the basis of UV, IR, mass spectrometry and NMR spectroscopic data.
Introduction
The Erythrina and Homoerythrina-type alkaloids, derived from two tyrosine units via oxidative coupling and intramolecular rearrangement, consist of more than 200 alkaloids from Erythrina and Cephalotaxus genus.1,2 The erythrinan alkaloids are ubiquitous compounds in the Erythrina genus of family Leguminosae. Special attention has been received in this field mainly by their curare-like neuro muscular blocking activities,3 anxiolytic-like activity,4 induced sleep,5 anticonvulsant activity,6 anticataract7 and antifeedant8 activity etc. Particularly noteworthy, the star molecule, dihydro-β-erythroidine, was used as tool to characterize neuronal nicotinic acetyl-choline receptors.9 Thus, pharmaceutical chemists paid much more attention to this type natural products. The erythrinan alkaloids possessed 6/5/6/6 spirocycle systems with a stable 5S-chiral center, seemingly exhibiting a not so diverse and fascinating molecular architecture. Nevertheless, the spirocyclic and aromatic skeleton in erythrinan alkaloids became challenging polycyclic molecular architectures.10–12 Generally speaking, skeleton rearrangement served as the main pathway to structural diversity of natural products. Analogously, besides aromatic erythrinan alkaloid (erysotramidine13), this class compound also included nonaromatic alkaloid, e.g. six-membered lactone (β-erythroidine14) and pyridine ring D (erymelanthine15). Both molecules attracted many interests in total synthesis.1,16,17 Under considerable efforts of our research group devoted to the phytochemical investigations on Erythrina species, several novel dimeric and trimeric erythrinan alkaloids some of which showed cytotoxicity were obtained.18,19 As part of an ongoing research for structural newly erythrinan alkaloids, phytochemical investigation of the flowers of Erythrina arborescens Roxb. led to eight new alkaloids erytharborines A–H (1–8) (Fig. 1) together with seventeen known alkaloids. Their isolation and structure elucidation were described in this study.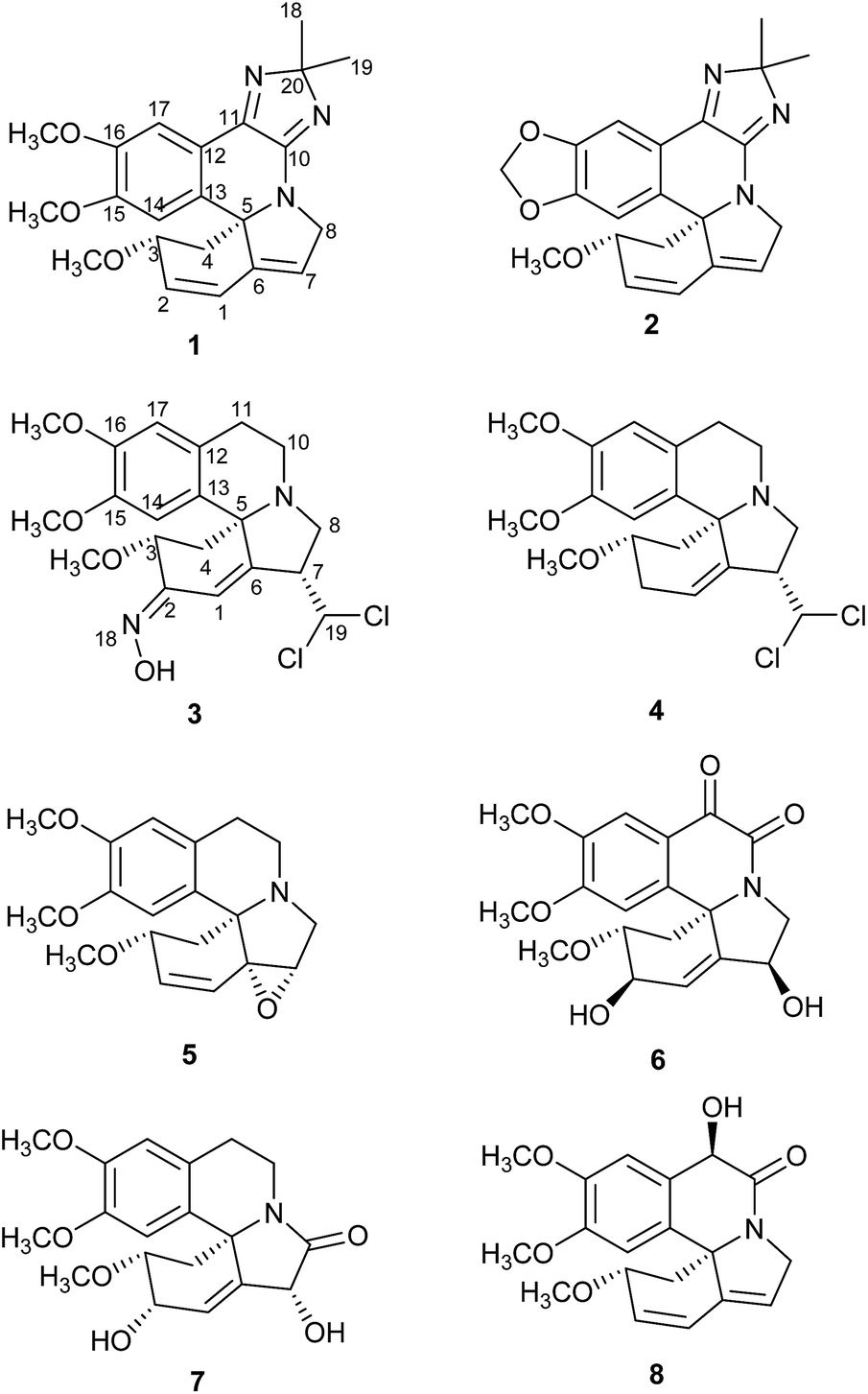 | ||
| Fig. 1 Structures of erytharborines A–H (1–8). | ||
Results and discussion
The alkaloid fraction of E. arborescens was separated to yield a total of 25 compounds by a combination of chromatographic procedures as described in the Experimental section. All compounds might be alkaloids since they showed positive response with Dragendorff's reagent on TLC.The UV absorptions (202, 227, 289 and 322 nm) and IR spectrum (1710, 1629, 1479 cm−1) of erytharborine A (1) indicated a good conjugated system. Presence of the typical conjugate olefin signals (δH 6.81, 6.04, 5.96), two aromatic singlet protons (δH 7.57 and 7.27) and three methoxyl groups (δH 3.90, 3.81 and 3.20) in the 1H NMR spectrum of 1, displayed the untapped A, B and D-rings of conjugated dienoid type erythrinan alkaloids. Two characteristic methylenes at δC 48.7 and 56.9 in the 13C NMR spectrum together with their HMBC correlations assigned themselves to C-4 and C-8, respectively. The untapped A, B and D-rings of 1 was further supported by its key correlations observed in the HMBC spectrum, δH 6.81 (H-1)/δC 76.8 (C-3) and 71.5 (C-5), δH 6.04 (H-2)/δC 48.7 (C-4), 140.4 (C-6), δH 5.96 (H-7)/δC 125.7 (C-1) and 71.5 (C-5), δH 7.27 (H-14)/δC 71.5 (C-5), 119.6 (C-12) and 149.9 (C-16), δH 7.57 (H-17)/δC 136.9 (C-13) and 152.4 (C-15) (Fig. 2). Its molecular formula C22H25N3O3 was deduced from HRESIMS at m/z = 380.1961 [M + H]+ (calcd. 380.1969), with three more carbons including two methyl groups (δC 25.0, 25.9) than general Erythrina alkaloid. In the HMBC spectrum, the correlations between H-17 and δC 155.4 (s) attributing the latter signal to C-11. Likewise, the correlations between H-8 (δH 4.56, 4.25) with δC 157.4 (s) attributing the latter signal to C-10. The HMBC correlations of δH 1.46 (3H) and 1.39 (3H) with δC 104.3 (s) established the linkage of the three carbons. Based on the molecular formula, 2H-imidazole ring was necessary in consideration of remainder unsaturation degrees of 1 (Fig. 2). In the ROESY spectrum, the NOE correlation of H-3/H-14 suggested H-3 was in β-orientation.
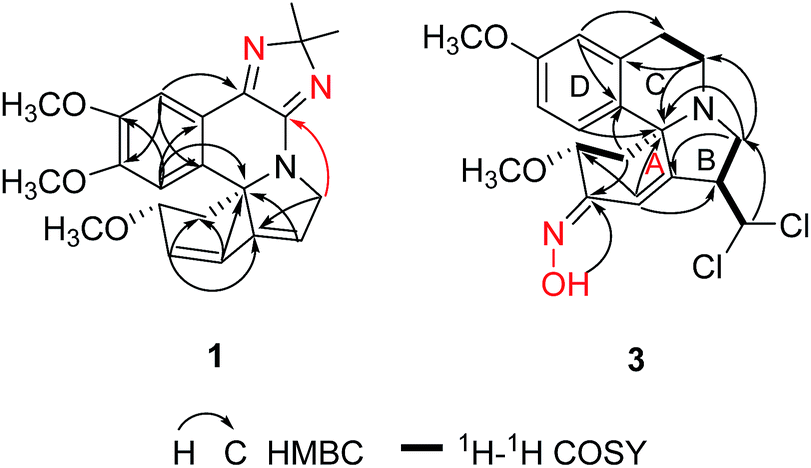 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of 1 and 3. | ||
Erytharborine B (2) was obtained as pale yellow amorphous powder with similar UV and IR absorption to 1. Its molecular formula was confirmed to be C21H21N3O3 by HRESIMS at m/z = 364.1658 [M + H]+ (calcd. 364.1656), with 14 daltons more than 1. Comparing their closely resembled 1H and 13C NMR data value (Table 1), compound 2 must possess a methylenedioxyl group (δH 6.12 and 6.09) at C-15 and C-16 in place with the two methoxyl groups (δH 3.90 and 3.81) in 1.
| Entry | δC (1) | δC (2) | δC (3)a | δC (4)a | δC (5)b | δC (6)a | δC (7)a | δC (8)b |
|---|---|---|---|---|---|---|---|---|
| a 13C NMR recorded in 150 MHz.b 13C NMR recorded in 125 MHz. Compound 3 was recorded in DMSO-d6. | ||||||||
| 1 | 125.7 d | 125.6 d | 114.5 d | 125.4 d | 128.6 d | 125.4 d | 123.8 d | 124.8 d |
| 2 | 132.6 d | 132.6 d | 151.6 s | 33.1 t | 136.9 d | 72.5 d | 62.8 d | 132.8 d |
| 3 | 76.8 d | 76.6 d | 73.4 d | 74.1 d | 76.5 d | 82.0 d | 76.9 d | 77.1 d |
| 4 | 48.7 t | 48.6 t | 42.9 t | 43.0 d | 46.5 t | 49.3 t | 35.6 t | 40.5 t |
| 5 | 71.5 s | 71.2 s | 63.7 s | 64.5 s | 66.1 s | 65.2 s | 62.3 s | 72.3 s |
| 6 | 140.4 s | 140.1 s | 150.1 s | 140.5 s | 71.2 s | 144.8 s | 140.7 s | 139.7 s |
| 7 | 120.9 d | 121.4 d | 51.3 d | 53.5 d | 64.6 d | 69.2 d | 70.7 d | 120.8 d |
| 8 | 56.9 t | 56.6 t | 49.9 t | 53.2 t | 56.4 t | 51.2 t | 172.6 s | 54.7 t |
| 10 | 157.5 s | 157.1 s | 39.8 t | 40.8 t | 51.6 t | 159.4 s | 35.8 t | 173.8 s |
| 11 | 155.4 s | 155.6 s | 20.6 t | 21.7 t | 29.4 t | 180.7 s | 25.9 t | 68.3 d |
| 12 | 119.6 s | 121.4 s | 125.5 s | 130.2 s | 130.9 s | 125.4 s | 126.8 s | 129.4 s |
| 13 | 136.9 s | 138.7 s | 126.3 s | 126.4 s | 131.2 s | 140.0 s | 131.9 s | 131.0 s |
| 14 | 108.2 d | 105.3 d | 110.9 d | 112.7 d | 110.1 d | 109.4 d | 110.3 d | 108.4 d |
| 15 | 152.4 s | 151.5 s | 147.8 s | 149.5 s | 148.0 s | 154.2 s | 147.9 s | 148.7 s |
| 16 | 149.9 s | 148.5 s | 146.1 s | 147.7 s | 148.8 s | 150.3 s | 149.9 s | 149.9 s |
| 17 | 109.9 d | 107.2 d | 112.7 d | 113.6 d | 112.7 d | 110.5 d | 114.3 d | 109.3 d |
| 18 | 25.0 q | 25.7 q | ||||||
| 19 | 25.9 q | 26.9 q | 74.3 d | |||||
| 20 | 104.3 s | 100.5 s | ||||||
| 3-OCH3 | 56.2 q | 56.6 q | 58, 5 q | 55.9 q | 56.0 q | 56.2 q | 56.0 q | 56.1 q |
| 15-OCH3 | 56.1 q | 55.4 q | 56.1 q | 56.3 q | 56.6 q | 56.2 q | 56.3 q | |
| 16-OCH3 | 56.1 q | 55, 3 q | 56.1 q | 56.4 q | 56.6 q | 56.3 q | 56.3 q | |
| OCH2O | 103.0 t | |||||||
The UV absorption of erytharborine C (3) at 204 and 289 nm indicated a tetrahydroisoquinoline chromophore.20 Meanwhile, its IR absorption bands at 3414 and 1611, 1513, and 1458 cm−1 resulted from the hydroxyls and aromatic rings, which was consistent with the characteristic of Erythrina alkaloid. Its molecular formula was determined to be C20H24N2O4Cl2 based on HRESIMS at m/z = 427.1197 [M + H]+, indicating nine degrees of unsaturation. The isotope peaks showed in the positive ESI-MS confirmed the presence of two chlorine atoms. The 1H-, 13C NMR and HSQC data for 3 indicated the presence of four methylenes, three methoxyls, three sp3 and three sp2 methines, one sp3 and six sp2 quaternary carbons. Above data further suggested 3 was similar to erthratidinone21 except for an additional carbon and nitrogen, and two chlorine atoms. In the HMBC spectrum, correlations from δH 6.71 (H-17) to δC 20.6 (C-11), δC 126.3 (C-13), and δC 147.8 (C-15), and from δH 6.28 (H-14) to δC 63.7 (C-5), δC 125.5 (C-12), and δC 146.1 (C-16) suggested D-ring was not changed.20 Its 1H–1H COSY correlations of H-11 (δH 2.98 and 2.49) to H-10 (δH 3.09 and 3.30), together with the correlation of H-10 with C-5 in the HMBC spectrum indicated C-ring was untapped. The coupling –CH2 (δH 2.94 and 3.18) and –CH (δH 3.66) correlated with C-6 in the HMBC spectrum, respectively, also assigned them to H-8 and H-7 and suggested ring-B was substituted. The methine δH 5.86 was attributed to newly CH-19 based on its 1H–1H COSY correlation with H-7, which was supported by the HMBC correlations from H-19 to C-8. Downfield proton and carbon signal of CH-19 meant linkage with two Cl atoms, also consideration of its molecular formula. Finally, singlet signal H-1 (δH 7.07) showed HMBC correlations with C-7 and C-5 suggested a double bond at C-1/6. Correlations of H-3 (δH 4.14)/H-4 (δH 2.71 and 1.68) in the 1H–1H COSY spectrum together with the HMBC correlations between H-4 with δC 151.6 assigned the signal to C-2. The remainder of a nitrogen atom and degree of unsaturation suggested there should be an E-oxime moiety as shown in Fig. 2,22 which was supported by HMBC correlation of δH 11.20 (OH) with C-2.
The molecular formula C20H25NO3Cl2 of alkaloid (4) was established by HRESIMS ([M + H]+ at m/z 398.1281) and was consisted with the 13C NMR spectrum, which revealed 20 carbonic resonance signal. The 1D NMR spectroscopic data of compound 4 were similar to those of compound 3 except for the following differentiations: in the 1H NMR spectrum, the signal displayed at δH 11.20 in 3 which was assigned to the active hydrogen in the oxime moiety was disappeared in compound 4. Correspondingly, the quaternary carbon signals at δC 151.6 (C-2) in compound 3 was replaced with a methylene (δC 33.1) in compound 4. Thus, compound 4 might be an analogue of 3 without the oxime moiety. The HMBC correlations of δH 2.05 (H-2)/δC 125.4 (C-1), δC 74.1 (C-3) and δC 140.5 (C-6) together with the HSQC data demonstrated directly that the methylene did belong to C-2. Relative configuration of H-3 in 3 and 4 was deduced as β from the coupling constants (J3,4eq. = 5.0 Hz, J3,4ax = 11.0 Hz) in the 1H NMR spectrum.23 This presumption was confirmed by the obvious NOE correlations of H-3/H-14. Likewise, the correlations of H-7/H-17 in the ROESY spectrum showed H-7 in 3 and 4 was β-oriented, too. The oxime of C2/N18 in 3 was determined as E via NOE between OH and H-1.
Erytharborine E (5) was isolated as an amorphous solid. Its molecular formula was deduced as C19H23N2O5 from the HRESIMS ([M + H]+ at 330.1699) and 13C NMR spectroscopic data, inferring nine degrees of unsaturation. In comparing with the 13C NMR data of erysotrine,13 compound 5 showed a oxygenated quaternary carbon (δC 71.2) and a oxygenated methine (δC 64.6) at up-field instead of olefinic signals of δC 143.4 (s, C-6) and δC 123.6 (d, C-7) of erysotrine, which suggested presence of an epoxide ring at C-6/7. The HMBC correlations of δH 2.87 (H-8)/δC 64.6 (C-7), δC 71.2 (C-6), δH 5.76 (H-1)/δC 64.6 (C-7) and δH 6.25 (H-2)/δC 71.2 (C-6) confirmed this conclusion. The epoxide was assigned as β-orientation on the base of molecule model.
Erytharborine F (6) was obtained as a white amorphous powder. Its molecular formula was determined to be C19H21NO7 based on its HRESIMS at m/z 398.1212 ([M + Na]+) and NMR spectra. The 1H NMR spectra (Table 2) confirmed the presence of two aromatic singlet protons (δH 7.36 and 7.18), one olefinic proton (δH 6.18) and three methoxyls (δH 3.93, 3.90 and 3.20). Its 1H- and 13C NMR data resembled those of (+)-10,11-dioxoepierythratidine24 with exception for an additional hydroxyl group, which was deduced from its molecular formula. Substitution of hydroxyl group at C-7 was supported by the HMBC correlations of δH 6.18 (H-1)/δC 69.2 (C-7), δH 4.31 (H-8)/δC 69.2 (C-7) and δH 4.98 (H-7)/δC 144.8 (C-6). The signals at δH 7.36 (H-17) showed correlation with the δC 180.7 (C-11) in the HMBC spectrum, while the signals at δH 4.31 (H-8) showed correlation with the δC 154.9, establishing dione at C-10/11. The hydroxyl groups at C-2 and C-7 were both β-oriented as deduced from the NOESY correlations of H-2/H-4ax, H-8ax/H-4ax, H-7/H-8ax.
| Entry | δH (1)a | δH (2)a | δH (3)a | δH (4)a | δH (5)b | δH (6)a | δH (7)a | δH (8)b |
|---|---|---|---|---|---|---|---|---|
| a 1H NMR recorded in 600 MHz.b 1H NMR recorded in 400 MHz; compound 3 was recorded in DMSO-d6. | ||||||||
| 1 | 6.81 (dd, 10.2, 2.4) | 6.78 (dd, 10.3, 2.4) | 7.07 (s) | 5.97 (t, 3.7) | 5.76 (brd, 10.4) | 6.18 (d, 4.9) | 6.20 (br, s) | 6.75 (br, d, 10.3) |
| 2 | 6.04 (d, 10.2) | 6.01 (d, 10.2) | 2.88 (overlap), 2.05 (overlap) | 6.25 (brd, 10.4) | 4.38 (dd, 4.9, 4.2) | 4.60 (dd, 4.3, 3.2) | 6.04 (d, 10.3) | |
| 3 | 3.76 (m) | 3.72 (m) | 4.14 (m) | 3.83 (m) | 3.79 (m) | 3.41 (dd, 12.0, 5.0) | 3.63 (dt, 11.9, 3.2) | 3.72 (dd, 11.5, 5.3) |
| 4 | 2.21 (dd, 11.3, 5.2), 1.95 (dd, 11.3, 10.2) | 2.21 (dd, 11.3, 5.3), 1.95 (d, 11.3) | 2.71 (dd, 11.0, 5.0) 1.68 (t, 11.0) | 2.29 (dd, 11.0, 5.0), 1.44 (t, 11.0) | 2.15 (dd, 12.6, 5.0), 1.94 (dd, 12.6, 10.0) | 2.16 (t, 12.0), 2.08 (dd, 12.0, 5.0) | 2.13 (dd, 11.9, 3.2), 1.94 (t, 11.9) | 2.76 (dd, 11.5, 5.3), 1.86 (t, 11.5) |
| 7 | 5.96 (d, 2.4) | 5.97 (d, 2.8) | 3.66 (dd, 7.0, 3.5) | 3.27 (m) | 3.61 (overlap) | 4.69 (dd, 7.8, 6.0) | 4.36 (d, 6.0) | 5.86 (s) |
| 8 | 4.56 (dd, 15.8, 2.4), 4.25 (d, 15.8) | 4.55 (dd, 15.8, 2.8), 4.24 (d, 15.8) | 2.94 (dd, 10.0, 3.5), 3.18 (dd, 10.0, 7.0) | 3.20 (overlap), 2.60 (dd, 9.9, 6.7) | 3.61 (overlap), 2.89 (d, 12.5) | 4.31 (dd, 10.2, 7.8), 3.10 (t, 10.2) | 4.31 (2H, br, s) | |
| 10 | 3.09 (m), 3.30 (overlap) | 3.44 (m), 3.09 (m) | 3.13 (m), 2.44 (m) | 4.03 (m), 3.47 (m) | ||||
| 11 | 2.98 (m), 2.49 (m) | 3.01 (m), 2.54 (m) | 2.73 (m), 2.60 (m) | 3.02 (2H, overlap) | 5.38 (s) | |||
| 14 | 7.27 (s) | 7.15 (s) | 6.28 (s) | 6.55 (s) | 7.12 (s) | 7.18 (s) | 6.33 (s) | 7.02 (s) |
| 17 | 7.57 (s) | 7.49 (s) | 6.71 (s) | 6.71 (s) | 6.78 (s) | 7.36 (s) | 6.81 (s) | 7.23 (s) |
| 18 | 1.46 (3H, s) | 1.45 (3H, s) | ||||||
| 19 | 1.39 (3H, s) | 1.38 (3H, s) | 5.86 (d, 6.0) | |||||
| 3-OCH3 | 3.20 (3H, s) | 3.21 (3H, s) | 3.27 (3H, s) | 3.22 (s) | 3.23 (3H, s) | 3.20 (3H, s) | 3.29 (3H, s) | 3.24 (3H, s) |
| 15-OCH3 | 3.81 (3H, s) | 3.65 (3H, s) | 3.77 (s) | 3.79 (3H, s) | 3.93 (3H, s) | 3.81 (3H, s) | 3.84 (3H, s) | |
| 16-OCH3 | 3.90 (3H, s) | 3.75 (3H, s) | 3.72 (s) | 3.71 (3H, s) | 3.90 (3H, s) | 3.79 (3H, s) | 3.72 (3H, s) | |
| OCH2O | 6.12 (br, s), 6.09 (br, s) | |||||||
| 2-OH | 4.84 (d, 4.2) | 3.59 (d, 4.3) | ||||||
| 7-OH | 4.98 (d, 6.0) | 4.96 (d, 6.0) | ||||||
| 11/18-OH | 11.20 (s) | 4.43 (s) | ||||||
Erytharborine G (7), a white amorphous powder, had the molecular formula C19H23NO6 as deduced from its HRESIMS ([M + Na]+ at m/z 384.1417) and NMR spectra. The pattern of 13C NMR data for 7 were similar to those of 6 except that the former contained only one carbonyl group in low-field. In the HMBC spectrum, correlations of δH 6.81 (H-17)/δC 25.9 (C-11, t) and δH 3.00 (H-11)/δC 35.8 (C-10, t) indicated that the carbonyl group was neither located at C-10 nor at C-11. The HMBC correlation between δH 4.36 (H-7) and δC 172.6 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) assigned the carbonyl group to C-8 position. Its ROESY spectrum gave correlations of H-3/H-14, H-2/H-14 and H-7/H-14, which demonstrated the relative configuration of H-2, H-3 and H-7 were β-oriented.
O) assigned the carbonyl group to C-8 position. Its ROESY spectrum gave correlations of H-3/H-14, H-2/H-14 and H-7/H-14, which demonstrated the relative configuration of H-2, H-3 and H-7 were β-oriented.
The molecular formula erytharborine H (8) was established as C19H21NO5 based on the HRESIMS ([M + Na]+ at 366.1310) and 13C NMR spectroscopic data. The 1H NMR spectrum showed the presence of two aromatic singlet protons (δH 7.23 and 7.02) and three conjugated olefinic protons (δH 6.75, 6.04, and 5.86), which were the characteristic signals to Erythrina alkaloid with a 1/2,6/7-diene system. When compared with 10-hydroxy-11-oxoerysotrine,25 compound 8 showed great similarity in 1H and 13C NMR data. In the HMBC spectra, correlations between H-8 (δH 4.31) and δC 173.8 (CO) attributed the carbonyl to C-10 position other than C-11. Likewise, the hydroxyl group was determined to be attached at C-11 by HMBC correlations of δH 7.23 (H-17) to δC 68.3 (C-11). Therefore, compound 8 was defined as 10-oxo-11-hydroxyerysotrine. On the basis of the ROESY experiment, a correlation of H-3/H-4 eq. and H-4 eq./H-11 assigned the 11-OH group as being β-oriented.
The positive optical rotation value of 1–8 suggested that they had same configuration at C-5.24,26 As the main constituent, alkaloid 23 showed same optical rotation ([α]23D + 206 (c = 0.36, CH3OH)) as previous reported erythrinine.16 So 1–8 should possess identical 5s-configuration, and named as erytharborines A-H, respectively. Additionally, all reported Erythrina and Homoerythrina-type alkaloids have this configuration so far.
The known alkaloids were identified as, erytharbine (9),25 8-oxoerthraline epoxide (10),27 erythratidinone (11),21 erythratine (12),28 erysotramidine (13),13 10,11-dioxoerysotrine (14),29 11β-hydroxyerysotramidine (15),30 erythratine (16),31 erythrartine N-oxide (17),31 erysotrine (18),13 8-oxoerythrinine (19),32 8-oxoerythraline (20),13 erythraline (21),13 erythraline N-oxide (22),33 erythrinine (23),20 erysovine (24),21 erysodine (25)34 on the basis of physical and spectrospopic comparison with published values.
Conclusions
To summary, twenty five erythrinan alkaloids were isolated from the flowers of E. arborescens Roxb. and among them eight novel ones, erytharborines A–H (1–8) have been elucidated. Alkaloids 1 and 2 were the first found erythrinan alkaloids with 2H-imidazole ring. In addition, 3 was an alkaloid containing an oxime group. Other alkaloids (9–25) were first obtained from E. Arborescens. The discovery of compounds 1–8 is a further addition to the diverse of alkaloids belonging to the Erythrina genus.Experimental section
General experimental procedures
Optical rotations were measured with a Jasco p-1020 digital polarimeter. UV spectra were recorded on a Shimadzu 2401PC spectrophotometer. IR spectra were obtained on a Bruker Tensor 27 infrared spectrophotometer with KBr pellets. 1H, 13C and 2D NMR spectra were obtained on Bruker AV-600, AVANCE III-500, and AVANCE III-400 MHz spectrometers with SiMe4 as an internal standard. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals. ESI and HRESIMS data were recorded on a Bruker HCT/Esquire and a Shimadzu UPLC-IT-TOF spectrometer, respectively. Column chromatography (CC) was performed on either silica gel (200–300 mesh, Qingdao Marine Chemical Co., Ltd., Qingdao, China) or RP-18 silica gel (20–45 μm, YMC Chemical Ltd., Japan). Fractions were monitored by TLC on silica gel plates (GF254, Qingdao Marine Chemical Co., Ltd., Qingdao, China), and spots were visualized with Dragendorff's reagent spray. MPLC was performed using a Buchi pump system coupled with RP-18 silica gel-packed glass columns (15 × 230 and 26 × 460 mm, respectively). HPLC was performed using Waters 1525EF pumps coupled with analytical semi-preparative or preparative Sunfire C18 columns (4.6 × 150 and 19 × 250 mm, respectively). The HPLC system employed a Waters 2998 photodiode array detector and a Waters fraction collector III.Plant material
Flowers of Erythrina arborescens Roxb. Hort. Beng were collected in September 2014 in Yunnan Province, P. R. China, and identified by Dr Chun-Xia Zeng. A voucher specimen (no. Cai20140907) was deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.Extraction and isolation
The dried flowers of E. Arborescens (6.5 kg) were powdered and extracted three times with MeOH at room temperature. After removing the solvent, the residue was dissolved in 2% HCl soln and filtered. The acidic soln was washed with EtOAc three times. The aqueous layer was then adjusted to pH 8–9 with NH3·H2O and extracted with EtOAc to obtain crude alkaloid extract (62.5 g). The extract was subjected to column chromatography (CC) over silica gel and eluted with gradient CHCl3/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–5:1) to afford seven fractions (I–VII).
:0–5:1) to afford seven fractions (I–VII).
Fraction II (10.4 g) was further chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (40:60–100:0, v/v) to give the five subfractions II-1–II-5. Subfraction II-2 (2.5 g) was subjected to C18 MPLC column once again using MeOH–H2O (40:60–70:30, v/v) as eluent to give the four subfractions (II-2-1–II-2-4). Fraction II-2-1 was further purified by a preparative column with a gradient flow from 40% to 55% aqueous methanol to give 19 (7 mg), 8 (50 mg), 5 (5 mg). Fraction II-2-2 was separated on a preparative C18 HPCL column with a gradient of MeOH–H2O (45:55–55:45, v/v) to afford 13 (4 mg) and 14 (7 mg). Fraction II-2-4 was purified by a preparative C18 HPCL column with a gradient of MeOH–H2O (50:50–65:35, v/v) to obtain 11 (20 mg) and 15 (10 mg). II-4 (1.7 g) was separated using C18 MPLC column with a gradient of MeOH–H2O (30:70–60:40, v/v) to afford five subfractions (II-4-1–II-4-5). Alkaloid 21 (500 mg) was crystallized from II-4-2. Fraction II-4-3 was purified by a preparative C18 HPCL column with a gradient of MeOH–H2O (35:65–45:55, v/v) to obtain 22 (5 mg). II-4-5 was purified by a preparative C18 HPCL column with a gradient of MeOH–H2O (50:50–60:40, v/v) to obtain 20 (5 mg). Compounds 1 (2 mg), 2 (2 mg), 3 (1.6 mg), 4 (1 mg), 9 (3 mg), 10 (2 mg) and 18 (7 mg) were obtained from fraction II-4-4 using C18 MPLC column with a gradient of MeOH–H2O (40:60–70:30, v/v), then followed by preparative HPLC with a gradient of MeOH–H2O (40:60–60:40, v/v).
Fraction III (0.9 g) was fractionated by C18 MPCL column with a gradient of MeOH–H2O (30:70–80:20, v/v) to give four subfractions (III-1–III-4). III-1 was subjected to a preparative C18 HPCL column with a gradient of MeCN–H2O (30:70–40:60, v/v) to afford 24 (20 mg). III-3 was further purified by a preparative C18 HPCL column with a gradient of MeCN–H2O (30:70–45:55, v/v) to afford 25 (18 mg).
Alkaloid 23 (1.5 g) was crystalized from fraction IV. The mother liquid of this fraction (3.0 g) was subjected to C18 MPCL column with a gradient of MeOH–H2O (20:80–70:30, v/v) to give four subfractions (IV-1–IV-4). IV-2 was separated on a preparative C18 HPCL column with a gradient of MeCN–H2O (20:80–35:65, v/v) to afford 16 (12 mg), 17 (7 mg).
Fraction V (1.6 g) was chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (20:80–60:40, v/v) to give five subfractions V-1–V-5. V-1 (910 mg) was subjected a C18 MPLC column once again with a gradient of MeOH–H2O (10:90–40:60, v/v) to give eight subfractions V-1-1–V-1-8. Compound 6 (2 mg) and 7 (2 mg) was obtained from V-1-4 using a preparative C18 HPCL column with a gradient of MeOH–H2O (30:70–45:55, v/v). Compound 12 (2 mg) was obtained from V-1-6 using a preparative C18 HPCL column with a gradient of MeOH–H2O (40:60–50:50, v/v).
Erytharborine A (1)
Pale yellow amorphous powder; [α]25D + 119.2 (c 0.1, MeOH); UV (MeOH) λmax (logε) 202 (4.03), 227 (3, 79), 289 (3.55), 322 (3.48) nm; IR (KBr) νmax 2927, 1710, 1629, 1479, 1383, 1252 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 1 and 2; positive HRESIMS m/z 380.1961 [M + H]+ (calcd. For C22H26N3O3, 380.1969).
Erytharborine B (2)
Pale yellow amorphous powder; [α]25D + 377.3 (c 0.1, MeOH); UV (MeOH) λmax (logε) 201 (3.99), 230 (3.80), 288 (3.59), 327 (3.50), nm; IR (KBr) νmax 3429, 2930, 1722, 1633, 1594, 1508, 1479, 1392, 1252 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 1 and 2; positive HRESIMS m/z 364.1658 [M + H]+ (calcd. For C21H22N3O3, 364.1656).
Erytharborine C (3)
White powder; [α]23D + 112.2 (c = 0.25, CH3OH); UV (CH3OH) λmax (logε) 204 (4.22) and 289 (3.46) nm; IR (KBr) νmax 3414, 2931, 1611, 1513, 1458, and 1256 cm−1; for 1H (600 Hz) and 13C (150 Hz) NMR data (DMSO-d6), see Tables 1 and 2; positive ESIMS m/z 427 [M + H]+, HRESIMS m/z 427.1197 [M + H]+ (calcd. For C20H25N2O4Cl2, 427.1191).
Erytharborine D (4)
White powder; [α]22D + 63.6 (c = 0.14, CH3OH); UV (CH3OH) λmax (logε) 206 (3.68), 232 (3.08) and 283 (2.69) nm; 1H (600 Hz) and 13C (150 Hz) NMR data (acetone-d6), see Tables 1 and 2; positive ESIMS m/z 398 [M + H]+, HRESIMS m/z 398.1281 [M + H]+ (calcd. For C20H26NO3Cl2, 398.1284).
Erytharborine E (5)
Colorless oil; [α]22D + 179.8 (c = 0.19, CH3OH); UV (CH3OH) λmax (logε) 203 (3.93), 223 (3.42) and 283 (2.95) nm; 1H (600 Hz) and 13C (150 Hz) NMR data (acetone-d6), Tables 1 and 2; positive ESIMS m/z 330 [M + H]+, HRESIMS m/z 330.1699 [M + H]+ (calcd. For C19H24NO7, 330.1700).
Erytharborine F (6)
White powder; [α]22D + 111.5 (c = 0.18, CH3OH); UV (CH3OH) λmax (logε) 203 (3.63), 248 (3.33), 289 (3.13) and 352 (2.94) nm; 1H (600 Hz) and 13C (150 Hz) NMR data (acetone-d6), Tables 1 and 2; positive ESIMS m/z 398 [M + Na]+, HRESIMS m/z. 398.1212 [M + Na]+ (calcd. For C19H21NO7Na, 398.1210).
Erytharborine G (7)
White powder; [α]22D + 311.1 (c = 0.05, CH3OH); UV (CH3OH) λmax (logε) 204 (4.33), 225 (3.80), and 283 (3.29) nm; 1H (600 Hz) and 13C (150 Hz) NMR data (acetone-d6), Tables 1 and 2; positive ESIMS m/z 384 [M + Na]+, HRESIMS m/z. 384.1417 [M + Na]+ (calcd. For C19H23NO6Na, 384.1418).
Erytharborine H (8)
Colorless oil; [α]22D + 161.1 (c = 0.25, CH3OH); UV (CH3OH) λmax (logε) 204 (3.91), 241 (3.47) and 283 (2.88) nm; 1H (400 Hz) and 13C (125 Hz) NMR data (acetone-d6), Tables 1 and 2; positive ESIMS m/z 366 [M + Na]+, HRESIMS m/z 366.1310 [M + Na]+ (calcd. For C19H21NO5Na, 366.1312).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is supported in part by the National Natural Science Foundation of China (31370377). The authors are grateful to Dr Chun-Xia Zeng for identification of plant samples.Notes and references
- Y. He and R. L. Funk, Org. Lett., 2006, 8, 3689–3692 CrossRef CAS PubMed.
- H. Abdelkafi and B. Nay, Nat. Prod. Rep., 2012, 29, 845–869 RSC.
- K. Folkers and K. Unna, J. Am. Chem. Soc., 1939, 61, 370–379 CAS.
- O. Flausino Jr, L. d. A. Santos, H. Verli, A. M. Pereira, V. d. S. Bolzani and R. L. Nunes-de-Souza, J. Nat. Prod., 2007, 70, 48–53 CrossRef PubMed.
- M. Ozawa, K. Honda, I. Nakai, A. Kishida and A. Ohsaki, Bioorg. Med. Chem. Lett., 2008, 18, 3992–3994 CrossRef CAS PubMed.
- S. A. Faggion, A. O. Siqueira Cunha, H. A. Fachim, A. S. Gavin, W. F. dos Santos, A. M. Soares Pereira and R. O. Beleboni, Epilepsy Behav., 2011, 20, 441–446 CrossRef PubMed.
- M. Umamaheswari, K. Asokkumar, A. T. Sivashanmugam, V. Subhadradevi and M. Neethu, Bangladesh Journal of Pharmacology, 2010, 5, 77–81 CrossRef.
- W. W. Cornelius, T. Akeng'a, G. O. Obiero and K. P. Lutta, Rec. Nat. Prod., 2009, 3, 96–103 CAS.
- S. Cheeta, S. Tucci and S. E. File, Pharmacol., Biochem. Behav., 2001, 70, 491–496 CrossRef CAS.
- D. Kalaitzakis, T. Montagnon, E. Antonatou and G. Vassilikogiannakis, Org. Lett., 2013, 15, 3714–3717 CrossRef CAS PubMed.
- L. F. Tietze, N. Toelle, D. Kratzert and D. Stalke, Org. Lett., 2009, 11, 5230–5233 CrossRef CAS PubMed.
- H. Umihara, T. Yoshino, J. Shimokawa, M. Kitamura and T. Fukuyama, Angew. Chem., Int. Ed., 2016, 55, 6915–6918 CrossRef CAS PubMed.
- A. S. Chawla, S. Chunchatprasert and A. H. Jackson, Org. Magn. Reson., 1983, 21, 39–41 CrossRef CAS.
- A. W. Hanson, Acta Crystallogr., 1963, 16, 939–942 CrossRef CAS.
- E. Dagne and W. Steglich, Tetrahedron Lett., 1983, 24, 5067–5070 CrossRef CAS.
- H. Fukumoto, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2006, 45, 2731–2734 CrossRef CAS PubMed.
- M. Paladino, J. Zaifman and M. A. Ciufolini, Org. Lett., 2015, 17, 3422–3425 CrossRef CAS PubMed.
- B.-J. Zhang, M.-F. Bao, C.-X. Zeng, X.-H. Zhong, L. Ni, Y. Zeng and X.-H. Cai, Org. Lett., 2014, 16, 6400–6403 CrossRef CAS PubMed.
- B.-J. Zhang, B. Wu, M.-F. Bao, L. Nia and X.-H. Cai, RSC Adv., 2016, 6, 87863–87868 RSC.
- K. Ito, H. Furukawa and H. Tanaka, J. Chem. Soc., Chem. Commun., 1970, 17, 1076–1077 RSC.
- D. R. Callejon, T. B. Riul, L. G. P. Feitosa, T. Guaratini, D. B. Silva, A. Adhikari, R. L. Shrestha, L. M. M. Marques, M. D. Baruffi, J. L. C. Lopes and N. P. Lopes, Molecules, 2014, 19, 5692–5703 CrossRef PubMed.
- M. Bacher, G. Brader, O. Hofer and H. Greger, Phytochemistry, 1999, 50, 991–994 CrossRef CAS.
- T. Sano, J. Toda and Y. Tsuda, Heterocycles, 1982, 18, 229–232 CrossRef CAS.
- T. Rukachaisirikul, P. Innok and A. Suksamram, J. Nat. Prod., 2008, 71, 156–158 CrossRef CAS PubMed.
- H. Tanaka, H. Hattori, T. Tanaka, E. Sakai, N. Tanaka, A. Kulkarni and H. Etoh, J. Nat. Med., 2008, 62, 228–231 CrossRef CAS PubMed.
- M. E. Amer, S. Elmasry, M. Shamma and A. J. Freyer, J. Nat. Prod., 1991, 54, 161–166 CrossRef CAS.
- H. Tanaka, T. Tanaka, H. Etoh, S. Goto and Y. Terada, Heterocycles, 1999, 51, 2759–2764 CrossRef CAS.
- D. H. R. Barton, R. James, G. W. Kirby, D. W. Turner and D. A. Widdowson, J. Chem. Soc. C, 1968, 12, 1529–1537 RSC.
- C. C. W. Wanjala, B. F. Juma, C. Bojase, B. A. Gashe and R. R. T. Majinda, Planta Med., 2002, 68, 640–642 CrossRef CAS PubMed.
- B. F. Juma and R. R. T. Majinda, Phytochemistry, 2004, 65, 1397–1404 CrossRef CAS PubMed.
- M. H. Sarragiotto, H. Leitao and A. J. Marsaioli, Can. J. Chem., 1981, 59, 2771–2775 CrossRef CAS.
- E. Dagne and W. Steglich, Phytochemistry, 1984, 23, 449–451 CrossRef CAS.
- M. Ozawa, S. Kawamata, T. Etoh, M. Hayashi, K. Komiyama, A. Kishida, C. Kuroda and A. Ohsaki, Chem. Pharm. Bull., 2010, 58, 1119–1122 CrossRef CAS PubMed.
- M. E. Amer, S. Elmasry, M. Shamma and A. J. Freyer, J. Nat. Prod., 1991, 54, 161–166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10827c |
| This journal is © The Royal Society of Chemistry 2017 |